
Abstract
DFT calculations show that a combination of an electric field and electronic excitation is a promising mechanism to force a molecular switch based on amino-imino tautomerisation into one of its two states. By calculating the effect of an electric field in the direction of the moving hydrogen on the shape of the barrier in the ground and low-lying excited states of previously proposed molecular switches consisting of 5- and 7-membered rings with adjacent amino and imino groups, we demonstrate that electric fields and photons in experimentally accessible ranges introduce sufficient asymmetry to push the switch into the desired configuration. Excitation to states with inverted order of the preferred geometry allows reversible switching without reversal of the electric field.
GRAPHICAL ABSTRACT

Statement of article significance
This manuscript demonstrates that a combination of electronic excitation and an electric field is a viable switching mechanism for previously proposed candidate switches for molecular electronics. Using DFT, it is shown that switching can be achieved with field strengths and photonic energies in experimentally accessible ranges.
In future, networks of such switches might act as transistors or other electronic devices at a size not accessible by current semiconductor-based microelectronics.
This will be of great interest to experimental groups working on molecular electronic devices and all researchers interested in molecular electronics. It may even bring the ‘molecular computer’ closer to reality.
Introduction
The incredible progress in the power of electronic devices seen over the past decades, starting with the first working silicon transistor in 1954 [Citation1], revolutionised almost all aspects of our life. The miniaturisation of integrated circuits allowed the creation of ever more complex electronic devices. It followed an apparent ‘law’ of doubling the number of transistors per area roughly every 18 months, dubbed Moore’s Law [Citation2]. This development has now reached its physical limits [Citation3], and alternative approaches to decrease the size of electronic components are being sought. One promising methodology is the use of molecular electronics, where single molecules act as switches or transistors.
Research on molecular switches has blossomed over the last few decades, and papers reviewing this area have started to appear in the literature [Citation4–6]. Molecular switches can be switched in a variety of ways, depending on the properties of the molecule, including conformational change, different spin states, charge states, dipole orientations, and bond formation. Switching can be initiated by external stimuli such as light, an electric field, temperature and chemical reactions. Usually a switch has two different states between which it can interconvert; however, recently a molecular switch that can photoswitch between eight different states via controlled rotations around three adjacent bonds has been presented [Citation7]. Wavelengths required for switching are typically in the UV range, although switches that are triggered by irradiation with visible light have been reported as well [Citation7,Citation8]. Light offers many advantages over other stimuli, such as the ability to focus on a small area and to be switched on and off on a short timescale using, for example, laser pulses.
We recently proposed a type of molecular switch based on amino-imino tautomerisation in ring molecules [Citation9,Citation10]. One advantage of this type of switch is that the switching mechanism is based on the movement of a very light particle (a single hydrogen atom) over a very short distance. This would enable high clock rates (number of possible switching events per second) in a molecular electronic device. While not competitive with the GHz range of switching frequencies common in current semiconductor chips, a single switching event based on the movement of just a hydrogen atom, triggered by a pulsed laser, should be feasible on a much shorter timescale than other, heavier, molecular switches, even with current technology. The switching mechanism is similar to that observed in molecules lying flat on a metal surface, such as naphthalocyanine [Citation11] and azophenine [Citation12]. To be useful in molecular electronics, it would be desirable if the direction of the bonds affected by the switching process were not parallel to a metal surface, as the surface would short-circuit any conduction in this direction.
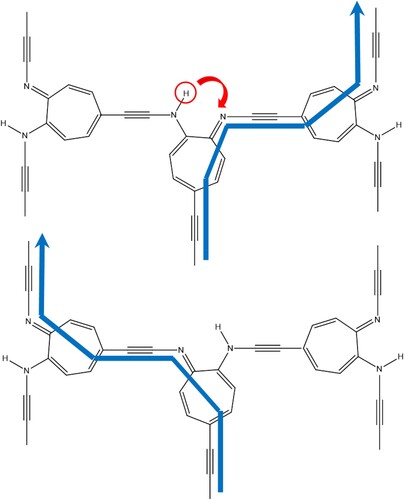
The prerequisites of a working, reversible, switch are: bistability (i.e. the existence of two minima of equal or similar depth on the potential energy surface); a difference in conductivity between those structures (for example, single versus double bonds in the pathway of a current, assuming that conjugated double bonds conduct electricity, whereas single bonds do not); and a means to switch between the two states. In previous work, we have shown that bistability can be achieved using molecules with an odd-membered ring, which have a symmetric transition state for the tautomerisation process [Citation10]. To trigger the switching process, this symmetry has to be disrupted to favour one of the two states. This can for example be done by the application of an electric field in the direction of the hydrogen movement during the tautomerisation [Citation9]. To build a molecular electronic device, switches need to be connected into a network by molecular wires. Molecular wires are chains that conduct electricity in one dimension [Citation13,Citation14]. Figure shows a proposed network of 7-membered ring molecules as switches and ethyne fragments as molecular wires. Tautomerisation on the central ring changes the location of the double bonds and changes the pathway comprised of alternating single and double bonds. This will change the pathway of current flow through conjugated double and triple bonds, as indicated in Figure . Ethyne is a simple model of a molecular wire used before in a computational study [Citation15]. In practical studies, molecular wires often consist of conjugated rings connected by ethyne linkages, such as oligo(phenylene ethynylene) [Citation16].
Figure 1. Network of molecular switches (aminotroponimine molecules), connected by ethyne molecular wires, showing the change in flow of current (indicated by the blue arrows), upon switching the circled hydrogen as indicated by the arching arrow.
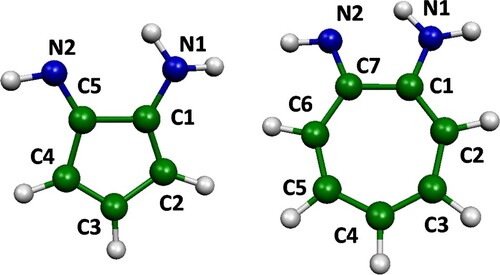
In the current paper, we consider the previously proposed odd-membered ring molecules aminotroponimine (IUPAC name 7-iminocyclohepta-1,3,5-trien-1-amine) and its five-membered ring analogue, 5-iminocyclopenta-1,3-dien-1-amine (see Scheme 1) [Citation10]. Aminotroponimine has been synthesised before [Citation17]. We will refer to these two ring molecules as the 7-ring and 5-ring, respectively, from here onwards. We propose combining an electric field with optical excitation to excite the molecule into an excited state with smaller or non-existent barrier to one of the two states of the switch. This may allow us to use molecules with a high barrier in the ground state or to use photons as a means of triggering the switch. The barrier heights in the ground and excited states will affect the function, stability and reversibility of the switch. The barrier in one direction should be sufficiently high to prevent spontaneous switching, whereas the energy of the excited state relative to the ground state determines the wavelengths needed for inducing the switching process. Earlier work showed that, in the ground state, the 5-ring has a slightly higher tautomerisation barrier than the 7-ring, and is therefore more resistant to spontaneous switching [Citation10]. Calculation of the switch rate at different temperatures showed that, for the 7-ring, some molecules can be expected to change configuration in a macroscopic assembly, even at liquid nitrogen temperatures. Whereas this is not desirable in a traditional electronic device, it may be acceptable in a system where the final state is measured statistically, e.g. in neural networks where fault tolerance is an intrinsic part of the method [Citation18–20]. Thus, both ring molecules are viable candidates for molecular switches and both will be further investigated in the current paper.
Scheme 1. Structures of the 5- and 7-ring, showing the atom labelling
Methodology
Calculations were carried out with the PBE0 [Citation21] hybrid density functional, augmented with the D3 empirical dispersion term [Citation22] and Becke–Johnson damping [Citation23], and the def2-TZVP basis set [Citation24], using Gaussian 16 [Citation25]. The structures of the 5-ring and 7-ring were optimised at this level of theory. We also optimised the two different rings with ethyne units attached to the two nitrogen atoms and the carbon on the opposite site of the ring from the nitrogens (C3 in the 5-ring; C4 in the 7-ring; see Scheme 1 for the atom labelling), to model molecular wires. Frequencies were calculated to confirm the optimised structures are true minima. This was done for both (symmetry-related) minima along the reaction coordinate of the tautomerisation. A relaxed scan was run, increasing the distance between the hydrogen atom initially attached to the amino nitrogen, starting at the optimised minimum geometry, in steps of 0.01 Å, from either minimum. For the 7-ring, 150 steps were taken, and 200 steps were taken for the 5-ring, to assure the energy minimum with the hydrogen attached to the other nitrogen was reached for the ground and all excited states considered. The geometries with the hydrogen atom beyond the energy maximum were combined to achieve a symmetric energy profile. To achieve an energy profile that reflects the symmetry of the molecule and is not biased towards one minimum, we use as the reaction coordinate the projection of the coordinate of the moving hydrogen onto the bond connecting the two carbon atoms attached to the nitrogens (the C1-C5 and C1-C7 bond for the 5-ring and 7-ring, respectively). Excited state calculations were run with TD-DFT, using the Tamm-Dancoff approximation [Citation26,Citation27] on the geometries obtained in the scan, using the same functional and basis set as before. Both singlet and triplet excited states were considered. The excited state calculations were repeated in the presence of an electric field of 1 V/Å, aligned along the bond connecting the two carbon atoms attached to the nitrogens.
Results
Figure shows the energy profiles of the ground state and five low-lying excited states of the 5- and 7-ring. The ground state barrier and most of the excited state barriers considered are larger for the 5-ring than for the 7-ring. The third and fourth excited state in the 5-ring (pink and blue lines), which are both triplets, show two avoided crossings. In the 7-ring the third and fourth excited state are of different multiplicity (excited state shown by blue line: singlet; pink line: triplet) and cross each other on both sides of the barrier. Due to the symmetry of the transition state, which is reflected in the energies of the ground and excited states, switching would have to be triggered by an external perturbation. In the remainder of this paper we investigate the effect of an electric field as a means to break the symmetry, with the objective to favour one of the two minima.
Figure 2. Energies (relative to the ground-state minimum) of the ground state (black line) and five low-lying excited states (coloured lines) of the 5-ring (left graph) and 7-ring (right graph) as a function of the reaction coordinate. The reaction coordinate is the projection of the moving hydrogen onto the C1-C5 (for the 5-ring) or C1-C7 bond (for the 7-ring).
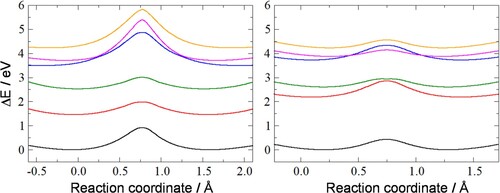
Figure shows the energy profiles of the ground state and five low-lying excited states of the 5- and 7-ring in the presence of an electric field of 1 V/Å, directed along the C1-C5 bond (for the 5-ring) or the C1-C7 bond (for the 7-ring), which is approximately the direction of movement of the hydrogen during the tautomerisation process.
Figure 3. Energies (relative to the ground-state minimum in the absence of an electric field) of the ground state (black line) and five low-lying excited states (coloured lines) of the 5-ring (left graph) and 7-ring (right graph) in the presence of the electric field as a function of the reaction coordinate. The reaction coordinate is the projection of the moving hydrogen onto the C1-C5 bond (for the 5-ring) or the C1-C7 bond (for the 7-ring).
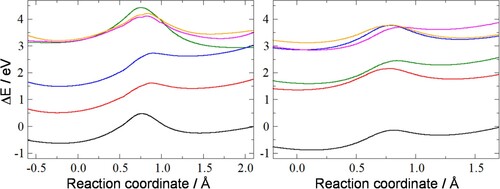
The presence of the electric field causes the profiles to be asymmetric, with one minimum favoured over the other. For some excited states the barrier has nearly disappeared. Thus, a combination of electric field and excitation could be used to switch the molecule from one tautomer to the other. For the 5-ring, without the electric field (see Figure ), the barrier is 0.92 eV. The electric field changes this to two unequal barriers of 1.10 and 0.72 eV, slightly favouring the minimum on the left in Figure . Excitation to the first or second excited state (red and blue lines) reduces the barrier towards the left minimum to 0.20 and 0.11 eV, respectively. For the 7-ring, the barrier is 0.43 eV without the electric field present. This changes to two unequal barriers of 0.74 and 0.19 eV. Two excited states have lower barriers towards the left minimum: the state represented by the green line (barrier: 0.16 eV) and the pink line (0.09 eV). Exciting to these states would make switching even easier. Reversing the direction of the electric field would allow switching back to the original state.
In a molecular electronic circuit a switch would be connected to other components via conducting wires that allow conjugation with double bonds in the switch molecule. The simplest example of a molecular wire is ethyne. To investigate the effect of attached wires on the energetics of the tautomerisation, we calculated the excitation profiles of the 5-ring and 7-ring with ethyne groups attached to the nitrogens and the carbon on the opposite side of the ring. The profiles without electric field present are included in the Supporting Information. Figure shows the profiles of the two molecules with wires in the presence of an electric field.
Figure 4. Energies (relative to the ground-state minimum in the absence of an electric field) of the ground state (black line) and five low-lying excited states (coloured lines) for the 5-ring (left graph) and six excited states for the 7-ring (right graph), with wires attached in the presence of the electric field as a function of the reaction coordinate. The reaction coordinate is the projection of the moving hydrogen onto the C1-C5 bond (for the 5-ring) or the C1-C7 bond (for the 7-ring).
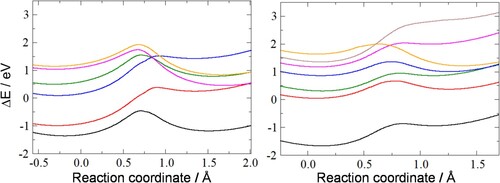
Compared to the molecules without wires, the range of energies of the excited states has decreased for the 5-ring. Qualitatively, the ground state and lower excited states are very similar. We have included an extra excited state for the 7-ring (the brown line) to show there are excited state profiles with no barrier for switching. Both molecules have excited states that favour the minimum on the left, as well as those favouring the minimum on the right. This opens the possibility of forcing the switch into the other minimum by purely optical means, without reversing the electric field.
Depending on the particular excited state desired, excitation energies are in the visible or near-UV range of the electromagnetic spectrum, and therefore easily experimentally accessible.
Conclusions
We have shown that the two investigated molecules, aminotroponimine and its 5-membered analogue, are promising candidates for single-molecule switches in molecular electronic devices. Their switching mechanism is amino-imino tautomerisation. Both show bistability, i.e. two states with equal stability. An electric field, combined with electronic excitation, introduces sufficient asymmetry to trigger a change of tautomeric state. Selective choice of field strength and excitation energy can be used to fine-tune the switching behaviour. In some cases an excited state reverses the order of preferred tautomeric state without reversal of the electric field, opening the possibility of switching purely based on photonic excitation in a constant field. This may allow devices switched with e.g. a pulsed laser, which may permit a faster clock rate, i.e. the number of switch events in a given time. The ultimate goal is to replace or complement electronic semiconductor devices with networks consisting of millions of molecular components. Initial studies may focus on anchoring individual switch candidates, or self-assembled monolayers on crystal surfaces, to allow investigating and manipulating them using established surface science methods. Excitation energies in the visible and near-UV range, as shown by our calculations, together with an electric field strength that is experimentally achievable, for example using a scanning tunnelling microscope (STM), indicate that experimental realisation should be feasible using current technology.
Supplemental Material
Download MS Word (58.6 KB)Acknowledgements
We thank EaStCHEM for computer resources supplied by the EaStCHEM Research Computing Facility.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Supplementary Materials: Figure S1: Relative energies of the ground state and five or six low-lying excited states of the 5-ring and 7-ring both with ethyne wires attached, as a function of the reaction coordinate. Table S1: Xyz files of the optimised 5-ring and 7-ring mimina and transition states with and without molecular wires. The research data supporting this publication can be accessed at https://doi.org/10.17630/fda73ef0-9944-4531-aed6-90b5c14a411a.
References
- M. Riordan, IEEE Spectr. May, 48 (2004).
- G.E. Moore, Electronics. 38, 114 (1965).
- M.M. Waldrop, Nature. 530, 144 (2016). doi:10.1038/530144a
- J.D. Harris, M.J. Moran and I. Aprahamian, Proc. Nat. Acad. Sci. 115, 9414 (2018). doi:10.1073/pnas.1714499115
- Y. Li; , K. Xu and X. Sun, Instrum. Sci. Technol. 48, 518 (2020). doi:10.1080/10739149.2020.1740247
- J.L. Zhang, J.Q. Zhong, J.D. Lin, W.P. Hu, K. Wu, G.Q. Xu, A.T.S. Wee and W. Chen, Chem. Soc. Rev. 44, 2998 (2015). doi:10.1039/C4CS00377B
- A. Gerwien, B. Jehle, M. Irmler, P. Mayer and H. Dube, J. Am. Chem. Soc. 144, 3029 (2022). doi:10.1021/jacs.1c11183
- A.-L. Leistner, S. Kirchner, J. Karcher, T. Bantle, M.L. Schulte, P. Gödtel, C. Fengler and Z.L. Pianowski, Chem. Eur. J. 27, 8094 (2021). doi:10.1002/chem.202005486
- H. Früchtl and T. van Mourik, Phys. Chem. Chem. Phys. 23, 1811 (2021). doi:10.1039/D0CP06250B
- T. van Mourik and H. Früchtl, Chem. Phys. Impact. 3, 100035 (6 pages) (2021). doi:10.1016/j.chphi.2021.100035
- P. Liljeroth, J. Repp and G. Meyer, Science. 317, 1203 (2007). doi:10.1126/science.1144366
- G.J. Simpson, S.W.L. Hogan, M. Caffio, C.J. Adams, H. Früchtl, T. van Mourik and R. Schaub, Nano Lett. 14, 634 (2014). doi:10.1021/nl4038517
- D.M. Guldi, H. Nishihara and L. Venkataraman, Chem. Soc. Rev. 44, 842 (2015). doi:10.1039/C5CS90010G
- D.K. James and J.M. Tour: Molecular Wires. In Molecular Wires and Electronics; edited by Luisa de Cola (Springer Berlin Heidelberg: Berlin, Heidelberg, 2005); Vol. 257; p. 33.
- R. Baer and R. Gould, J. Chem. Phys. 114, 3385 (2001). doi:10.1063/1.1342761
- G. Liu and J.J. Gooding, Langmuir. 22, 7421 (2006). doi:10.1021/la0607510
- T. Machiguchi, T. Takeno, T. Hasegawa and Y. Kimura, Chem. Lett. 21, 1821 (1992). doi:10.1246/cl.1992.1821
- V. Piuri, J. Parallel Distrib. Comput. 61, 18 (2001).
- C.H. Sequin and R.D. Clay, Proc. Int. Joint Conf. Neural Netw. (IJCNN). 1, 703 (1990).
- C. Torres-Huitzil and B. Girau, IEEE. Access. 5, 17322 (2017). doi:10.1109/ACCESS.2017.2742698
- C. Adamo and V. Barone, J. Chem. Phys. 110, 6158 (1999). doi:10.1063/1.478522
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys. 132, 154104 (19 pages)(2010). doi:10.1063/1.3382344
- A.D. Becke and E.R. Johnson, J. Chem. Phys. 123, 154101 (2005). doi:10.1063/1.2065267
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys. 7, 3297 (2005). doi:10.1039/b508541a
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, G.A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A.V. Marenich, J. Bloino, B.G. Janesko, R. Gomperts, B. Mennucci, H.P. Hratchian, J.V. Ortiz, A.F. Izmaylov, J.L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V.G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J.A. Montgomery, Jr., J.E. Peralta, F. Ogliaro, M.J. Bearpark, J.J. Heyd, E.N. Brothers, K.N. Kudin, V.N. Staroverov, T.A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A.P. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, J.M. Millam, M. Klene, C. Adamo, R. Cammi, J.W. Ochterski, R.L. Martin, K. Morokuma, O. Farkas, J.B. Foresman and D.J. Fox, Gaussian 16 Rev. C.01 (Wallingford, CT, 2016).
- S.M. Dancoff, Phys. Rev. 78, 382385 (1950). doi:10.1103/PhysRev.78.382
- I. Tamm, J. Phys. USSR. 9, 449 (1945).