
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The increasing awareness of the adverse effects of exposure to UV radiation in combination with the conclusion that presently employed sunscreen agents are far from optimal has led to the need to develop novel UV filters with improved absorption and stability characteristics. Studies of natural sunscreens can provide fundamental insight into strategies to come to a rational design of such filters. Here, we use high-resolution laser spectroscopic methods to study the spectroscopy and excited-state dynamics of methyl sinapate, a prominent plant sunscreen derivative. We find that our experimental approach based on two-color Resonance Enhanced Two-Photon Ionisation spectroscopy enables us to observe a hitherto not observed decay pathway leading to a long-lived state, albeit with a significantly lower yield than in cinnamates and coumarates. In combination with extended excitation spectra and employing the results of quantum chemical calculations a comprehensive picture is obtained of the internal conversion pathways that are accessible to this compound. Similar high-resolution studies on clusters of methyl sinapate with water show how solvent-solute interactions affect the electronic structure and excited-state decay dynamics of the chromophore.
GRAPHICAL ABSTRACT
1. Introduction
While solar radiation is essential to all lifeforms on our planet, exposure is also potentially harmful to all forms of life. Adverse health effects in humans include various types of skin cancer, photoaging and photoallergy [Citation1–3]. Nature has developed a finely balanced inventory of photoactive molecules to protect us against overexposure, but today’s society has urged the need for further protection [Citation4–6]. A range of artificial sunscreens has therefore been developed and incorporated into skin care cosmetics [Citation7,Citation8]. Such sunscreen formulations are generally composed of inorganic scatters and several organic filters to provide a broad-band and efficient protection.
The primary purpose of the organic filters is to efficiently absorb harmful UV radiation. The absorption of UV photons is, however, only the first step in a complex cascade of follow-up processes in which the absorbed photon energy is dissipated from an electronically excited state into harmless vibrational energy in the electronic ground state. Ideally, this occurs with an efficiency of 100%, but in practice this ‘photon-to-heat’ pathway has to compete with several other dissipation pathways such as internal conversion to long-lived reactive states, isomerisation and dissociation. These pathways not only reduce the efficacy of the sunscreen but may also lead to harmful side effects [Citation9]. For a further -rational- development of filters [Citation10–12] it is therefore key not only to optimise their absorption characteristics, but especially to come to a fundamental understanding of their electronically excited-state dynamics [Citation13].
As yet, the interest in UV filters has by and large been guided by their sunscreen applications. Their final ‘product’, namely heat, has largely been ignored but has now started to be of interest for other applications as well. One of these applications is the development of ‘molecular heaters’ for agricultural purposes [Citation14]. These ‘molecular heaters’ are aimed to extend growth seasons and the geographical locations suitable for agriculture, increase crop yield at high crop density and, concomitantly, reduce greenhouse energy costs. Ideally, one would like to employ for this purpose nature-inspired molecules that absorb light at wavelengths that are either harmful to the plant or not used in photosynthesis, and convert this photon energy into heat.
An important class of compounds that are used both as natural as well as artificial UV-B protectants are based on cinnamic acid and its derivatives. Plants employ sinapate esters featuring methoxy substituents meta to the vinyl group and a para hydroxy group, while commercial sunscreens use alkyl-esters with only a p-methoxy substituent on the phenyl ring. Although these compounds have been known for a long time, high-resolution laser spectroscopic studies on 2-ethylhexyl-4-methoxycinnamate (EHMC) -the most common UV-B filter found in commercial sunscreens- led to the realisation that high-resolution studies in the frequency domain and time-resolved studies in the time domain can contribute significantly to the development of novel sunscreens [Citation15–17]. As a result, recent years have witnessed a rapidly increasing interest in the detailed understanding of the spectroscopic properties of the electronically excited states of these compounds and their dynamics, and how to employ this knowledge to design and develop UV filters with improved properties [Citation18–33].
Central to these properties is the manifold of the lower-lying electronically excited singlet states consisting of the so-called V(ππ*), V’(ππ*) and 1nπ* states [Citation34,Citation35]. The V(ππ*) state is associated with the HOMO → LUMO excitation and has a large oscillator strength while the V’(ππ*) state has a small oscillator strength and is described by contributions from the HOMO → LUMO+1 and HOMO-1 → LUMO excitations. The 1nπ* state is for all practical purposes forbidden and arises from excitation of a lone-pair electron on the carbonyl oxygen atom to the LUMO. These three states are energetically close by, leading to intricate potential energy surfaces and internal conversion pathways. For EHMC, for example, the 1nπ* state is vertically S3 but adiabatically becomes S1 [Citation15,Citation21,Citation26,Citation36]. As a result, excitation of the bright ππ* states does not lead to fast internal conversion to the ground state as one ideally would like to have. Instead, the dominant decay pathway is internal conversion to the 1nπ* state, giving rise to the observation of a decay on a time scale of tens of nanoseconds of an electronically excited state that has been identified by Ebata et al. as the lowest excited triplet state [Citation33,Citation37]. Such long-lived states are clearly detrimental to the purpose of these filters, and can give rise to adverse side reactions.
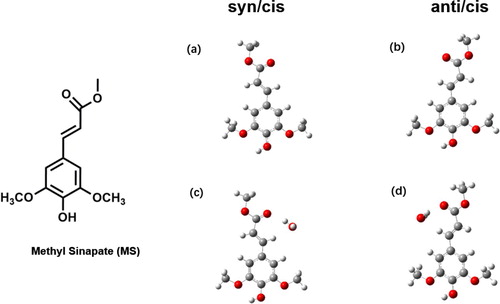
The natural UV-B filter used by plants is sinapoyl malate. The foundation for disentangling the properties of this compound has been set in high-resolution laser spectroscopic molecular beam studies on a series of sinapate esters of increasing complexity [Citation18]. Interestingly, no indication was found in these studies of internal conversion pathways leading to long-lived states. At first sight, it might thus appear that nature has succeeded to avoid trapping of energy in long-lived electronically excited states. Further studies on the excited-state properties of sinapate esters are therefore of considerable interest. In the present studies, we focus on methyl sinapate (MS, Figure ), a compound that features the pertinent chromophoric parts of sinapoyl malate while at the same time allowing for detailed high-resolution studies. Dean et al. reported for this compound one-color Resonance Enhanced Two-Photon Ionisation (R2PI) excitation spectra and dispersed emission spectra for relatively low excited-state vibrational energies, but, as stated above, did not report any triplet state [Citation18]. Subsequent time domain studies of MS in the gas phase and solution phase with femtosecond time-resolved spectroscopic techniques showed species that lasted beyond the nanosecond probe window [Citation22,Citation28]. For the solution phase this species was attributed to a photoisomerization product, while for the gas phase it was tentatively attributed to a triplet species. However, since in both the gas and solution phase excitation took place at the absorption maximum it is not possible to say whether at such energies decay channels become available that at lower energies would not be not present.
Figure 1. Molecular structure of conformers of methyl sinapate and its complex with a single water molecule. Conformers (a) and (c) correspond to the syn/cis conformation of the bare molecule and its complex with H2O in a configuration designated as II, respectively. Conformers (b) and (d) correspond, respectively, to the anti/cis conformation and its complex with H2O in the II configuration.
In the present studies, we employ a two-color R2PI approach for which our previous studies have shown that it allows for the observation of internal conversion pathways from the initially excited electronic state to lower-lying electronically excited states that cannot efficiently be ionised using one-color R2PI [Citation15,Citation36,Citation38–40]. We report extended excitation spectra as well as pump–probe experiments that provide detailed insight into the energy dissipation pathways at work in MS. We also investigate how these pathways are influenced by interactions with solvent molecules. We do this by performing similar experiments on the MS-H2O complex, but also by extending the previously reported time-resolved studies in the femtosecond time domain to the nanosecond time domain.
2. Methods
2.1. Experimental
Methyl sinapate was synthesised according to previously reported methods [Citation18,Citation41,Citation42], further details being provided in the Supplemental Material. The REMPI, depletion and lifetime measurements used a molecular beam set-up that has been described in detail before [Citation43]. For the present experiments MS samples were put into a sample holder that was heated to 160°C. A pulsed supersonic expansion with a pulse duration of 180 µs was obtained by expanding a mixture of MS vapour with 1.5 bars of Ne using a General Valve pulsed nozzle with an orifice diameter of 0.5 mm. Studies of hydrated MS were performed by mixing the carrier gas with small amounts of water vapour. After skimming the molecular beam with a 2.5 mm skimmer, the beam entered the ionisation region where mass-resolved ion detection took place using a reflectron type time-of-flight spectrometer (R.M. Jordan Co.).
One- and two-color Resonance Enhanced Two-Photon Ionisation (R2PI) experiments have been performed using a frequency-doubled Sirah Cobra-Stretch dye laser operating on DCM or Rhodamine 101, and pumped by a Spectra Physics Lab 190 Nd:YAG laser. In the two-color R2PI and pump–probe experiments ionisation took place with a Neweks PSX-501 ArF excimer laser (193 nm, 6.42 eV). Typical excitation and ionisation pulse energies were 10–50 µJ and 1 mJ, respectively. Pump–probe experiments that were performed to study the decay dynamics of MS and its hydrated clusters used a delay generator (Stanford Research Systems DG645) to scan the delay between the excitation and ionisation lasers with steps of 1 ns. Finally, depletion experiments were performed by depleting the ground state population with a frequency-doubled Sirah Precision Scan dye laser operating on DCM and pumped by a Spectra Physics Lab 190 Nd:YAG laser. These experiments typically used depletion pulse energies of 2 mJ and a time delay between the depletion laser and the excitation-ionisation probe lasers of 150 ns.
2.2. Theoretical
Ground-state geometries of MS and MS-H2O were optimised by Density Functional Theory (DFT) while vertical and adiabatic excitation energies of the lower electronically excited singlet and triplet states were obtained by Time Dependent (TD)-DFT calculations. Initially, calculations were performed using the B3LYP functional with the 6-311G(d) basis set, but in particular the calculation of the adiabatic excitation energy of the S2(ππ*) state turned out to be critically dependent on the employed functional. Additional calculations have therefore been performed at the CAM-B3LYP/cc-pVDZ, M06-2X/cc-pVDZ, M05-2X/6-31+G(d), and PBE0/cc-pVDZ levels. Simulations of vibrationally-resolved excitation spectra have been performed by calculation of the force fields in ground and electronically excited states followed by calculations of the pertinent Franck-Condon factors. For comparison with the experimental spectra theoretically calculated vibrational frequencies were scaled with a uniform scaling factor of 0.966. All calculations have been performed with the Gaussian16, Rev.A.03 suite of programmes [Citation44].
3. Results and discussion
3.1. Methyl sinapate
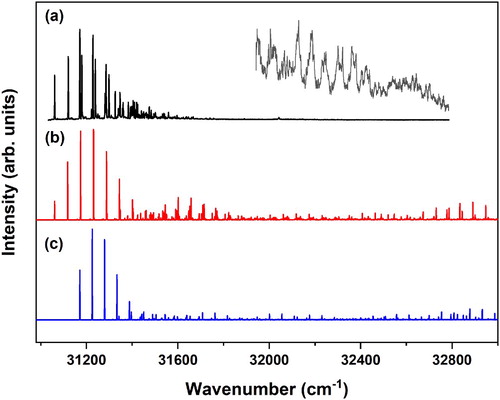
Figure shows the two-color R2PI excitation spectrum of MS in the 30900–33000 cm−1 region. This spectrum nicely reproduces and extends the one-color R2PI spectrum reported previously [Citation18], although for the high-intensity bands the present spectrum appears to be less saturated. Previously, it has been shown that this spectrum contains dominant contributions of the syn/cis (A) and anti/cis (B) conformations with 0–0 transitions that we find at 31059.8 and 31170.1 cm−1, and a very minor contribution from the anti/trans (C) conformation with a 0–0 transition at 31288.3 cm−1 [Citation45]. In agreement with previous calculations [Citation18] we find at the B3LYP/6-311G(d) that the anti/cis (B) conformation has the lowest energy, the syn/cis (A) and anti/trans (C) conformations being higher in energy by 0.04 and 1.07 kcal/mol, respectively. Figure shows that the Franck-Condon activity in the excitation spectrum rapidly decreases for vibrational energies higher than ∼800 cm−1. Spectra recorded at higher excitation energies that are displayed in the inset of Figure make clear that at these excitation energies there is still activity. However, bands in this region are significantly broader and should most probably be attributed to transitions to a dense manifold of dark vibrational levels that obtain oscillator strength by coupling to a bright Franck-Condon allowed vibrational level in the excited state.
Figure 2. (a) (1 + 1’) R2PI excitation spectrum of methyl sinapate. The inset shows part of the spectrum that has been recorded with higher laser intensities in order to determine the presence of vibrational activity at higher vibrational energies. (b and c) Franck-Condon simulations of the V(ππ*) ← S0 excitation spectrum of the syn/cis (A) (b) and anti/cis (B) conformations (c) obtained at the B3LYP/6-311G(d) level and employing a scaling factor for vibrational frequencies of 0.966.
Interestingly, we find that the signal intensities under one-color R2PI conditions are significantly smaller than under similar two-color ionisation conditions, indicating that the cross section for ionisation from the electronically excited state with 193 nm is considerably larger. One of the possible explanations for such a wavelength dependence is that ionisation does not only take place from the electronically excited state that is resonantly excited, but also from lower-lying electronically excited states that are populated upon internal conversion from this initially excited state. Since the ionisation energy of the lower-lying electronically excited states is higher than from the initially excited state, they will be more efficiently ionised when ionisation takes place with 193 nm instead of ∼320 nm. Such a situation is quite similar to what has been observed for cinnamates and coumarates for which it was concluded that after excitation of the bright 1ππ* state internal conversion to a dark 1nπ* state and intersystem crossing to the triplet manifold occurred [Citation15,Citation26,Citation30,Citation33,Citation37,Citation46]. We thus conclude that internal conversion pathways to other electronically excited states also play a significant role for MS.
The red and blue traces in Figure display the Franck-Condon spectra predicted at the B3LYP/6-311G(d) level for the transition to the V(ππ*) state of the syn/cis and anti/cis conformations, respectively. For the further discussion that will follow later it is useful to notice that calculations at other levels of theory predict essentially the same spectrum. Up to ∼600 cm−1 excess vibrational energy experiment and theory show excellent agreement, but at higher energies the experimental spectrum shows hardly any activity while according to the calculations such activity should still be present. This pertains in particular to the C = C stretch region (∼1600–1700 cm−1 above the origin) for which significant activity is predicted. That such an activity is not an artefact of the calculation is confirmed by the dispersed emission spectra reported previously for MS which, as one would expect on the basis of the present calculations, show significant activity in the C = C stretch and to a lesser extent in the vinyl CH bend region [Citation18]. As noticed above, vibrational activity becomes visible in this region under significantly higher pulse energy conditions albeit that the bands are significantly broader. If this broadening would only be due to a coupling to a dense manifold of dark vibrational states, one would expect that the integrated band intensities to be still in line with the theoretically predicted intensities. However, -also taking the higher pulse energies employed for recording the inset of Figure into account- one nevertheless comes to the conclusion that intensity is missing. We therefore tentatively conclude that ∼3 kcal/mol above the zero-point level of the V(ππ*) state an additional decay channel starts to open up that leads to a reduction of the R2PI signal.
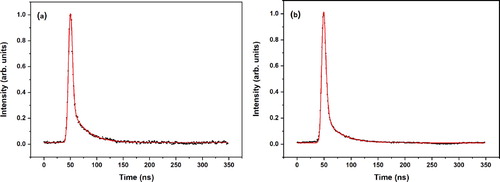
Figure displays pump–probe traces obtained at the origin transitions of conformers A and B. These traces immediately make clear that they are not associated with a simple monoexponential decay since in that case the sharp drop after the maximum should have been absent. Convoluting a Gaussian profile with a width of about 3 ns with a biexponential decay, on the other hand, leads to adequate fits that nicely reproduce the experimental traces. For conformer A we find for excitation of the vibrationless level decay rates of conformer A (2 ns)−1 and (27 ns)−1 with relative preexponential factors of 0.93 and 0.07, respectively, while for the same transition of conformer B decay rates of (3 ns)−1 and (29 ns)−1 with the same relative preexponential factors are obtained. The fast decay rates observed in the present experiments are puzzling since previous Laser Induced Fluorescence (LIF) and Dispersed Fluorescence (DFL) experiments reported fluorescence lifetimes of 12 and 14 ns at the electronic origins of conformers A and B, respectively [Citation18]. On the other hand, ps pump–probe R2PI experiments on sinapic acid with a probe laser wavelength of 243 nm found for the syn/cis conformer at the origin transition a decay rate of (1.9 ns)−1 [Citation29], which nicely follows what would be expected on the basis of the present experiments [Citation47]. A further consideration is based on the radiative decay rate that is expected on the basis of Fermi’s golden rule for vertical emission at an energy from the stable minimum of the S1 state with an oscillator strength
, which is given by
with
,
,
, and
being the elementary charge, the vacuum electric permittivity, the mass of the electron, and the speed of light, respectively. Using an emission energy of 31060 cm−1, a radiative decay rate of (12 ns)−1 would imply an oscillator strength of 0.046 for the S1 → S0 transition. However, our calculations discussed below predict -in agreement with calculations performed previously [Citation18,Citation32]- that the oscillator strength of the V(ππ*) → S0 transition is at least an order of magnitude larger.
Figure 3. (1 + 1’) R2PI decay curves of the (a) syn/cis (A) and (b) anti/cis (B) conformations of methyl sinapate after excitation at their vibrationless origin transition (31059.8 and 31170.1 cm−1, respectively). The red traces are fits of the decays to the convolution of a Gaussian profile with a biexponential decay with decay times (relative contribution) for syn/cis (a) of 1.8 ns (0.93) and 27.0 ns (0.07), and for anti/cis (b) 3.1 ns (0.93) and 29.2 ns (0.07).
We therefore conclude that the emission decay characteristics of MS require further research, and that the pump–probe traces as measured in the present R2PI studies reflect the true dynamics of the system. Given this, we interpret the observed traces as showing a fast decay from the excited V(ππ*) state with a rate on the order of 109 s−1. This decay populates at least in part a state that can still be ionised, and which decays with the slower decay rate
. At the same time we notice that part of the V(ππ*) population might also decay directly nonradiatively to the ground state [Citation48,Citation49]. This fraction of the population would not be visible anymore since ionisation of the populated ground state levels would be extremely inefficient due to unfavourable Franck-Condon factors. The present experiments formally do not allow us to determine the branching ratio over these two pathways. However, if we assume that the cross section for ionisation with 193 nm from the long-lived state and from the V(ππ*) state are similar (see Supplemental Material for a further discussion), the observed decays would indicate that on the order of 10% of the population of the initially excited V(ππ*) state decays to the long-lived state and 90% nonradiatively to the ground state. To investigate the energy dependence of the excited state dynamics we compare the R2PI excitation spectrum obtained with temporally overlapping excitation and ionisation lasers, and with a 10 and 20 ns delay between them (Figure S1 of the Supplemental Material). Normalising these spectra on the signal intensity of the 0–0 transition of the syn/cis conformation, we find for all practical purposes the same intensity distribution. We thus conclude that in the energy region where Franck-Condon activity can be observed vibrational levels in the excited state display the same decay dynamics.
In previous work on coumarates and cinnamates we found that the excited-state dynamics are dominantly determined by the fact that adiabatically the 1nπ* state is the lowest excited singlet state, while for vertical excitation it is S3 with the V’(ππ*) and V(ππ*) states being S1 and S2, respectively. As a result, the initially excited bright ππ* states undergo rapid internal conversion to the 1nπ* state from which rapid intersystem crossing occurs to the triplet manifold because of a favourable spin–orbit coupling between the 1nπ* state and an energetically close-lying higher 3ππ* state. In order to determine whether a similar situation is also present here we have performed TD-DFT calculations on the vertical and adiabatic excitation energies of the lower excited singlet states of MS. Initially, such calculations have been performed at the B3LYP/6-311G(d) level for which we find vertical excitation energies of the S1(V(ππ*)), S2(V’(ππ*)), and S3(nπ*) states (Table ) and oscillator strengths that are in excellent agreement with the values reported previously [Citation18,Citation22,Citation32]. Geometry optimisation of the V(ππ*) and 1nπ* states shows that the relaxation energy for the 1nπ* state is substantially larger than for the V(ππ*) state (0.83 eV vs. 0.34 eV) but that the V(ππ*) state nevertheless remains also adiabatically the lowest excited singlet state. Interestingly, we find, however, that geometry optimisation of the S2(V’(ππ*)) state brings the molecule to geometries where S1(V(ππ*)) and S2(V’(ππ*)) become quasi degenerate after which the geometry optimisation switches to that of the S1(V(ππ*)) state. We thus conclude that at this level of theory a conical intersection between the S1(V(ππ*)) and S2(V’(ππ*)) states is predicted to occur. TD-DFT is not capable to provide a proper description of conical intersections, but a rough estimate of the energy of this conical intersection has been obtained by taking the energy where switching occurs as that of the conical intersection. We then find that it lies about 6 kcal/mol above the minimum of the S1(V(ππ*)) state. It is interesting to notice that as a ballpark figure this number is in line with the energy that was calculated for the transition state in the S1 state of MS in methanol (∼3 kcal/mol) [Citation32], and for the transition state in the S1 state of isolated sinapic acid (∼6 kcal/mol) [Citation29], certainly when associating such a transition state with an (avoided) crossing with a higher-lying electronically excited state. It is also interesting to notice that this energy is roughly speaking in the range of the C=C stretch region for which we concluded above it has a reduced intensity to what was expected on the basis of Franck-Condon simulations.
Table 1. Vertical and adiabatic TD-DFT excitation energies (eV) of the lower electronically excited states of the syn/cis and anti/cis conformations of methyl sinapate obtained at the B3LYP/6-311G(d) level. Oscillator strengths for vertical excitation are given in parentheses. Experimental adiabatic excitation energies for the syn/cis and anti/cis conformations are 3.851 and 3.865 eV, respectively.
Comparison of the present calculations with previous TD-DFT calculations on sinapic acid [Citation18] indicates that calculations on this class of compounds are quite sensitive to the level of theory that is employed. These calculations were performed at the M05-2X/6-31+G(d) level and found that at this level both the V(ππ*) and V’(ππ*) state have a stable minimum. In fact, Franck-Condon simulations of the excitation spectra of these two states led at that time to the conclusion that the observed excitation spectrum should be assigned to the V(ππ*) state. In view of these results, we have repeated our TD-DFT calculations on MS with various functionals and basis sets that have in the recent past been employed on (substituted) cinnamates and coumarates. Table S1 in the Supplemental Material shows that in all cases the V(ππ*) state is predicted to be the lowest excited state for both vertical as well as adiabatic excitation. The Table also shows that with respect to the higher-lying excited states the situation is considerably less clear-cut and that the level of theory here dominantly influences the conclusions that are drawn on the topology of their potential energy surfaces. For the V’(ππ*) state the long-range corrected functionals find a stable minimum, while the other functionals optimise the state to a near-degeneracy with the V(ππ*) state after which state switching occurs. With respect to the 1nπ* state the situation is reversed. For this state a stable minimum is predicted by the B3LYP and PBE0 functionals, while for the long-range corrected functionals geometry optimisation under Cs restrictions find the state to become S2 and thus to feature a conical intersection with the V’(ππ*) state, which is S2 for vertical excitation. However, a frequency analysis in the planar minimum of the 1nπ* state finds one imaginary frequency along an out-of-plane coordinate. Non-planar geometry optimisation of this state brings the molecule to geometries where it becomes nearly degenerate with either of the ππ* states, after which state switching occurs.
The overall picture that emerges from these calculations is one in which the V(ππ*) state is the lowest excited singlet state for both vertical as well as adiabatic excitation. Internal conversion to the 1nπ* state, which in the cinnamates and coumarates dominates the decay dynamics, is thus absent for low excitation energies. At somewhat higher energies it is quite likely that conical intersections occur between the lower excited singlet states. These conical intersections are important for obtaining a quantitative understanding of the excited-state decay dynamics including photoisomerization pathways, in particular at room temperature under solution conditions, but need multireference methods for a proper description.
The conclusion that the V(ππ*) state is the lowest excited singlet state has several implications. Firstly, it means that the slow decay rate observed in the present study must be associated with the decay of the lowest triplet state. Secondly, in cinnamates and coumarates the 1nπ* state is adiabatically the lowest excited singlet state and close in energy to a higher-lying 3ππ* state. As a result, ISC is favoured and takes place on a time scale of ps. In MS, on the other hand, ISC must either occur by spin–orbit coupling of the V(ππ*) state to a higher-lying 3nπ* state, or, less likely, by indirect coupling to a 3ππ* state via coupling to the 1nπ* state. In line with the conclusion that ISC in MS is less favoured than in the cinnamates and coumarates we indeed observe (i) decay rates of V(ππ*) vibronic levels that are several orders of magnitude slower (on the order of 109 s−1) and (ii) competition with direct internal conversion pathways to the ground state. Ebata et al. have shown for several coumarates and cinnamates including sinapic acid that in the lowest triplet state the molecule adopts a perpendicular geometry for the vinyl double bond [Citation26,Citation29,Citation30,Citation33,Citation37]. In line with these results we find a similar geometry for the lowest triplet state of MS. It is interesting to notice that in all these compounds -and now also for MS- quantitatively similar nanosecond decay times are observed (20–30 ns) even though the ordering of the V(ππ*), V’(ππ *) and 1nπ* singlet states sensitively depends on the detailed substituent pattern. This observation provides further support for the conclusion that these decays are associated with the lowest excited triplet state since in all these compounds this is invariably the triplet analogue of the V(ππ*) state, that is, the state dominantly described by the HOMO → LUMO excitation.
3.2. (Micro)solvated methyl sinapate
Figure (a) displays the (1 + 1’) R2PI excitation spectrum recorded for the MS-H2O complex obtained by monitoring the mass of MS ions. The same spectra but with a considerably lower signal-to-noise ratio are obtained when the mass of MS-H2O ions is monitored. Such a difference can in principle be explained by dissociation of the complex in the electronically excited state or upon ionisation which both would lead to a reduction of the signal at the mass of MS-H2O ions. Below we will argue that pump–probe experiments demonstrate that dissociation occurs upon electronic excitation of the neutral complex.
Figure 4. (a) (1 + 1’) R2PI excitation spectrum of the methyl sinapate-H2O cluster monitoring the mass of the methyl sinapate ion. The asterisks in the spectrum indicate the probe frequencies used in (b-d) to obtain conformation-specific absorption spectra with UV-UV depletion spectroscopy at 30238.0 (b), 30374.8 (c) and 30324.6 (d) cm−1.
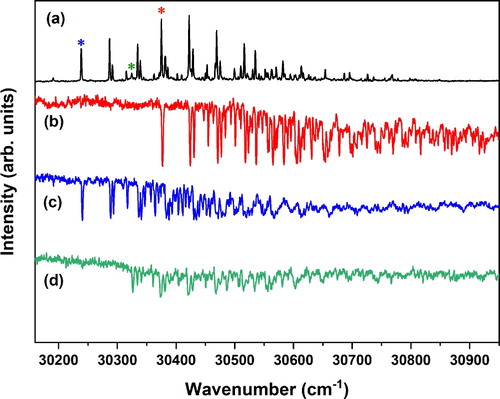
UV-UV depletion spectroscopy (Figure (b,c)) shows that this spectrum is built upon the excitation spectra of two major conformers with 0–0 transitions at 30238.0 and 30374.8 cm−1, and a very minor contribution from a conformer with a 0–0 transition at 30324.6 cm−1 which we will not consider in the rest of the discussion. Calculations on the MS-H2O complex find that the water molecule can either bind to the phenolic part of the molecule (conformer I) where hydrogen bonds are formed with the hydroxy group and one of the methoxy groups, or to the carbonyl group (conformer II) where the OH group of water is hydrogen-bonded to the carbonyl oxygen as a proton donor and the water oxygen weakly bound to the CH of the propenyl group (Figure ). A third possibility in which the OH group of water is hydrogen-bonded to the carbonyl oxygen as a proton donor and the water oxygen weakly bound to the CH of the methyl ester group is much higher in energy and will not further be considered here. Interestingly, we find for both the syn/cis and anti/cis conformer that conformer II has a significantly lower energy (∼1 kcal/mol) than conformer I, with anti/cis being slightly more stable than syn/cis, similar to the non-complexed molecule. The H2O---HO hydrogen bond is stronger than the H-O-H---O=C hydrogen bond, and one might therefore have expected conformer I to be more stable than conformer II. Apparently, however, the intermolecular hydrogen bonds of the water molecule with the hydroxy and methoxy groups of MS cannot compete with the intramolecular hydrogen bond between these two groups. Based on these results we assign the 30238 and 30375 cm−1 origins to the syn/cis (II) and anti/cis (II) conformations, respectively.
It is interesting to notice (i) that the R2PI excitation spectrum of the MS-H2O complex resembles qualitatively to a large extent the spectrum of the bare molecule, and (ii) that the depletion spectra of the complex also mirror the intensities of bands observed in the R2PI spectrum. This contrasts with what is observed for coumarate-based compounds for which vibrational activity rapidly disappears in the R2PI spectrum for vibrational energies above ∼500 cm−1 while vibrational activity remains in the depletion spectra [Citation15,Citation36]. This was attributed to a lowering of the E-Z isomerisation barrier of the vinyl double bond in the electronically excited state of the complex. Apparently, such a lowering of the isomerisation barrier does not occur for MS.
We thus find that complexation of MS with a water molecule red-shifts the energy of the S1 ← S0 origin transition by about 800 cm−1. It is instructive to compare this shift with the shifts observed for methyl 4-hydroxycinnamate (about 650–700 cm−1) [Citation35,Citation39,Citation40], and sinapic acid (about 150 cm−1). In methyl 4-hydroxycinnamate the water molecule is bound to the phenolic part of the molecule, while for sinapic acid water is bound to the COOH site where hydrogen bonds are formed with the C=O and OH groups. Electronic excitation of these molecules predominantly involves the π system of the phenyl group and the central double bond. One would therefore expect that water complexation on the phenolic site would have a larger influence on the electronic excitation energies than complexation on the carboxylic site, as is indeed observed experimentally when comparing the shifts of methyl 4-hydroxycinnamate and sinapic acid. From that perspective the large shift of 800 cm−1 observed for hydration of MS -where complexation occurs on the carboxylic site- is noteworthy since the calculations clearly indicate that for MS complexation occurs on the carbonyl site for which one would in first instance have expected a relatively small influence on electronic excitation energies. The key difference between sinapic acid and MS is that for MS a weak hydrogen bond is present with a CH of the propenyl group, which is not the case for sinapic acid. Apparently, this hydrogen bond affects the electronic structure of the π system significantly. More insight into the influence of water complexation on the electronic structure of MS might be obtained by IR absorption studies [Citation39,Citation40]. Such studies are presently being pursued.
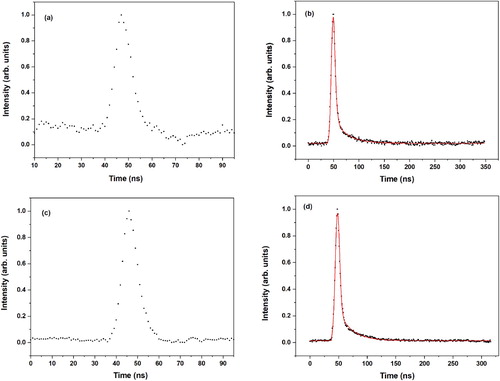
Figure shows pump–probe traces recorded for excitation at the 30238 and 30375 cm−1 origin transitions monitoring ions at the mass of the MS-H2O complex and at the mass of MS. Traces obtained at the mass of the MS-H2O complex suffer significantly from one-color contributions to the signal but do not give an indication for an exponential decay. We therefore interpret these traces as arising from the convolution of the two laser pulses and one-color contributions and conclude that the electronically excited state of the MS-H2O complex decays within the duration of the excitation pulse. In contrast, when the mass of MS is monitored, decay traces are seen that are very similar to the ones observed when exciting the bare molecule. In fact, fits of these decays with the convolution of a Gaussian with a biexponential decay lead for all practical purposes to the same preexponential factors and the same decay rates as found for the bare molecule (see Figures and ). For the bare molecule we have concluded that the decay of the electronically excited state involved intersystem crossing to the triplet manifold followed by a slower decay of the lowest triplet state. The observation that the same decay is observed when exciting the MS-H2O cluster implies that the excited cluster dissociates within the excitation pulse into an electronically excited MS molecule in its V(ππ*) state and a water molecule. Importantly, the present observations rule out an alternative scenario in which dissociation occurs upon intersystem crossing to and internal conversion within the triplet manifold. This would indeed release a significant amount of internal energy into the complex and facilitate dissociation. However, in such a case the (2–3 ns)−1 fast decay rate would not have been observed in the pump–probe traces obtained at the mass of MS.
Figure 5. (1 + 1’) R2PI decay curves of the syn/cis (AII) and anti/cis (BII) conformations of the MS-H2O complex after excitation at their vibrationless origin transition (30238.0 and 30374.8 cm−1, respectively). Panels (a) and (b) display decay curves of the syn/cis (AII) conformation obtained at the mass of MS-H2O and MS, respectively. Panels (c) and (d) display similar decay curves for the anti/cis (BII) conformation. The red traces are fits of the decays to the convolution of a Gaussian profile with a biexponential decay with decay times (relative contribution) for syn/cis (a) of 2.5 ns (0.97) and 26.2 ns (0.03), and for anti/cis (b) 2.7 ns (0.94) and 29.1 ns (0.06).
The present experiments have shown that under isolated and microsolvated conditions excitation of MS leads to the population of the lowest triplet state which decays on a tens of nanosecond time scale to the electronic ground state. One might therefore expect that such a decay would also be observed under conditions in which MS is fully solvated. Femtosecond transient absorption experiments of MS in cyclohexane indeed give evidence for a transient species with a lifetime longer than the ns delay that could be reached in these experiments, but this species was quite convincingly attributed to the formation of the cis isomer [Citation28]. We have performed nanosecond transient absorption experiments on MS dissolved in cyclohexane, methanol, dioxane, or acetonitrile [Citation50]. In all solvents we find that within the sensitivity of these experiments no transient signals can be observed beyond the bleaching and stimulated emission signals present during the excitation pulse. This could either indicate that under solvated conditions the triplet state decays faster, or that intersystem crossing from the excited singlet state is a much less efficient decay pathway than under isolated conditions. Since a faster triplet decay would have been observed in the femtosecond transient absorption experiments [Citation28], we conclude that it is a significant reduction of the intersystem crossing rate that is responsible for the absence of a triplet nanosecond transient in solution. From a sunscreen point of view such a conclusion is quite positive as it implies that under non-isolated conditions participation of a longer-lived reactive electronically excited state in dissipating harmful UV radiation is reduced.
4. Conclusions
High-resolution nanosecond laser spectroscopic studies have been employed to elucidate the excited-state dynamics of methyl sinapate whose chromophore is used in nature as an agent to protect plants from harmful UV-B radiation. Using a two-color R2PI excitation scheme with a 193 nm laser to ionise excited-state molecules has enabled us to extend significantly the range of electronically excited states that can be ionised efficiently in such experiments and that might be involved in the dissipation of the absorbed photon energy. Extended R2PI excitation spectra covering up to 2000 cm−1 vibrational energy in the electronically excited V(ππ*) state indicate that ∼3 kcal/mol above the zero-point level of this state an additional decay channel starts to open up. On the basis of quantum chemical calculations this pathway has tentatively been assigned to a (near-)conical intersection between the V(ππ*) and V’(ππ*) states.
One of the key issues in assessing the suitability of molecules as UV screening agents is the extent to which harmful long-lived electronically excited states are involved in the decay of the electronically excited state that is initially excited. Pump–probe experiments have shown that in methyl sinapate long-lived states are indeed involved but far less than observed for cinnamate-based UV-B absorbers. Quantum chemical calculations of adiabatic excitation energies have provided an appealing rationale for this observation as they show that in sinapates the 1nπ* state is no longer the lowest excited state. As a result, the efficient 1nπ*–3ππ* intersystem crossing pathway that is available in cinnamates after internal conversion of the bright 1ππ* states to the dark 1nπ* state is closed down in the sinapates. Interestingly, we find that in both sinapate-based and coumarate-based, and most recently also in ferrulate-based agents [Citation51], very similar decay rates of (20–30 ns)−1 are observed. This is in line with the conclusion that this decay occurs from the lowest triplet state which in all these cases arises from the HOMO → LUMO excitation, and that in this state all these molecules adopt a geometry with a perpendicular vinyl bond.
Our studies on the MS-H2O complex have provided detailed insight into how complexation with water influences the electronic structure and excited-state dynamics of the bare chromophore. Remarkably, we find that the lowest-energy conformations of the complex involve hydrogen bonds with the carbonyl group and the CH of the propenyl group. Normally one would assume that such a configuration would not influence the electronic structure of the isolated chromophore to a large extent, but this assumption is contradicted by the observed shifts in excitation energy which are even larger than in coumarates. Pump–probe studies of the complex have shown that excitation of the complex leads to rapid dissociation within the exciting laser pulse, leading to an excited MS in its V(ππ*) state that decays back to the ground state in the same way as in the non-complexed molecule.
The present studies have provided a comprehensive view on the electronic structure and excited-state dynamics of methyl sinapate. In combination with the many other high-resolution and time-resolved studies that have been performed on sinapates and related compounds a picture is arising that shows how seemingly small changes in structure may have far reaching consequences. At the same time, these studies pave the way for a rational design of novel compounds with improved characteristics for UV protection purposes. Such efforts are clearly of interest from both an academic as well as commercial point of view, and are indeed presently being pursued in many laboratories around the world.
TMPH-2020-0378-File001.docx
Download MS Word (133.7 KB)Acknowledgements
We thank Ivan Romanov and Melania Kozdra for experimental assistance. Jiayun Fan acknowledges a doctoral fellowship from the China Scholarship Council (No. 201808440365). This project has received funding from the European Union's Horizon 2020 research and innovation programme under the grant agreement No. 828753.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- G. Fisher, Z. Wang, S. Datta, J. Varani, S. Kang, and J. Voorhees, N. Engl. J. Med. 337, 1419 (1997). doi: 10.1056/NEJM199711133372003
- R.P. Gallagher and T.K. Lee, Prog. Biophys. Mol. Biol. 92, 119 (2006). doi: 10.1016/j.pbiomolbio.2006.02.011
- L. Marrot and J. Meunier, J. Am. Acad. Dermatol. 58, S139 (2008). doi: 10.1016/j.jaad.2007.12.007
- C. Cockell and J. Knowland, Biol. Rev. Camb. Philos. Soc. 74, 311 (1999). doi: 10.1017/S0006323199005356
- N.D. Paul and D. Gwynn-Jones, Trends Ecol. Evol. 18, 48 (2003). doi: 10.1016/S0169-5347(02)00014-9
- U. Osterwalder and B. Herzog, Photochem. Photobiol. Sci. 9, 470 (2010). doi: 10.1039/b9pp00178f
- A.S. Aldahan, V.V. Shah, S. Mlacker, and K. Nouri, JAMA Dermatol. 151, 1316 (2015). doi: 10.1001/jamadermatol.2015.3011
- N.N. Rosic, Mar. Drugs. 17, 638 (2019). doi: 10.3390/md17110638
- M. Loden, H. Beitner, H. Gonzalez, D.W. Edstrom, U. Akerstrom, J. Austad, I. Buraczewska-Norin, M. Matsson, and H.C. Wulf, Br. J. Dermatol. 165, 255 (2011). doi: 10.1111/j.1365-2133.2011.10298.x
- R. Losantos, I. Funes-Ardoiz, J. Aguilera, E. Herrera-Ceballos, C. Garcia-Iriepa, P.J. Campos, and D. Sampedro, Angew. Chem. Int. Ed. 56, 2632 (2017). doi: 10.1002/anie.201611627
- M.D. Horbury, E.L. Holt, L.M.M. Mouterde, P. Balaguer, J. Cebrian, L. Blasco, F. Allais, and V.G. Stavros, Nat. Commun. 10 (2019). doi: 10.1038/s41467-019-12719-z
- J. Krutmann, T. Passeron, Y. Gilaberte, C. Granger, G. Leone, M. Narda, S. Schalka, C. Trullas, P. Masson, and H.W. Lim, J. Eur. Acad. Dermatol. Venereol. 34, 447 (2020). doi: 10.1111/jdv.16030
- L.A. Baker, B. Marchetti, T.N.V. Karsili, V.G. Stavros, and M.N.R. Ashfold, Chem. Soc. Rev. 46, 3770 (2017). doi: 10.1039/C7CS00102A
- V. Stavros, M.N.R. Ashfold, K. Franklin, W.J. Buma, and T. Munnik, GB Patent nr 2018 0015 020 (14 September 2018).
- E.M.M. Tan, M. Hilbers, and W.J. Buma, J. Phys. Chem. Lett. 5, 2464 (2014). doi: 10.1021/jz501140b
- V.G. Stavros, Nat. Chem. 6, 955 (2014). doi: 10.1038/nchem.2084
- K. Glusac, Nat. Chem. 8, 734 (2016). doi: 10.1038/nchem.2582
- J.C. Dean, R. Kusaka, P.S. Walsh, F. Allais, and T.S. Zwier, J. Am. Chem. Soc. 136, 14780 (2014). doi: 10.1021/ja5059026
- T.N.V. Karsili, B. Marchetti, M.N.R. Ashfold, and W. Domcke, J. Phys. Chem. A. 118, 11999 (2014). doi: 10.1021/jp507282d
- Y. Miyazaki, K. Yamamoto, J. Aoki, T. Ikeda, Y. Inokuchi, M. Ehara, and T. Ebata, J. Chem. Phys. 141 (2014). doi: 10.1063/1.4904268
- X. Chang, C. Li, B. Xie, and G. Cui, J. Phys. Chem. A. 119, 11488 (2015). doi: 10.1021/acs.jpca.5b08434
- L.A. Baker, M.D. Horbury, S.E. Greenough, F. Allais, P.S. Walsh, S. Habershon, and V.G. Stavros, J. Phys. Chem. Lett. 7, 56 (2016). doi: 10.1021/acs.jpclett.5b02474
- Y. Peperstraete, M. Staniforth, L.A. Baker, N.D.N. Rodrigues, N.C. Cole-Filipiak, W. Quan, and V.G. Stavros, Phys. Chem. Chem. Phys. 18, 28140 (2016). doi: 10.1039/C6CP05205C
- N.D.N. Rodrigues, M. Staniforth, J.D. Young, Y. Peperstraete, N.C. Cole-Filipiak, J.R. Gord, P.S. Walsh, D.M. Hewett, T.S. Zwier, and V.G. Stavros, Faraday Discuss. 194, 709 (2016). doi: 10.1039/C6FD00079G
- X. Xie, C. Li, Q. Fang, and G. Cui, J. Phys. Chem. A. 120, 6014 (2016). doi: 10.1021/acs.jpca.6b05899
- K. Yamazaki, Y. Miyazaki, Y. Harabuchi, T. Taketsugu, S. Maeda, Y. Inokuchi, S. Kinoshita, M. Sumida, Y. Onitsuka, H. Kohguchi, M. Ehara, and T. Ebata, J. Phys. Chem. Lett. 7, 4001 (2016). doi: 10.1021/acs.jpclett.6b01643
- M.D. Horbury, L.A. Baker, N.D.N. Rodrigues, W. Quan, and V.G. Stavros, Chem. Phys. Lett. 673, 62 (2017). doi: 10.1016/j.cplett.2017.02.004
- L.A. Baker, M. Staniforth, A.L. Flourat, F. Allais, and V.G. Stavros, ChemPhotoChem. 2, 743 (2018). doi: 10.1002/cptc.201800060
- S. Kenjo, Y. Iida, N. Chaki, S. Kinoshita, Y. Inokuchi, K. Yamazaki, and T. Ebata, Chem. Phys. 515, 381 (2018). doi: 10.1016/j.chemphys.2018.07.007
- S. Kinoshita, Y. Inokuchi, Y. Onitsuka, H. Kohguchi, N. Akai, T. Shiraogawa, M. Ehara, K. Yamazaki, Y. Harabuchi, S. Maeda, and T. Ebata, Phys. Chem. Chem. Phys. 21, 19755 (2019). doi: 10.1039/C9CP02914A
- X. Zhao, J. Luo, Y. Liu, P. Pandey, S. Yang, D. Wei, and K. Han, J. Phys. Chem. Lett. 10, 5244 (2019). doi: 10.1021/acs.jpclett.9b02175
- X. Zhao, J. Luo, S. Yang, and K. Han, J. Phys. Chem. Lett. 10, 4197 (2019). doi: 10.1021/acs.jpclett.9b01651
- S. Muramatsu, S. Nakayama, S. Kinoshita, Y. Onitsuka, H. Kohguchi, Y. Inokuchi, C. Zhu, and T. Ebata, J. Phys. Chem. A. 124, 1272 (2020). doi: 10.1021/acs.jpca.9b11893
- E. Gromov, I. Burghardt, H. Koppel, and L. Cederbaum, J. Phys. Chem. A. 109, 4623 (2005). doi: 10.1021/jp0447791
- M. de Groot, E.V. Gromov, H. Koppel, and W.J. Buma, J. Phys. Chem. B. 112, 4427 (2008). doi: 10.1021/jp7101308
- E.M.M. Tan, S. Amirjalayer, B.H. Bakker, and W.J. Buma, Faraday Discuss. 163, 321 (2013). doi: 10.1039/c2fd20139a
- S. Kinoshita, Y. Miyazaki, M. Sumida, Y. Onitsuka, H. Kohguchi, Y. Inokuchi, N. Akai, T. Shiraogawa, M. Ehara, K. Yamazaki, Y. Harabuchi, S. Maeda, T. Taketsugu, and T. Ebata, Phys. Chem. Chem. Phys. 20, 17583 (2018). doi: 10.1039/C8CP00414E
- S. Smolarek, A. Vdovin, D.L. Perrier, J.P. Smit, M. Drabbels, and W.J. Buma, J. Am. Chem. Soc. 132, 6315 (2010). doi: 10.1021/ja101668v
- S. Smolarek, A. Vdovin, E.M.M. Tan, M. de Groot, and W.J. Buma, Phys. Chem. Chem. Phys. 13, 4393 (2011). doi: 10.1039/c0cp02221g
- E.M.M. Tan, S. Amirjalayer, S. Smolarek, A. Vdovin, A.M. Rijs, and W.J. Buma, J. Phys. Chem. B. 117, 4798 (2013). doi: 10.1021/jp312624e
- F. Allais, S. Martinet, and P. Ducrot, Synthesis. 3571 (2009). doi: 10.1055/s-0029-1216983
- Y. Liu, X. Zhao, J. Luo, and S. Yang, J. Lumin. 206, 469 (2019). doi: 10.1016/j.jlumin.2018.10.111
- S. Smolarek, A. Vdovin, A. Rijs, C.A. van Walree, M.Z. Zgierski, and W.J. Buma, J. Phys. Chem. A. 115, 9399 (2011). doi: 10.1021/jp111127g
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 16, Revision A.03 (Gaussian, Inc., Wallingford, CT, 2016).
- In [18] the 0-0 transitions of conformations A, B, and C were reported to be at 31065, 31174 cm−1, and 31285 cm−1, respectively. In the present experiments wavelengths have been determined using a HighFinesse WS5 wavelength meter.
- Y. Iida, S. Kinoshita, S. Kenjo, S. Muramatsu, Y. Inokuchi, C. Zhu, and T. Ebata, J. Phys. Chem. A. 124, 5580 (2020). doi: 10.1021/acs.jpca.0c03646
- The pump-probe experiments reported in [48] monitored the decay of the R2PI signal up to a delay of about 1.2 ns. As a result, a second component in the decay associated with the slower decay rate will not have been visible and has indeed not been reported.
- Fluorescence quantum yields show that radiative decay to the electronic ground state only forms a very minor decay pathway [51] and is therefore not relevant for the present analysis.
- B. Smyk, G. Mędza, A. Kasparek, M. Pyrka, I. Gryczynski, and M. Maciejczyk, J. Phys. Chem. B. 121, 7299 (2017). doi: 10.1021/acs.jpcb.7b05508
- M.W.H. Hoorens, M. Medved’, A.D. Laurent, M. Di Donato, S. Fanetti, L. Slappendel, M. Hilbers, B.L. Feringa, W. Jan Buma, and W. Szymanski, Nat. Commun. 10, 2390 (2019). doi: 10.1038/s41467-019-10251-8
- I. Romanov (personal communication).