
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The solubility of water in liquid n-perfluorohexane and in an equimolar mixture of n-hexane + n-perfluorohexane is experimentally determined as a function of temperature. The solubility of water in the equimolar mixture is significantly higher than the average of the solubilities in the pure solvents suggesting, for the first time, that mixing hydrogenated and perfluorinated chains enhances the solubility of water. The solubility in the equimolar mixture of n-hexane + n-perfluorohexane is also determined theoretically with the SAFT-γ Mie group-contribution approach, allowing a direct quantitative estimate of how much the large deviations from ideality contribute to the solubility of water in the mixture. In addition, the SAFT-γ Mie approach is used to represent the solubility of water in a number of n-perfluoroalkylalkanes, covering a range of relative lengths of the hydrogenated and perfluorinated chains. The theory can be used to predict the relative extent of the solubility of water in the different solvents, in good agreement with the experimental data. This is accomplished by using a single parameter to describe the strong attractive interaction between water and the CH2CF2 group at the junction between the hydrogenated and perfluorinated segments, which is known to be responsible for the increased solubility of water in these substances.
GRAPHICAL ABSTRACT
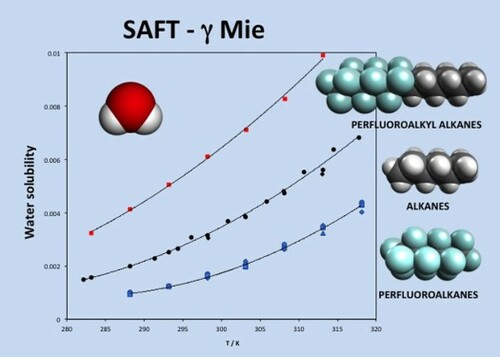
1. Introduction
Alkanes are highly hydrophobic fluids. Perfluoroalkanes are recognised as being even more hydrophobic than alkanes. The solubility of water in both types of substances confirms these observations. For example, the solubility of water in n-perfluorohexane is approximately one third of that in n-hexane at ambient conditions. Furthermore, alkanes and perfluoroalkanes are also known to be highly incompatible. Their mixtures display clear signs of weak unlike intermolecular forces, such as large positive deviations from Raoult’s law, liquid–liquid immiscibility, and large positive excess enthalpies and volumes [Citation1–7]. Anomalies in the transport [Citation8], surface [Citation9], and conformational [Citation10] properties of these mixtures are also known.
One of the main purposes of our current work is to understand how the solubility of water in mixtures of alkanes and perfluoroalkanes is affected by the large deviations from ideality. At the molecular level, nano-segregation in (n-hexane + n-perfluorohexane) mixtures, above the upper critical solution temperature (UCST), has been inferred from 129Xe NMR spectra and confirmed by molecular-dynamics simulation [Citation11,Citation12]. The global reorganisation of the fluid resulting from the mixing process and, in particular, the extremely large excess volume (∼ 5cm3mol−1 [Citation13]) can be expected to have an impact on the solubility of small solutes, such as water, in alkane + perfluoroalkane mixtures. To the best of our knowledge this is the first time that this impact has been quantified or even considered in a systematic manner.
The semifluorinated n-perfluoroalkylalkanes (PFAAs) can be considered as ‘chemical mixtures’ of n-alkanes and n-perfluoroalkanes. In these linear chain molecules, the perfluorinated and hydrogenated n-alkyl segments are covalently bonded together (represented here by the general formula F(CF2)n(CH2)mH or FnHm for conciseness); the existence of the mutually phobic segments gives rise to a marked amphiphilic character [Citation14]. Accordingly, PFAAs form micelles and reverse micelles [Citation15,Citation16], smectic liquid-crystalline phases and layered solids [Citation17–23], surface crystallised structures [Citation24,Citation25], and remarkable nano-patterned molecular films at the surface of water [Citation14,Citation26,Citation27]. Nano-segregation in liquid PFAAs has been recently demonstrated with 129Xe NMR spectra [Citation28].
In recent work [Citation29] we reported experimental data for the solubility of water in liquid PFAAs as a function of temperature, which demonstrated the existence of a specific interaction between the water molecule and the electric dipole that exists at the link between the hydrogenated and perfluorinated n-alkyl segments. The molecular organisation in the aqueous solutions of PFAAs solutions was further investigated using atomistic molecular-dynamics simulations, providing information about the location of water molecules within the complex nanostructures of PFAAs. These findings are an important contribution to understanding the spontaneous formation of nanostructured Langmuir films of PFAAs at the surface of water and wet silicon wafers, which consist of highly organised arrays of hemi-micelles [Citation14,Citation30].
It should be emphasised that our current work is part of a systematic experimental, computational, and theoretical study on the thermophysical properties of liquid PFAAs, either pure or mixed with n-alkanes, n-perfluoroalkanes, and water. We have previously reported various properties of pure liquid PFAA including the liquid density [Citation31,Citation32], vapour pressure [Citation33], viscosity [Citation34], and surface tension [Citation35]. The partial molar volumes of PFAA in n-octane at infinite dilution [Citation36,Citation37] and the water-PFAA liquid-liquid interfacial tension [Citation38] have also been measured over a range of temperatures, pressures, and relative length of the hydrogenated and fluorinated n-alkyl chains. These data were crucial for the development, parameterisation, and testing of the molecular models and force fields used in computer simulations and theoretical calculations.
In our current paper we report new experimental data for the solubility of water in liquid n-perfluorohexane as a function of temperature. Additionally, the solubility of water in an equimolar mixture of n-hexane + n-perfluorohexane is also experimentally determined over the same temperature range. The solubility of water in the equimolar mixture is found to be significantly higher than the average of the solubilities in the pure solvents. We thus show, for the first time, that mixing hydrogenated and perfluorinated chains, enhances the solubility of water in these semifluorinated systems.
Additionally, the SAFT-γ Mie group-contribution approach is used to model the experimental results: model parameters describing the interactions between water and the groups forming the n-alkanes and n-perfluoroalkanes are first developed; these are then used to determine the water solubility in the equimolar mixture of n-hexane + n-perfluorohexane; this calculation can be carried out by taking into account, or ignoring, the large deviations to ideality displayed by the n-hexane + n-perfluorohexane mixture [Citation39]. The comparison provides a quantitative assessment of the effect of deviations from ideality on the solubility of water in the n-hexane + n-perfluorohexane mixture. In both cases, the theory can be used in a fully predictive manner. The theoretical model incorporating the non-ideality is found to be in excellent agreement with the experimental data, while larger differences are found when deviations from ideality are not accounted for.
The SAFT-γ Mie group-contribution approach is also used to model the recently reported [Citation29] experimental solubility of water in a number of PFAAs systems with varying relative lengths of the hydrogenated and perfluorinated chains. In order to model these systems, it is necessary to characterise the interaction between water and the CH2CF2 junction (group), which is known to be responsible for the increased solubility of water in these substances. Using a single unlike group-interaction parameter, the relative order of the solubility of water in the different PFAA solvents is predicted, in good agreement with the experimental findings.
2. Experimental
2.1. Materials
Pure n-hexane and n-perfluorohexane are acquired from Sigma-Aldrich. Both compounds have stated purities greater than 99% and are used without further purification. The water is distilled and then deionised and filtered through a Millipore system.
2.2. Equipment
The solubility of water in the studied compounds is determined by the Karl-Fischer coulometric method, using a Metrohm 831 coulometer equipped with a generator electrode and diaphragm. The Karl-Fischer reagents used are Hydranal – Coulomat AG and Hydranal – Coulomat CG, supplied by Fluka, as the anolyte and catolyte, respectively. The accuracy of the instrument is regularly checked by titrating certified samples of Hydranal – Water Standard 0.1 (100 mg H2O / g) from Fluka.
2.3. Procedure
The solubility of water in n-perfluorohexane and in an equimolar mixture of n-hexane and n-perfluorohexane are determined following the same procedure used in previous work from our laboratory [Citation29,Citation40]. In summary, the equilibration between the aqueous and organic phases is achieved after a minimum of 24 h of direct contact between the phases, inside thermostated glass flasks. The stability of the thermostat is expected to be better than ±0.02 K and the total temperature uncertainty is ±0.05 K.
For each temperature, at least five independent samples of between 0.3 and 0.5 ml are extracted and analyzed, determining by weight the amount of sample added to the coulometer. The sampling is performed using a Hamilton gas-tight glass and PTFE syringe, which is kept warmer than the equilibration temperature during the sampling and weighing process to avoid the loss of water by phase separation on cooling. As in the previous studies [Citation29,Citation40], frequent verification of the solubility of water in n-heptane ensures the stability of the measurements and the internal consistency of the obtained data. The careful evaluation of these results is consistent with an intrinsic relative dispersion of the method of up to 5%.
The n-hexane + n-perfluorohexane equimolar mixture is prepared by weight to a mole fraction of x = 0.5000 ± 0.0004. Since this mixture is denser than water it remains at the bottom of the equilibration flask; care is taken to maintain the flask as full as possible during the equilibration period (by replacing the sampled volume with water), in order to avoid changes in liquid composition by differential evaporation. Due to the very low solubility of the organic compounds in water, changes in the alkane/perfluoroalkane mole ration due to dissolution in the aqueous phase is considered to be negligible.
3. SAFT-γ Mie model
The systems studied here are modelled with the SAFT-γ Mie group-contribution approach [Citation41–43]. Each molecule is represented as a heteronuclear chain of fused spherical segments, which correspond to its different constituent chemical groups. Each segment is taken to interact through a Mie potential [Citation44], which can also be decorated with short-range associative sites to mediate specific directional interactions such as hydrogen bonds. The underlying thermodynamic description is based on the SAFT-VR Mie equation of state [Citation45]; a high level of accuracy in the description of fluid-phase equilibria and thermodynamic derivative properties of the reference Mie fluid is provided, as a result of taking the high-temperature perturbation expansion up to third order in the Helmholtz free energy and to second order in the corresponding radial distribution function.
A given group k is modelled as one or more identical spherical segments, and a shape factor Sk is used to characterise the contribution of each segment to the overall thermodynamic properties of the molecule. The interactions between two segments k and l are represented with Mie potentials of variable repulsive and attractive range:
(1)
(1) where rkl is the distance between the centres of the segments, σkl the segment diameter, ϵkl the depth of the potential well (the dispersion energy), and
and
the repulsive and attractive exponents of the segment−segment interaction, respectively. The prefactor
is a function of the exponents, ensuring that the minimum of the interaction is at − ϵkl:
(2)
(2)
In common with other SAFT approaches, specific associative interactions such as hydrogen bonding can be treated by incorporating short-range square-well association sites, placed on any given segment. A segment can have any number of different site types (a, b, …). The association interaction between two square-well sites, one of type a on segment k and another of type b on segment l, is given by
(3)
(3) where
is the center−center distance between association sites a and b,
is the association energy, and
the cutoff range of the association interaction. The cutoff range
can be equivalently described in terms of a bonding volume
[Citation46].
The molecular parameters that characterise the repulsive, dispersive, and associative attractive interactions are usually refined using target experimental data of pure compounds or mixtures. In order to reduce the complexity of the parameter estimation procedure, a number of like parameters, typically the number of segments of each group, the number and type of associating sites and the attractive exponent of the dispersive interactions, are pre-assigned fixed values based on the chemical nature of each group. The unlike interaction parameters can either be determined from the like interaction parameters by means of appropriate combining rules or refined by considering relevant experimental data.
The unlike size parameter (segment diameter) is typically determined from the arithmetic-mean (Lorentz) rule,
(4)
(4) the unlike dispersion energy can be estimated with a modified geometric-mean rule that takes into account the size asymmetry of the groups [Citation47],
(5)
(5) and the unlike repulsive and attractive exponents of the Mie potentials are determined as
(6)
(6) which is derived from a geometric mean of the van der Waals integrated energy of the respective Sutherland potentials [Citation41,Citation45]. No unlike association interactions are needed to treat the mixtures of interest in our current work.
Once the parameters for the chemical functional groups characterising the system have been established, the Helmholtz free energy can be determined following the expressions presented in previous work [Citation41,Citation42,Citation48]. Other thermodynamic properties are then obtained from the free energy using standard thermodynamic relations.
4. Results and discussion
A SAFT-γ Mie model suitable for the description of the solubility of water in perfluoroalkylalkanes is developed here. The modelling of the water-PFAA interactions is undertaken in a systematic and sequential way, in order to allow for the assessment of the various effects that govern the solubility in these mixtures.
As discussed in more detail in previous work [Citation40], the experimental determination of the solubility of water in alkanes, and by extension in other essentially non-polar organic liquids, is prone to a very large uncertainty: discrepancies of more than 100% between authors are not uncommon, and a joint compilation [Citation49] by IUPAC and NIST includes data with deviations of up to 30% between different recommended sources. This intrinsic uncertainty makes the task of performing a detailed interpretation of the solubility very difficult, as the rationalisation of the different contributions to the thermodynamics of the mixtures can easily be lost in the large discrepancies in the values from different sources. One would expect that a series of solubility measurements performed in the same laboratory, by the same researchers and using the exact same procedure, has a degree of internal consistence that allows a more detailed analysis. For this reason, we previously performed a series of experimental measurements of the temperature dependence of the solubility of water in four n-alkanes [Citation40] and five n-perfluoroalkylalkanes [Citation29]; furthermore, in our current work we report new solubility measurements of water in n-perfluorohexane and in an equimolar mixture of n-hexane + n-perfluorohexane.
The n-alkane and n-alkyl segments of the PFAA molecules are modelled here using the SAFT-γ Mie description of n-alkanes reported by Papaioannou et al. [Citation41], while their n-perfluorinated counterparts are described using the parameters recently developed by Morgado et al. [Citation50]. The corresponding model for water was presented by Dufal et al. [Citation48]. Water is described as a single Mie spherical segment with four association sites (two representing the hydrogen atoms and two the lone electron pairs) to mimic hydrogen bonding. All of the SAFT-γ Mie interaction parameters used in our current work are summarised in Table .
Table 1. Functional group parameters of the SAFT-γ Mie group-contribution approach used in this work.
The first step to model the solubility of water in a system comprising fluorinated and hydrogenated alkyl chains is the parameterisation of the interaction between the water molecule and the n-alkyl and n-perfluoroalkyl constituent groups (i.e., CH2, CH3, CF2 and CF3).
The fluid-phase equilibria of aqueous n-alkanes has been modelled using the SAFT-γ Mie equation of state by Hutacharoen et al. [Citation51]: experimental three-phase vapor–liquid-liquid equilibria (VLLE) data at various temperatures and mutual n-alkane + water solubility data at 298 K were used to estimate the methyl-water (CH3 – H2O) and methylene-water (CH2 – H2O) unlike dispersion energy (ϵkl) and repulsive exponent () parameters of the SAFT-γ Mie models. With this approach, a very good global description of the n-alkane + water fluid-phase behaviour was achieved. In our current work, however, we re-parameterise the unlike interaction between water and the n-alkane (CH3 and CH2) groups to quantitatively reproduce our recently published experimental solubility data [Citation40] of water in n-hexane, n-heptane, n-undecane, and n-hexadecane over the temperature range of 283–318 K, thus focusing on the organic hydrocarbon-rich phase. Only the unlike interaction energies (ϵkl) are re-parameterized, with all the remaining interaction parameters fixed by the combining rules (equations 4 and 6). The SAFT-γ Mie description of the solubility of water in these n-alkanes is shown in Figure , along with the corresponding experimental values. As can be seen, the theory allows one to reproduce the experimental solubility data [Citation40,Citation49,Citation52–54] within the scatter of the experimental results; the representation of the solubility of water in the hydrocarbon-rich phase is much improved with respect to the previous parameterisation where the focus was to obtain a compromise in the simultaneous description of the two coexisting liquid phases [Citation51]. The solubility of the n-alkanes in the aqueous phase with the reparametrized model, over the same temperature range is also shown in Figure . The theory correctly predicts the order of magnitude of the n-alkane solubilities, which are much lower than the corresponding values for water in the organic-rich phase, as well as the steep decrease of solubility with the chain length of the hydrocarbon [Citation51], even though the behaviour of this phase was not taken into account in the parameter estimation.
Figure 1. Solubility of water in selected n-alkanes (top) and solubility of the same n-alkanes in water (bottom) at atmospheric pressure. The symbols represent the experimental data from references [Citation40,Citation49,Citation52–54] and the continuous curves the description obtained with the SAFT-γ Mie group-contribution approach with the reparametrized unlike interactions between water and the methyl and methylene groups of the hydrocarbons; the corresponding description obtained with the parameters reported in reference [Citation51] are represented by dashed curves.
![Figure 1. Solubility of water in selected n-alkanes (top) and solubility of the same n-alkanes in water (bottom) at atmospheric pressure. The symbols represent the experimental data from references [Citation40,Citation49,Citation52–54] and the continuous curves the description obtained with the SAFT-γ Mie group-contribution approach with the reparametrized unlike interactions between water and the methyl and methylene groups of the hydrocarbons; the corresponding description obtained with the parameters reported in reference [Citation51] are represented by dashed curves.](/cms/asset/4ca2f264-3744-4e86-ad94-f4cbb010f922/tmph_a_1910743_f0001_oc.jpg)
The temperature dependence of the solubility of water in n-perfluorohexane at atmospheric pressure is determined experimentally, and the results are presented in Table and Figure . As can be seen, this solubility is markedly lower than the solubility of water in n-alkanes, with values for n-perfluorohexane more than 30% lower than the corresponding values for n-hexane. This system was previously studied by Freire et al. [Citation55] reporting values 20% to 30% higher than our current measurements. This deviation is within the IUPAC-NIST [Citation49] criterion for good agreement and should be regarded as acceptable, especially considering that the low solubility of water in n-perfluorohexane is very challenging to determine accurately. Moreover, it is important to note that the standard molar thermodynamic functions of solution at 298.15 K ( = 20.5 ± 0.2 kJ.mol−1,
= 39.7 ± 0.8 kJ.mol−1 and
= 64.5 ± 2.5 J.mol−1.K−1), calculated from the temperature dependence of the solubility data as described in our previous study [Citation40], agree with the results of Freire et al. [Citation54] within the respective uncertainties.
Figure 2. Solubility of water in n-perfluorohexane (left) and solubility of n-perfluorohexane in water (right) at atmospheric pressure. The symbols represent the experimental data obtained in our current work and the continuous curve the description obtained with SAFT-γ Mie group-contribution approach. The dotted curves represent the SAFT-γ Mie predictions using the combining rule values for the unlike interactions with water.
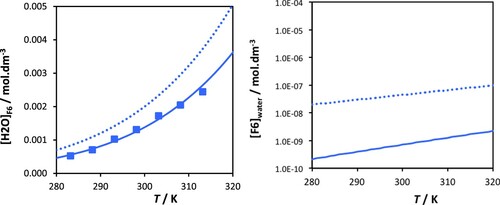
Table 2. Experimental solubility of water in n-perfluorohexane and in an equimolar mixture of n-hexane and n-perfluorohexane, at atmospheric pressure as a function of temperature.Table Footnotea
The experimental values of the solubility determined here are used to parameterise the unlike interactions between water and the n-perfluoroalkyl groups (CF3 – H2O and CF2 – H2O), within the SAFT-γ Mie group-contribution framework. Both the dispersion energy (ϵkl) and repulsive exponent () are adjusted; the corresponding values estimated from the experimental data are reported in Table . The SAFT-γ Mie description of the solubility of water in n-perfluorohexane is seen to reproduce the experimental data within the uncertainty in Figure . In the case of the solubility for n-perfluorohexane in water, no experimental data is available for comparison. We find that our SAFT-γ Mie predictions for the solubilities are around 5 orders of magnitude lower than the corresponding solubility of n-hexane in water (Figure ). As can be seen in Table , the optimised interaction parameters that describe the n-perfluoroalkane + water mixture are characterised by lower unlike dispersion energies and higher repulsive exponents than the corresponding values for their n-alkane + water counterparts, reflecting the enhanced hydrophobicity of perfluoroalkyl groups.
As previously discussed, mixtures of hydrogenated and perfluorinated molecules are highly non-ideal, exhibiting large excess properties, which highlight the ‘antipathy’ between the alkyl and perfluoroalkyl segments. We also study the solubility of water in (n-hexane + n-perfluorohexane) mixtures to assess the effect of the non-ideality of the mixture on the solubility of water. In essence water is used here as a probe of the unusual behaviour of these mixtures.
The temperature dependence of the solubility of water in an equimolar mixture of n-hexane + n-perfluorohexane is determined experimentally in the same series of measurements as the other compounds of this study. It is important to mention that the temperature range of the reported data is somewhat limited, as the measurements could only be performed in the small temperature interval where a homogeneous (stable) liquid phase is observed; i.e., between a few degrees above the upper critical solution temperature (UCST) of the n-hexane + n-perfluorohexane mixture and a few degrees below the normal boiling point of the mixture. The solubility data measured is presented in Table and included in Figure .
Figure 3. Temperature dependence of the solubility of water in an equimolar mixture of n-hexane + n-perfluorohexane, in n-hexane, and in n-perfluorohexane at atmospheric pressure. The symbols represent experimental data from reference [Citation40] and from our current work; the curves the description obtained with the SAFT-γ Mie group-contribution approach: the thin continuous curves represent calculations in the pure compounds (as in Figures and ); the dashed curve represents the prediction of the solubility in the solvent mixture using only combining rules (equations 4–6) for the alkyl-perfluoroalkyl (H6 – F6) interactions (model I); the thick continuous curve represents the description using the optimised parameterisation of the interactions for the solvent mixture [Citation39] (model II); the dashed gray line represents the average of the solubilities in the pure solvents. See text for details.
![Figure 3. Temperature dependence of the solubility of water in an equimolar mixture of n-hexane + n-perfluorohexane, in n-hexane, and in n-perfluorohexane at atmospheric pressure. The symbols represent experimental data from reference [Citation40] and from our current work; the curves the description obtained with the SAFT-γ Mie group-contribution approach: the thin continuous curves represent calculations in the pure compounds (as in Figures 1 and 2); the dashed curve represents the prediction of the solubility in the solvent mixture using only combining rules (equations 4–6) for the alkyl-perfluoroalkyl (H6 – F6) interactions (model I); the thick continuous curve represents the description using the optimised parameterisation of the interactions for the solvent mixture [Citation39] (model II); the dashed gray line represents the average of the solubilities in the pure solvents. See text for details.](/cms/asset/7a67528d-4d69-407d-b082-5a1aa8f6652a/tmph_a_1910743_f0003_ob.jpg)
It is apparent from Figure that the solubility of water in the equimolar mixture is higher than the average of the solubilities in the pure solvents, and is closer to the solubility of water in pure n-hexane, suggesting that mixing hydrogenated and perfluorinated chains enhances the solubility of water in the mixture. This is especially remarkable, considering that the volume fraction of perfluorohexane (with the larger molar volume) in the equimolar mixture is larger than that of n-hexane.
The SAFT-γ Mie group-contribution framework can be used in to further analyze this interesting phenomenon. We proceed to examine the predicted solubility of water in the equimolar H6 + F6 mixture using the parameterisation described for the interactions between the water molecule and the alkyl and perfluoroalkyl constituent segments (CH2, CH3, CF2 and CF3), and compare two options for the description of unlike alkyl-perfluoroalkyl groups (i.e., the CH2-CF2, CH2-CF3, CH3-CF2, CH3-CF3) group-group interactions. We consider first the use of unlike parameters determined with the combining rules presented in equations (4) – (6); this model is refered to as model as Model I. This model does not lead to the large excess volume and excess enthalpy of the solvent mixture. We then consider a second model (Model II) using the binary interaction parameters determined in previous work [Citation38], which reproduce the large excess volume and excess enthalpy of the solvent mixture. The results of the predictions obtained with both models are represented in Figure together with the experimental data. In Model I, the large non-ideality characteristic of mixtures of alkanes and perfluoroalkanes is not accounted for. As a result, the SAFT-γ Mie prediction of the solubility of water in the mixed phase is close to the average of the solubilities in the pure compounds. When Model II is used, the predicted solubility increases significantly (∼10%), and the values are found to be in close agreement with the experimental data. The difference between the two calculations is a direct measure of the influence of the non-ideality of the mixture on the solubility of water, possibly a consequence of the increased free volume of the H6 + F6 mixture, which results from the large positive excess volume of the equimolar mixture (∼5cm3.mol−1 [Citation13]).
Linear perfluoroalkylalkanes have been modelled with the SAFT-γ Mie group-contribution method in previous work [Citation39] where, as well as representing the CH2, CH3, CF2, and CF3 segments, it was necessary to consider a link group (CH2CF2) to account for the increased cohesion of the liquid imparted by the dipole moment at the alkyl – perfluoroalkyl junction. This representation of the PFAA molecules is used in our current study to predict the solubility of water in these compounds, taking into account the previously parameterisation presented in [Citation39] for the pure compounds, and determining the unlike interactions between water and the CH2CF2 group through combining rules (equations (4) and (6)). The SAFT-γ Mie predictions are compared with the corresponding experimental data [Citation29] in Figure . The theoretical predictions lead to solubilities of water which are slightly higher in liquid PFAAs compared to those in pure n-alkanes or n-perfluoroalkanes with the same number of carbons. We note however that in comparison to the experimental data, the model underestimates the water solubility in PFAAs by about 50%, and predicts a very similar solubility in all of the PFAAs, in contrast to what is observed experimentally.
Figure 4. Solubility of water in linear PFAAs (top) and solubility of the same PFAAs in water (bottom) at atmospheric pressure. The symbols represent experimental data [Citation29], the dashed curves the predictions obtained with the SAFT-γ Mie group-contribution approach using combining rules for the interaction between water and the CH2CF2 link group, and the continuous curves the SAFT-γ Mie calculations with the optimised CH2CF2 – H2O unlike interaction. See text for details.
![Figure 4. Solubility of water in linear PFAAs (top) and solubility of the same PFAAs in water (bottom) at atmospheric pressure. The symbols represent experimental data [Citation29], the dashed curves the predictions obtained with the SAFT-γ Mie group-contribution approach using combining rules for the interaction between water and the CH2CF2 link group, and the continuous curves the SAFT-γ Mie calculations with the optimised CH2CF2 – H2O unlike interaction. See text for details.](/cms/asset/a9bc8680-2048-4c6f-8467-2cc565d81b35/tmph_a_1910743_f0004_oc.jpg)
The underestimation of the water solubility in the PFAAs strongly suggests that these systems exhibit a specific interaction between water and the link group (CH2CF2), which needs to be accounted for in addition to the other unlike interactions. As discussed in a previous work [Citation29], this specific interaction is associated with Keesom forces between the permanent dipole of water and the localised dipole at the alkyl – perfluoroalkyl junction. In our work, we model this specific interaction by estimating the unlike interaction energy parameter (ϵkl) between water and the CH2CF2 group to reproduce the experimental solubility of water in PFAAs. The solubilities obtained with the SAFT-γ Mie group-contribution approach using the optimised H2O – CH2CF2 unlike interaction are also shown in Figure , where it can be seen that good agreement with the experimental data is achieved. The theory now correctly reproduces the magnitude and temperature dependence of the experimental values, as well as the order of relative solubility for the various systems. The value estimated for the unlike interaction energy parameter ( = 613.48 K) is quite large when compared to the unlike interaction energy parameters for the alkyl (
= 235.67 K;
= 327.22 K) and perfluoroalkyl (
= 212.07 K;
= 315.60 K) groups. This is further evidence, obtained independently through the theoretical SAFT-γ Mie description, of the relevance of the significant interaction between water and the dipole at the alkyl-perfluoroalkyl bond. By analyzing the water-rich phase it is apparent that the solubility of PFAAs in water is predicted to be significantly higher than that of n-alkanes (cf. Figure ) or n-perfluoroalkanes (cf. Figure ) of the same chain length, indicative the influence of these polar interactions also in the behaviour of the aqueous phase; unfortunately, no experimental results are yet available to assess these predictions.
5. Conclusions
New experimental data for the temperature dependence of the solubility of water in liquid n-perfluorohexane and in an equimolar mixture of n-hexane + n-perfluorohexane are reported. The solubility of water in the equimolar mixture is found to be higher than the average of the solubility in the pure solvents. A detailed analysis of these results suggests, for the first time, that mixing hydrogenated and perfluorinated chains enhances the solubility of water in such systems. Furthermore, new SAFT-γ Mie parameters describing the interactions between water and the groups forming the n-alkanes and n-perfluoroalkanes are developed to best represent the target experimental data.
The solubility of water in the equimolar mixture of n-hexane + n-perfluorohexane is also determined with the SAFT-γ Mie group-contribution approach, both by applying standard combing rules to determine the unlike interactions, and by recurring to parameters that describe the marked non-ideal behaviour exhibited by the mixture. In both cases, the theory is used in a fully predictive manner. The theoretical predictions confirm the importance of accounting for the deviations from ideality in the solvent mixture, allowing for a quantitative estimation of the contribution of these deviations to the solubility of water in the n-hexane + n-perfluorohexane mixture.
The SAFT-γ Mie group-contribution approach is also used to model the recently reported experimental solubility of water in a number of linear perfluoroalkylalkanes covering a range of relative lengths of the hydrogenated and perfluorinated segments. As expected, we find it is necessary to refine the interaction between water and the CH2CF2 group, which is known to be responsible for the increased solubility of water in these semifluorinated substances [Citation29]. Using a single unlike interaction energy parameter, one is able to correctly predict the relative order of the solubility of water in the different solvents with the theory, in good agreement with the experimental data.
We believe the present results can be an important contribution to understanding the large deviations to ideality in alkane + perfluoroalkane mixtures. The subject will be further addressed in future work, in particular the energetics of the mixing process.
Acknowledgments
We dedicate this paper to the memory of our esteemed friend and colleague Gerhard Findenegg. Centro de Química Estrutural acknowledges the financial support of Fundação para a Ciência e Tecnologia (UIDB/00100/2020). A. Galindo and G. Jackson acknowledge additional support from the Engineering and Physical Sciences Research Council (EPSRC) of the UK through grants (EP/E016340 and EP/J014958) to the Molecular Systems Engineering group at Imperial College London.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- J.H. Simons, and R.D. Dunlap, J. Chem. Phys. 18, 335–346 (1950). doi:https://doi.org/10.1063/1.1747628.
- J.S. Rowlinson, and F.L. Swinton, Liquids and Liquid Mixtures, 3rd ed. (Butterworth Scientific, London, 1982).
- E.M.D. Siebert, and C.M. Knobler, J. Phys. Chem. 75, 3863–3870 (1971). doi:https://doi.org/10.1021/j100694a014.
- S. Brode, and I.R. McDonald, Mol. Phys. 65, 1007–1012 (1988). doi:https://doi.org/10.1080/00268978800101561.
- M. Schoen, C. Hoheisel, and O. Beyer, Mol. Phys. 58, 699–709 (1986). doi:https://doi.org/10.1080/00268978600101511.
- C. Duce, M.R. Tinè, L. Lepori, and E. Matteoli, Fluid Phase Equilib. 199, 197–212 (2002). doi:https://doi.org/10.1016/S0378-3812(01)00815-9,
- G.M. Schneider, Fluid Phase Equilib. 199, 307–317 (2002). doi:https://doi.org/10.1016/S0378-3812(01)00803-2.
- P. Morgado, J. Black, J.B. Lewis, C.R. Iacovella, C. McCabe, L.F.G. Martins, and E.J.M. Filipe, Fluid Phase Equilib. 358, 161–165 (2013). doi:https://doi.org/10.1016/j.fluid.2013.07.060.
- T. Handa, and P. Mukerjee, J. Phys. Chem. 85, 3916–3920 (1981). doi:https://doi.org/10.1021/j150625a042.
- P. Morgado, A.R. Garcia, L.F.G. Martins, L.M. Ilharco, and E.J.M. Filipe, Langmuir. 33, 11429–11435 (2017). doi:https://doi.org/10.1021/acs.langmuir.7b02516.
- P. Morgado, L.F.G. Martins, and E.J.M. Filipe, PCCP. 21, 3742–3751 (2019). doi:https://doi.org/10.1039/C8CP06509H.
- P. Morgado, R.P. Bonifácio, L.F.G. Martins, and E.J.M. Filipe, J. Phys. Chem. B. 117, 9014–9024 (2013). doi:https://doi.org/10.1021/jp4060507.
- R.G. Bedford, and R.D. Dunlap, J. Am. Chem. Soc. 80, 282–285 (1958). doi:https://doi.org/10.1021/ja01535a007.
- G.M.C. Silva, P. Morgado, P. Lourenço, M. Goldmann, and E.J.M. Filipe, PNAS. 116, 14868–14873 (2019). doi:https://doi.org/10.1073/pnas.1906782116.
- M.P. Turberg, and J.E. Brady, J. Am. Chem. Soc. 110, 7797–7801 (1988). doi:https://doi.org/10.1021/ja00231a034.
- B.P. Binks, P.D.I. Fletcher, S.N. Kotsev, and R.L. Thompson, Langmuir. 13, 6669–6682 (1997). doi:https://doi.org/10.1021/la970408i.
- W. Mahler, D. Guillon, and A. Skoulios, Mol. Cryst. Liq. Cryst. 2, 111–119 (1985).
- C. Viney, T.P. Russell, L.E. Depero, and R.J. Twieg, Mol. Cryst. Liq. Cryst. 168, 63–82 (1989).
- C. Viney, R.J. Twieg, T.P. Russell, and L.E. Depero, Liq. Cryst. 5, 1783–1788 (1989). doi:https://doi.org/10.1080/02678298908045688.
- J.F. Rabolt, T.P. Russell, and R.J. Twieg, Macromolecules. 17, 2786–2794 (1984). doi:https://doi.org/10.1021/ma00142a060.
- T.P. Russell, J.F. Rabolt, R.J. Twieg, R.L. Siemens, and B.L. Farmer, Macromolecules. 19, 1135–1143 (1986). doi:https://doi.org/10.1021/ma00158a035.
- J. Höpken, and M. Möller, Macromolecules. 25, 2482–2489 (1992). doi:https://doi.org/10.1021/ma00035a029.
- P. Marczuk, and P. Lang, Macromolecules. 31, 9013–9018 (1998). doi:https://doi.org/10.1021/ma981130j.
- Y. Hayami, and G.H. Findenegg, Langmuir. 13, 4865–4869 (1997). doi:https://doi.org/10.1021/la9702446.
- P. Marczuk, P. Lang, G.H. Findenegg, S.K. Mehta, and M. Moller, Langmuir. 18, 6830–6838 (2002). doi:https://doi.org/10.1021/la025596d.
- M. Maaloum, P. Muller, and M.P. Krafft, Angew. Chem. Int. Ed. 41, 4331–4334 (2002). doi:https://doi.org/10.1002/1521-3773(20021115)41:22<4331::AID-ANIE4331>3.0.CO;2-J.
- A.L. Simões Gamboa, E.J.M. Filipe, and P. Brogueira, Nano Lett. 2, 1083–1086 (2002). doi:https://doi.org/10.1021/nl025698o.
- P. Morgado, J. Barras, and E.J.M. Filipe, PCCP. 22, 14736–14747 (2020). doi:https://doi.org/10.1039/D0CP02123G.
- P. Morgado, J. Barras, and E.J.M. Filipe, Fluid Phase Equilib. (2020). doi:https://doi.org/10.1016/j.fluid.2020.112754.
- L. Bardin, M.C. Faure, E.J.M. Filipe, P. Fontaine, and M. Goldmann, Thin Solid Films. 519 (1), 414–416 (2010). doi:https://doi.org/10.1016/j.tsf.2010.07.065.
- P. Morgado, H. Zhao, F.J. Blas, C. McCabe, L.P.N. Rebelo, and E.J.M. Filipe, J. Phys. Chem. B. 111, 2856–2863 (2007). doi:https://doi.org/10.1021/jp063136d.
- P. Morgado, J.B. Lewis, C.M.C. Laginhas, L.F.G. Martins, C. McCabe, F.J. Blas, and E.J.M. Filipe, J. Phys. Chem. B. 115, 15013–15023 (2011). doi:https://doi.org/10.1021/jp207567y.
- P. Morgado, G. Das, C. McCabe, and E.J.M. Filipe, J. Phys. Chem. B. 119, 1623–1632 (2015). doi:https://doi.org/10.1021/jp5109448.
- P. Morgado, C.M.C. Laginhas, J.B. Lewis, C. McCabe, L.F.G. Martins, and E.J.M. Filipe, J. Phys. Chem. B. 115, 9130–9139 (2011). doi:https://doi.org/10.1021/jp201364k.
- P. Morgado, O. Lobanova, E.A. Müller, G. Jackson, M. Almeida, and E.J.M. Filipe, Mol. Phys. 114 (18), 2597–2614 (2016). doi:https://doi.org/10.1080/00268976.2016.1218077.
- P. Morgado, R. Tomás, H. Zhao, M.C. Dos Ramos, F.J. Blas, C. McCabe, and E.J.M. Filipe, J. Phys. Chem. C. 111 (43), 15962–15968 (2007). doi:https://doi.org/10.1021/jp073758e.
- P. Morgado, H. Rodrigues, F.J. Blas, C. McCabe, and E.J.M. Filipe, Fluid Phase Equilib. 306, 76–81 (2011). doi:https://doi.org/10.1016/j.fluid.2011.02.020.
- P. Morgado, J. Gaspar, and E.J.M. Filipe, J. Mol. Liq. 312, 113385 (2020). doi:https://doi.org/10.1080/00268976.2020.1722270.
- P. Morgado, J. Barras, A. Galindo, G. Jackson, and E.J.M. Filipe, J. Chem. Eng. Data. 65, 5909–5919 (2020). DOI:https://doi.org/10.1021/acs.jced.0c00785.
- P. Morgado, J. Barras, P. Duarte, and E.J.M. Filipe, Fluid Phase Equilib. 503, 112322 (2020). doi:https://doi.org/10.1016/j.fluid.2019.112322.
- V. Papaioannou, T. Lafitte, C. Avendaño, C.S. Adjiman, G. Jackson, E.A. Müller, and A. Galindo, J. Chem. Phys. 140, 054107 (2014). doi:https://doi.org/10.1063/1.4851455.
- S. Dufal, V. Papaioannou, M. Sadeqzadeh, T. Pogiatzis, A. Chremos, C.S. Adjiman, G. Jackson, and A. Galindo, J. Chem. Eng. Data. 59, 3272–3288 (2014). doi:https://doi.org/10.1021/je500248h.
- A.J. Haslam, A. Gonzalez-Perez, S. DiLecce, S.H. Khalit, F.H. Perdomo, S. Kournopoulos, M. Kohns, T. Lindeboom, M. Wehbe, S. Febra, G. Jackson, C.S. Adjiman, and A. Galindo, J. Chem. Eng. Data. 65, 5862–5890 (2020). doi:https://doi.org/10.1021/acs.jced.0c00746.
- G. Mie, Ann. Phys. (Berlin, Ger.). 316, 657–697 (1903). doi:https://doi.org/10.1002/andp.19033160802.
- T. Lafitte, A. Apostolakou, C. Avendaño, A. Galindo, C.S. Adjiman, E.A. Müller, and G. Jackson, J. Chem. Phys. 139, 154504 (2013). doi:https://doi.org/10.1063/1.4819786.
- G. Jackson, W.G. Chapman, and K.E. Gubbins, Mol. Phys. 65, 1–31 (1988). doi:https://doi.org/10.1080/00268978800100821.
- A.J. Haslam, A. Galindo and G. Jackson, Fluid Phase Equilib. 266, 105 (2008). doi:https://doi.org/10.1016/j.fluid.2008.02.004
- S. Dufal, T. Lafitte, A.J. Haslam, A. Galindo, G.N.I. Clark, C. Vega, and G. Jackson, Mol. Phys. 113, 948–984 (2015). doi:https://doi.org/10.1080/00268976.2015.1029027.
- A. Maczynski, D.G. Shaw, M. Goral, B. Wisniewska-Goclowska, A. Skrzecz, Z. Maczynska, I. Owczarek, K. Blazej, M.-C. Haulait-Pirson, F. Kapuku, G.T. Hefter, and A. Szafranski, J. Phys. Chem. Ref. Data. 34, 441–476 (2005). doi:https://doi.org/10.1063/1.1790005.
- P. Morgado, B. Colaço, V. Santos, G. Jackson, and E.J.M. Filipe, Mol. Phys. (2020). doi:https://doi.org/10.1080/00268976.2020.1722270.
- P. Hutacharoen, S. Dufal, V. Papaioannou, R.M. Shanker, C.S. Adjiman, G. Jackson, and A. Galindo, Ind. Eng. Chem. Res. 56 (38), 10856–10876 (2017). doi:https://doi.org/10.1021/acs.iecr.7b00899.
- A. Maczynski, D.G. Shaw, M. Goral, B. Wisniewska-Goclowska, A. Skrzecz, I. Owczarek, K. Blazej, M.-C. Haulait-Pirson, G.T. Hefter, F. Kapuku, Z. Maczynska, A. Szafranski, and C.L. Young, J. Phys. Chem. Ref. Data. 34 (3), 1399–1487 (2005). doi:https://doi.org/10.1063/1.1840737.
- D.G. Shaw, A. Maczynski, M. Goral, B. Wisniewska-Goclowska, A. Skrzecz, I. Owczarek, K. Blazej, M.-C. Haulait-Pirson, G.T. Hefter, F. Kapuku, Z. Maczynska, and A. Szafranski, J. Phys. Chem. Ref. Data. 35 (1), 153–203 (2006). doi:https://doi.org/10.1063/1.2134730.
- D.G. Shaw, A. Maczynski, M. Goral, B. Wisniewska-Goclowska, A. Skrzecz, I. Owczarek, K. Blazej, M.-C. Haulait-Pirson, G.T. Hefter, F. Kapuku, Z. Maczynska, and A. Szafranski, J. Phys. Chem. Ref. Data. 35 (2), 687–784 (2006). doi:https://doi.org/10.1063/1.2132315.
- M.G. Freire, L.R. Gomes, L.M.N.B.F. Santos, I.M. Marrucho, and J.A.P. Coutinho, J. Phys. Chem. B. 110, 22923–22929 (2006). doi:https://doi.org/10.1021/jp0622942.