
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Proton transfer reaction mass spectrometry has a so far little exploited potential for the analysis of high-purity process and inert gases. In order to protonate impurities like saturated hydrocarbons, precursor ions with very low proton affinity must be used, which in turn leads to a large energy release upon protonation of molecules with high proton affinity, potentially causing fragmentation of the product ions. To explore the potential of low-proton affinity precursor ions like ArH+ and N2H+ for analytical purposes, we studied their gas-phase reactions and kinetics with acetone by Fourier transform ion cyclotron resonance (FT–ICR) mass spectrometry. The dominant product ion in both cases is protonated acetone, but fragment ions formed include C2H3O+, C3H5+ and CH3O+, which are formed in higher abundance with ArH+ compared to N2H+. The reaction efficiencies are determined to be close to 100%. Quantum chemical calculations reveal energetically favorable reaction pathways and explain the loss of water leading to the formation of C3H5+, as well as loss of methane to yield C2H3O+ via dissociative proton attachment, which corresponds to the main product in the EI mass spectrum of acetone. Formation of protonated formaldehyde CH3O+ involves rearrangement of the C–C bonds to eliminate ethylene.
GRAPHICAL ABSTRACT
Introduction
Propanone, commonly known as acetone, is a valuable by-product of the cumene process [Citation1]. It is used as a solvent and as chemical feedstock. Almost seven million tons of acetone have been produced worldwide in 2021 [Citation2]. In the troposphere, it is formed in the hydroxyl radical initiated oxidation of α-pinene [Citation3], and it is an important precursor [Citation4–6] for tropospheric HOx. Home use include nail polish removers or varnishes. Acetone is taken up by the human body via inhalation, raising the concentration in urine and exhaled breath where it is excreted [Citation7–9]. It also occurs in the human body as a metabolite when the body uses fat instead of glucose to synthesise ATP, and it serves as a biomarker for diabetes [Citation10,Citation11].
In the gas phase, the proton transfer reaction (PTR) of acetone with H3O+ proceeds with a rate coefficient of 3.9·10−9 cm³ s−1, observed with the flowing afterglow technique [Citation12]. For N2H+, PTR was studied with formaldehyde CH2O, resulting in a rate coefficient [Citation13] of 3.3·10−9 cm³ s–1, and with formic acid HCOOH yielding a rate coefficient [Citation13] of 1.7·10−9 cm³ s–1. Note that for reactions with H3O+ instead of N2H+, both the measured rate coefficient and the calculated average dipole orientation (ADO) collision rate coefficient kADO are even higher, although the proton affinity (PA) of N2 is significantly lower [Citation13,Citation14] than of H2O. However, PTR to acetone has so far been studied neither from ArH+ nor from N2H+.
For trace gas analysis in air by proton transfer reaction mass spectrometry (PTRMS), H3O+ is the most widely used proton donor [Citation15–17], with PA(H2O) = 7.16 eV [Citation18]. It is capable of ionising many volatile organic compounds (VOCs) in the atmosphere, such as isoprene or acetone, while the main air components including N2, O2, Ar and CO2 remain uninvolved, as their PAs are 5.12, 4.36, 3.83, and 5.61 eV, respectively [Citation18]. Also saturated hydrocarbons like methane or ethane have significantly smaller proton affinities than water and are therefore not ionised.
For the analysis of technical gases, however, detection of these compounds on trace levels is highly desirable. In these dry environments, the standard PTRMS methods using H3O+ or NH4+ as chemical ionisation agents will not allow for the detection of most contaminants, which have much lower proton affinities than H2O or NH3. Precursor ions with very low proton affinity, on the other hand, might be suitable to detect traces of air or hydrocarbons, including nitrogen, oxygen or CO2. In order to assess the suitability of PTR mass spectrometry for this purpose, the reactivity of proton donors with low proton affinity, like ArH+ or N2H+, must be understood in more detail. Due to the very low proton affinity of argon, ArH+ is capable of transferring the proton to almost any gaseous molecule, except for helium, neon and gaseous fluorine [Citation19]. On the other hand, the energy release upon proton transfer from ArH+ is substantial. With PA(Ar) = 3.83 eV, an additional 3.34 eV are released compared to PTR from H3O+. PTR from ArH+ may cause fragmentation of compounds with high proton affinity, such as acetone, making the mass spectra more complex and assignment of neutral precursors more complicated. The situation is less severe for N2H+, with PA(N2) = 5.12 eV. Compared to PTR from H3O+, the additional energy release is still 2.03 eV, potentially causing fragmentation. This implies that PTR from ArH+ to N2 and from both ArH+ and N2H+ to water are also possible. Any application using ArH+ as a proton donor would therefore require an environment with only minimal abundances of N2 and H2O such as high purity argon gas, the same holds true for N2H+ and H2O.
To learn more about potential pitfalls, we here choose acetone CH3COCH3 as an example of a highly volatile organic solvent with relatively high proton affinity. For PTR from argonium ArH+ or diazenylium N2H+ to acetone, no rate coefficients are available in the literature, and fragmentation pathways are not known. Since acetone is sometimes used to remove organic residue from metal surfaces, it constitutes a potential contaminant in process gas, as fittings, gas bottles or tubing may contain traces of acetone. In particular for the analysis of high purity gases on the pptv level, minimal residues are relevant. We here report proton transfer reaction rate coefficients kabs of ArH+ and N2H+ to acetone measured by FT–ICR mass spectrometry, analyze the product branching and determine the rate coefficients. Fragmentation mechanisms are examined by quantum chemical calculations.
Experimental and theoretical methods
The experiments were carried out on a modified Bruker/Spectrospin CMS47X Fourier transform ion cyclotron resonance mass spectrometer (FT–ICR–MS), equipped with a laser evaporation ion source [Citation20,Citation21]. A detailed description of the setup was published previously [Citation22,Citation23]. ArH+ or N2H+ cations are generated by laser vaporisation of a solid magnesium disk, followed by supersonic expansion of the hot plasma in a short gas pulse of helium seeded with H2 and Ar or N2, respectively, into high vacuum. Supersonic expansion stabilises ArH+ or N2H+, among other ions, and cools the ions well below room temperature. Laser vaporisation is certainly not the most straightforward way of making these ions, but it is actually advantageous for the present study. The very short gas pulses of the source reduce the gas load in the vacuum system and allow for excellent ultra high vacuum conditions, which benefits the determination of absolute pressure and rate coefficients, and minimises side reactions with impurities from the source region.
The ions are further transferred by ion optics via differential pumping stages into the ICR cell [Citation24]. Here the ions are stored, and unwanted ions are removed by resonant excitation, leaving either ArH+ or N2H+ as the primary ion [Citation25]. For a defined period of time, the mass selected ions are exposed to the collision gas, which is introduced at a constant backing pressure via a leak valve. Before opening the leak valve, the acetone is degassed by several freeze–pump–thaw cycles.
Reaction kinetics are generated from mass spectra recorded at varying reaction delays and reaction gas pressures. Pseudo-first order rate coefficients are individually fitted to each recorded kinetic using a genetic algorithm, which treats initial ion intensities and unimolecular rate coefficients as fit parameters. The absolute rate coefficients kabs can be derived after pressure correction, which accounts for the polarizability of acetone for the cold cathode gauge pressure reading as well as the relative positioning of the ICR cell and the pressure gauge with respect to the position of the turbomolecular pump and the leak valve [Citation26]. Averaging over three measurements for N2H+ and 24 measurements for ArH+ yields the values listed below. Errors are given as the experimental standard deviation in Table . Additionally, the main source of systematic error in the setup is assigned to the pressure reading from the cold cathode gauge and the geometry correction, whereby its uncertainty is estimated to be 30% [Citation27,Citation28]. The noise level shown in the kinetics is calculated as the noise level of the respective mass spectrum normalised against the ion intensity. Since the ArH+ ions need approximately 1 s to thermalise to room temperature, the respective data points of the kinetics were discarded. Due to the smaller rotational constant of 3.12 cm-1 [Citation29] and the additional degrees of freedom, the problem is less severe for N2H+, thermalisation happens in approximately 0.1 s. Since the time needed for thermalisation is pressure dependent, the time frame that was used for the numerical fit was adjusted separately for each kinetics.
Table 1. Average of absolute rate coefficients kabs of reactions (1)–(5) are compared with calculated collision rate coefficients kADO, resulting in the efficiency ϕ = kabs/kADO. Errors correspond to the standard deviation of all measuremts. The energy release of each reaction in eV was calculated as the CCSD/aug-cc-pVDZ level of theory single point energy combined of the MP2/aug-cc-pVDZ structures and zero-point energies.
Here it is worthwhile to point out that the thermalisation in this case does not imply that the trapped ions are hot and need collisional cooling. On the contrary, ArH+ ions are formed in a supersonic expansion of helium, which efficiently cools rotations to temperatures below 10 K. With the rotational constant of ArH+ calculated as B = 10.27 cm-1 [Citation30], the J = 0 rotational ground state is most populated at this low rotational temperature. Rivero et al. have recently shown that rotational excitation can have a significant influence on the dynamics and thus on the rate of chemical reactions in the gas phase using simulations [Citation31]. This is further corroborated by experiments, which suggest a link between a molecule’s rotational state and its reactive cross section [Citation32,Citation33]. In collisions with acetone at room temperature, however, the two rotational degrees of freedom of ArH+ are efficiently thermalised, so that the pseudo-first order behavior is established in less than 1 s.
In order to determine the efficiency of the reaction, the respective ADO collision rates are calculated as
where µ is the reduced mass, q the charge of the ion, α describes the polarizability of the neutral molecule, c the dipole locking constant and µD the dipole moment of the neutral collision partner [Citation34,Citation35]. The trajectory method put forward by Su and Chesnavich [Citation36] may be slightly more accurate than ADO theory, but most efficiencies in the gas phase ion chemistry literature have been determined with respect to the ADO collision rate. Since the major source of systematic error is the uncertainty of the pressure measurement, we decided to stay with ADO theory.
Quantum chemical calculations were performed at the MP2/aug-cc-pVDZ level of theory including zero-point energy corrections from harmonic frequency calculations, using the Gaussian 16 software suite [Citation37]. In order to benchmark the results, single-point calculations were performed applying the CCSD method with the MP2 optimised structures.
Table S1 compares optimised and zero-point corrected energy levels at MP2/aug-cc-pVDZ level of theory and the single-point energy of the MP2-optimised structures at CCSD/aug-cc-pVDZ level of theory, with zero-point corrections on the MP2/aug-cc-pVDZ level. In order to gauge the difference between the zero-point energies calculated with MP2 and CCSD, a full geometry optimisation with subsequent calculation of the zero-point energy was performed for two transition states that showed the highest difference in energy between MP2 and single-point CCSD calculations. The most significant difference was found to be only 5 meV. Similarly, the single-point and fully optimised CCSD energies without zero-point correction applied differ by 3 meV or less. This indicates that the differences arise not from disparities of the geometries of MP2 and CCSD or their respective zero-point energy corrections, but rather from the methods. While MP2 and single-point CCSD values differ by up to 0.2 eV, the conclusions for the proposed reaction pathways are not affected, except for the final isomerisation of C3H5+ for PTR from N2H+, where the transition state for isomerisation exceeds the energy of the entrance channel by 0.04 eV. Energies are given in eV with a precision of 0.01 eV throughout the text. Sums and differences were calculated before rounding to this precision, which leads to minor rounding errors.
Furthermore, to gauge whether the MP2 or CCSD results are more reliable, the calculated proton affinities of Ar, N2, and acetone were benchmarked against experimental results [Citation18]. The results are summarised in Table , which shows that CSSD comes closer to the experimental values. Finally, all optimised geometries used are listed at the end of the supporting information.
Table 2. Comparison of experimental [Citation18] and calculated proton affinities of argon, nitrogen, and acetone. The calculated values contain MP2/aug-cc-pVDZ, and CCSD/aug-cc-pVDZ optimised values including their respective thermal enthalpy corrections. Furthermore, CCSD/aug-cc-pVDZ single-point energies using MP2/aug-cc-pVDZ optimised structures and thermal enthalpy corrections are listed.
Results and discussion
The reaction of N2H+ with acetone leads mostly to protonated acetone, while products involving dissociation of acetone are barely above noise level, as seen in Figure (a). Therefore, rate coefficients of the formation of such fragments can only be determined for measurements with sufficient signal to noise ratio (S/N). Protonated acetone is also the most abundant product upon PTR from ArH+, as depicted in Figure (b). However, the high excess energy released during PTR from ArH+ leads to a higher contribution of dissociative proton transfer via loss of methane, as well as water and ethylene loss, reactions (2)-(4), respectively. Each contribute roughly 10% of the product ions after 3.2 s reaction delay in reaction with ArH+, while the reaction with N2H+ yields only 3-7%, respectively, after the same reaction time at a higher pressure. Loss of ethylene leads to formation of protonated formaldehyde, a primary product with significantly higher abundance in the reaction with ArH+ compared to N2H + . However, its intensity is quickly depleted due to a secondary PTR to acetone, reactions 4 and 5. In reactions (1)-(4), X denotes either N2 or Ar.
(1)
(1)
(2)
(2)
(3)
(3)
(4)
(4)
(5)
(5)
Figure 1. Reaction kinetics of (a) N2H+ and (b) ArH+ with acetone at a backing pressure of 1.9 × 10−8 mbar and 1.3 × 10−8 mbar, respectively. For (a) and (b) the first 0.15 and 0.8 s were neglected for the fit, respectively, as thermalisation effects conflict with the pseudo-first order reaction model used for fitting the results.
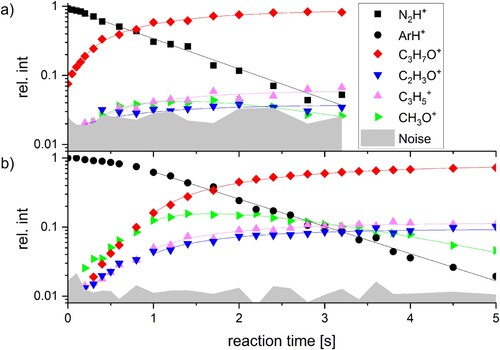
Both N2H+ and ArH+ react with acetone close to collision rate, with 2.5·10−9 cm³ s–1 and 2.4·10−9 cm³ s−1, respectively, see Table . Fragmentation in the reaction with N2H+ is only observable in half of the measurements due to poor S/N, combined with relatively low rate coefficients ranging from 0.6–2.7·10−10 cm³ s–1, reactions 2–4. However, the reaction of ArH+ with acetone exhibits significantly more fragmentation, with only 58% resulting directly in the proton transfer product. Here, the formation of C2H3O+ and C3H5+ proceeds with a partial rate coefficient of kpart(2,Ar) = 1.9·10−10 cm³ s−1 and kpart(3,Ar) = 2.2·10−10 cm³ s-1, respectively. Notably C2H3O+ is also the main fragment in EI mass spectra of acetone [Citation38]. Furthermore, the formation of protonated formaldehyde proceeds relatively fast by PTR from ArH+ with kpart(4,Ar) = 6.1·10−10 cm³ s-1. However, as the PA of formaldehyde corresponds to 7.39 eV, which is significantly lower [Citation18] than the PA(CH3COCH3) = 8.42 eV, a very fast secondary reaction leads again to C3H7O+ with a rate coefficient of kabs(5’’) = 2.2·10−9 cm³ s–1. The same reaction is also observed after PTR from N2H+, here with a rate constant of kabs(5’) = 1.2·10−9 cm³ s–1. The rate coefficients for reaction (5) in the ArH+ and N2H+ experiments agree within error limits, but the error margins are quite large, see Table . We attribute this behavior to the complex interplay of ion internal energy after the initial proton transfer reaction and radiative cooling before the next collision can take place, which leads to a pressure dependence of the secondary reaction.
The contribution of 42% fragmentation upon proton transfer from ArH+ compared to 21% for N2H+ is most likely due to the different excess energy, with minor influence of the additional degrees of freedom of the CH3COCH3 + N2H+ collision complex.
The absolute rate coefficients are compared to their corresponding calculated collision rate coefficients kADO, collected in Table . Due to its lower mass, N2H+ captures acetone slightly faster than ArH+, with kADO(N2H+) = 2.8·10−9 cm³ s–1 and kADO(ArH+) = 2.5·10−9 cm³ s–1, respectively. Comparison of the sum of all partial rate coefficients for N2H+ + C3H6O and for ArH+ + C3H6O with their ADO rates provides the corresponding efficiencies of ϕ = 90–93% and ϕ = 96%, respectively. Within error limits, proton transfer proceeds with collision rate in both cases.
The potential energy surfaces of reactions 1–5 were investigated using quantum chemical calculations at the MP2/CCSD/aug-cc-pVDZ level of theory. Figure shows energy levels of stationary points including zero-point correction in harmonic approximation. The structures were optimised on MP2/aug-cc-pVDZ level of theory, displayed energies were determined using single-point CCSD/aug-cc-pVDZ calculations and MP2/aug-cc-pVDZ zero-point energy. PTR from linear N2H+ or ArH+ to acetone proceeds without barrier, with an energy release of –3.35 eV and –4.57 eV, respectively, Figure (a,b), for protonation at the oxygen atom. Protonated acetone may stabilise by IR emission and, likely to a smaller extent, kinetic energy release of the products, resulting in protonated acetone, the main product. However, the available energy is sufficient to afford hydrogen atom transfer from a CH3 group to OH, leading to elimination of H2O, as shown in detail in Figure (a). The calculations predict the structure of the ionic product C3H5+ as either CH2CCH3+ at –2.14 eV, or the energetically preferred CH2CHCH2+ at –2.51 eV, with a barrier of 0.82 eV for the isomerisation from CH2CCH3+. For ArH+, this barrier lies still significantly below the entrance channel, with -1.18 eV (MP2) or -1.31 eV (CCSD), depending on the theory level. For N2H+, however, the transition state is almost isoenergetic to the entrance channel, and a mixture of the two C3H5+ isomers is most likely obtained.
Figure 2. Reaction profiles of PTR from N2H+ and ArH+ to acetone and formation of (a) C3H5+ with loss of H2O; (b) CH2OH+ with loss of C2H4; (c) C2H3O+ with loss of CH4. Structures are calculated at the MP2/augcc-pVDZ level of theory, the energies are calculated using CCSD/aug-cc-pVDZ single-point energies and the zero-point corrections derived from MP2/aug-cc-pVDZ level of theory. The trivial neutrals (Ar or N2) are not shown after the PTR, but their energies were considered.
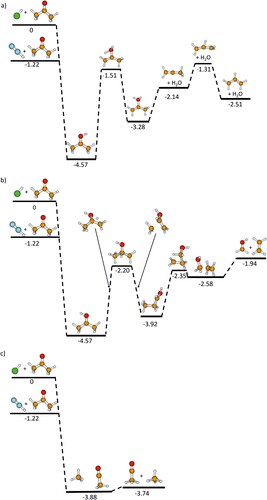
Hydrogen atom transfer to the central carbon atom, as shown in Figure (b), does not lead to a stable structure. The corresponding stationary point on the potential energy surface is a well characterised transition state, with one imaginary frequency that connects the protonated acetone structure with the CH3CH2CHOH+ intermediate, which is formed via a transient cyclopropane structure. The structures shown to the left and right of the transition state are geometry optimised intermediate structures along the reaction path from an IRC calculation to illustrate the rearrangement. Subsequent β-hydrogen atom transfer from the CH3 group to CHOH through a 4-membered cyclic transition state leads to ethylene and protonated formaldehyde. Thus, the initially central carbon atom ends up in the formaldehyde moiety.
Dissociative proton attachment to a terminal methyl group leads to elimination of methane and CH3CO+, illustrated in Figure (c). In reactions of H3O+ with acetone, Bohme and co-workers [Citation12] exclusively observed proton transfer with formation of protonated acetone. The higher proton affinity of water, PA(H2O) = 7.16 eV, shifts the energetics by 3.32 eV relative to the ArH+ entrance channel. This renders the pathways in Figure (a,b) thermochemically inaccessible, and makes dissociative proton transfer in Figure (c) only 0.47 eV exothermic. Since already the much smaller shift from ArH+ to N2H+ substantially suppresses reaction (2), it is not surprising that this pathway disappears completely for H3O+, although being thermochemically accessible. While proton transfer from CH3CO+ to acetone has been reported in earlier work [Citation39–44], the low intensity of this product in our study combined with the high intensity of protonated acetone as primary product does not warrant an unambiguous identification of this reaction channel.
Conclusions
Within error limits, PTR from N2H+ as well as ArH+ to acetone proceeds with collision rate. Protonated acetone is the dominant product in both cases, with branching ratios of 79% and 58%, respectively. Thus the presence of protonated acetone allows for an unambiguous identification of this compound in trace gas analysis. The high excess energy, however, leads to substantial fragmentation. Protonated ketene CH3CO+ is not a big problem from an analytical point of view, since it is not an abundant contaminant in high-purity gases, while protonated formaldehyde CH2OH+ and protonated propadiene C3H5+ may hamper the quantification of the respective neutrals, especially in cases where acetone concentrations are substantially higher than those of formaldehyde or propadiene. CH3CO+ originates from protonation of a methyl group, followed by methane elimination, via dissociative proton transfer. Protonated formaldehyde CH2OH+ and C3H5+ are formed after protonating the carbonyl oxygen atom. If the initial H atom migration takes place from a terminal CH3 group to OH in protonated acetone, H2O elimination leads to C3H5+. Conversely, if a methyl H atom is transferred to the carbonyl carbon atom, extensive rearrangements lead to ethylene elimination and detection of protonated formaldehyde. The additional energy introduced into the system by proton transfer from ArH+ affords a higher branching ratio of the fragmentation products than N2H+.
Acknowledgements
We gratefully acknowledge financial support from the European Regional Development Fund (ERDF) and the Province of Tyrol via K-Regio Projekt GALANT, grant number EFRE 2018-8. The computational results presented have been achieved using the HPC infrastructure LEO of Universität Innsbruck. The authors thank Dr. Milan Ončák for helpful discussion.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- H. Hock and S. Lang, Ber. Dtsch. Chem. Ges. A/B. 77, 257–264 (1944). doi:10.1002/cber.19440770321
- AP News, Global Acetone Industry. (2020). https://apnews.com/press-release/business-wire/coronavirus-pandemic-business-health-2019-2020-coronavirus-pandemic-e71147f2b7814d58b1885395384352fa.
- A. Wisthaler, N.R. Jensen, R. Winterhalter, W. Lindinger and J. Hjorth, Atmos.: Environ. 35, 6181–6191 (2001). doi:10.1016/S1352-2310(01)00385-5 .
- H.B. Singh, M. Kanakidou, P.J. Crutzen and D.J. Jacob, Nature. 378, 50–54 (1995). doi:10.1038/378050a0 .
- L. Jaeglé, Atmos. Environ. 35, 469–489 (2001). doi:10.1016/S1352-2310(00)00376-9.
- S. Canneaux, N. Sokolowski-Gomez, E. Henon, F. Bohr and S. Dóbé, Phys. Chem. Chem. Phys. 6, 5172–5177 (2004). doi:10.1039/B409900A .
- T. Kawai, T. Yasugi, K. Mizunuma, S. Horiguchi, H. Iguchi and M. Ikeda, Toxicol. Lett. 62, 85–91 (1992). doi:10.1016/0378-4274(92)90081-T .
- M.P. Kalapos, Acetone, in: P. Wexler (Ed.), Encyclopedia of Toxicology, Third Edition, Academic Press an imprint of Elsevier, Amsterdam, Boston, Heidelberg, London, New York, Oxford, Paris, San Diego, San Francisco, Singapore, Sydney, Tokyo, 2014, pp. 36–39.
- E. Wigaeus, S. Holm and I. Åstrand, Scand. J. Work Environ. Health. 7, 84–94 (1981). https://www.jstor.org/stable/40964883.
- A.J. Wein, L.R. Kavoussi, A.W. Partin and C.A. Peters, in Campbell-Walsh Urology. Elsevier, Philadelphia, PA, 2016.
- C. Turner, VOC Analysis by SIFT-MS, GC-MS, and Electronic Nose for Diagnosing and Monitoring Disease, edited by A. Amann, D. Smith (Eds.), Volatile Biomarkers: Non-Invasive Diagnosis in Physiology and Medicine, first edition, Elsevier, Amsterdam, 2013, 343–357. doi:10.1016/B978-0-44-462613-4.00018-0.
- G.I. Mackay, S.D. Tanner, A.C. Hopkinson and D.K. Bohme, Can. J. Chem. 57, 1518–1523 (1979). doi:10.1139/v79-248.
- C.G. Freeman, P.W. Harland and M.J. McEwan, Aust. J. Chem. 31, 2157–2160 (1978). doi:10.1071/CH9782157 .
- S.D. Tanner, G.I. Mackay and D.K. Bohme, Can. J. Chem. 57, 2350–2354 (1979). doi:10.1139/v79-378 .
- W. Lindinger, A. Hansel and A. Jordan, Int. J. Mass Spectrometry Ion Processes. 173, 191–241 (1998). doi:10.1016/S0168-1176(97)00281-4.
- W. Lindinger and A. Jordan, Chem. Soc. Rev. 27, 347–375 (1998). doi:10.1039/a827347z .
- R.S. Blake, P.S. Monks and A.M. Ellis, Chem. Rev. 109, 861–896 (2009). doi:10.1021/cr800364q .
- E.P.L. Hunter and S.G. Lias, J. Phys. Chem. Ref. Data. 27, 413–656 (1998). doi:10.1063/1.556018 .
- Experimental Proton Affinities, 2022. https://cccbdb.nist.gov/palistx.asp.
- V.E. Bondybey and J.H. English, J. Chem. Phys. 74, 6978–6979 (1981). doi:10.1063/1.441064 .
- T.G. Dietz, M.A. Duncan, D.E. Powers and R.E. Smalley, J. Chem. Phys. 74, 6511–6512 (1981). doi:10.1063/1.440991.
- C. Berg, T. Schindler, G. Niedner-Schatteburg and V.E. Bondybey, J. Chem. Phys. 102, 4870–4884 (1995). doi:10.1063/1.469535 .
- R.F. Höckendorf, O.P. Balaj, C. van der Linde and M.K. Beyer, Phys. Chem. Chem. Phys. 12, 3772–3779 (2010). doi:10.1039/b921395c .
- C. Berg, M. Beyer, T. Schindler, G. Niedner-Schatteburg and V.E. Bondybey, J. Chem. Phys. 104, 7940–7946 (1996). doi:10.1063/1.471510 .
- A.G. Marshall, C.L. Hendrickson and G.S. Jackson, Mass Spectrom. Rev. 17, 1–35 (1998). doi:10.1002/(SICI)1098-2787(1998)17:1<1:AID-MAS1>3.0.CO;2-K.
- R.F. Höckendorf, C. van der Linde, O.P. Balaj, I. Herber and M.K. Beyer, Int. J. Mass Spectrometry. 300, 44–49 (2011). doi:10.1016/j.ijms.2010.12.007 .
- T. Schindler, C. Berg, G. Niedner-Schatteburg and V.E. Bondybey, Ber. Bunsen-Ges. Phys. Chem. 96, 1114–1120 (1992). doi:10.1002/bbpc.19920960906 .
- D. Schröder, H. Schwarz, D.E. Clemmer, Y.M. Chen, P.B. Armentrout, V.I. Baranov and D.K. Bohme, Int. J. Mass Spectrom. Ion Process. 161, 175–191 (1997). doi:10.1016/S0168-1176(96)04428-X .
- P. Thaddeus and B.E. Turner, Astrophys. J. 201, L25–L26 (1975). doi:10.1086/181932.
- J.M. Brown, D.A. Jennings, M. Vanek, L.R. Zink and K.M. Evenson, J. Mol. Spectrosc. 128, 587–589 (1988). doi:10.1016/0022-2852(88)90173-7.
- U. Rivero, O.T. Unke, M. Meuwly and S. Willitsch, J. Chem. Phys. 151, 104301 (2019). doi:10.1063/1.5114981.
- S.L. Anderson, P.R. Brooks, J.D. Fite and O. van Nguyen, J. Chem. Phys. 72, 6521–6528 (1980). doi:10.1063/1.439154.
- A.A. Viggiano and R.A. Morris, J. Phys. Chem. 100, 19227–19240 (1996). doi:10.1021/jp962084x .
- T. Su and M.T. Bowers, J. Chem. Phys. 58, 3027–3037 (1973). doi:10.1063/1.1679615 .
- G. Kummerlöwe and M.K. Beyer, Int. J. Mass Spectrom. 244, 84–90 (2005). doi:10.1016/j.ijms.2005.03.012 .
- T. Su and W.J. Chesnavich, J. Chem. Phys. 76, 5183–5185 (1982). doi:10.1063/1.442828 .
- M.J. Frisch et al., Gaussian 16 Revision A.03, 2016.
- Acetone Mass Sepctrum (electron ionization), 2014. https://webbook.nist.gov/cgi/cbook.cgi?ID=C67641%Mask=200#Mass-Spec.
- W.J. van der Hart and H.A. van Sprang, J. Am. Chem. Soc. 99, 32–35 (1977). doi:10.1021/ja00443a007 .
- P. Ausloos and S.G. Lias, Chem. Phys. Lett. 51, 53–56 (1977). doi:10.1016/0009-2614(77)85353-0 .
- L.W. Sieck and P. Ausloos, Radiat. Res. 52, 47–58 (1972). doi:10.2307/3573587 .
- K.A.G. MacNeil and J.H. Futrell, J. Phys. Chem. 76, 409–415 (1972). doi:10.1021/j100647a020 .
- A.S. Blair and A.G. Harrison, Can. J. Chem. 51, 703–717 (1973). doi:10.1139/v73-107 .
- M. Kumakura and T. Sugiura, J. Phys. Chem. 82, 639–643 (1978). doi:10.1021/j100495a003 .