
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Intramolecular radical and proton migration in the gas phase are important processes driving the dissociation reactions underlying common tandem mass spectrometry processes, such as collision-induced and electron-induced dissociation reactions (CID and ExD). Mechanistic insights in these processes requires experiments that probe the molecular structures of ions along the reaction pathways, usually combined with quantum-chemical calculations. The combination of ion trap mass spectrometry with IR laser spectroscopy, pioneered among others by Dieter Gerlich, provides a particularly effective method to explore details of the ion structures. In this work, we employ infrared multiple-photon dissociation (IRMPD) spectroscopy and density functional theory (DFT) calculations to probe the reactant and product ion structures of a homolytic bond cleavage reaction. First, we employ IRMPD spectroscopy to establish that protonation of the 4-bromoaniline precursor occurs on the amine moiety and then that C-Br homolytic cleavage produces the π-radical cation of aniline. Transition-state calculations are performed to compare the various pathways that connect reactant and product ions, including both proton and radical transfer mechanisms.
GRAPHICAL ABSTRACT

1. Introduction
Both proton transfer and radical transfer are among the most commonly invoked processes to understand the reaction mechanisms of gaseous ions in mass spectrometry. The high chemical reactivity of open-shell radicals is well appreciated throughout chemistry in their role as intermediates in a plethora of chemical processes, from the synthesis of polycylic aromatic hydrocarbons in the interstellar medium [Citation1] to the formation of degradation products in cellular processes [Citation2]. In the field of ion chemistry, there has been a continuous interest in the structure and chemistry of radical cations [Citation3–9]. Early on, ionisation relied strongly on electron ionisation, producing predominantly radical cation species. Although the advent of softer ionisation methods has shifted this paradigm to closed-shell ions, electron-induced dissociation methods (ExD) [Citation10–13] have become popular tandem mass spectrometry (MS/MS) methods and generate radical cation and anion products from the closed-shell precursors. Most common implementations of ExD MS/MS involve a 1-electron reduction of multiply protonated species, and numerous studies have addressed details of the underlying dissociation reaction mechanisms. Radical migration through H-atom transfers plays a central role in these mechanisms.
In contrast, collision-induced dissociation (CID) of closed-shell protonated species formed in electrospray ionisation (ESI) has been shown to be driven predominantly by proton transfer processes. The dissociation mechanisms of protonated peptides have been especially well studied in this regard in attempts to gain insight into the mechanistics underlying MS-based peptide and protein sequencing. Indeed, the “mobile proton model” of peptide dissociation [Citation14–17] emphasises the role of proton transfer in these mechanisms. Note that efforts to understand ExD dissociation mechanisms have also focussed particularly on peptides because of the use of ExD in protein sequencing applications.
Mechanistic insights into radical and proton migration processes have been obtained from quantum-chemical computations [Citation16,Citation18], as well as from experimental investigations. These experimental mass spectrometry studies must be able to distinguish isomeric ion structures that differ only in the location of a single H or H
, i.e. they must be sensitive to the regioselectivity of protonation and radical formation. Distinguishing structures of the same m/z-value is obviously a challenge in mass spectrometry [Citation19,Citation20], and classical structural elucidation tools rely on tandem mass spectrometry methods, such as collision induced dissociation (CID) [Citation20] or ion-molecule reactions [Citation21]. An alternative, relatively new structural method in MS involves different implementations of ion spectroscopy, which have been extensively employed to characterise CID and ExD reaction products of small peptides [Citation21–30]. These methods provide unique IR fingerprints that relate sensitively to the product ion isomeric structure, including the location of protons and radicals.
Proteins and peptides are extremely complex molecules from a physical and chemical standpoint: they contain a plethora of nucleophilic sites and are conformationally very flexible, which is often instrumental in the H/H
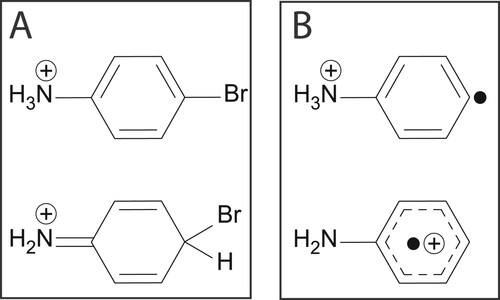
-transfer pathways. In contrast, in this study we investigate a system with no conformational flexibility, so that the intrinsic radical/proton mobility along the path from precursor to product can be evaluated. We study the homolytic bond cleavage reaction in a protonated Br-substituted aniline derivative. Using ion spectroscopy [Citation22,Citation29,Citation31], we establish the reactant and product ion structures, which have both been under discussion [Citation33,Citation34]. Whether aniline and its halo-derivatives preferably protonate on the amine nitrogen or on the para-position of the aromatic ring has been an issue of debate and may depend on the ion source conditions [Citation33–40]. Homolytic expulsion of the bromine radical may then result in the aniline radical cation or in a distonic phenylenyl-ammonium (
C
H
-NH
) isomer of aniline
(Figure ) [Citation38,Citation41]. With the reactant and product ion structures established, we model the conceivable reaction pathways connecting reactant and product, which include both proton and radical migration pathways, by quantum-chemical computations.
Figure 1. (A) Possible tautomeric structures of protonated 4-bromoaniline. (B) Isomers of the ion CH
N
attainable upon homolytic loss of bromine from protonated 4-bromoaniline: distonic phenylenyl-ammonium (top) or aniline radical cation (bottom).
2. Experimental methods
Experiments are performed at room temperature in a modified 3D ion trap mass spectrometer (Bruker AmaZon Speed ETD) coupled to the beamline of the Free-Electron Laser for Infrared eXperiments (FELIX) [Citation42]. A detailed description of the setup has been reported elsewhere [Citation43]. In order to produce the protonated species, an atmospheric pressure chemical ionisation (APCI) source was employed [Citation44]. Protonated ions as well as radical cations are typically formed and observed in the mass spectrum.
4-Bromoaniline was purchased from Sigma-Aldrich (St. Louis, USA) and used without further purification. A few mg were dissolved in methanol yielding a mM concentration. This stock solution was further diluted with methanol until a concentration of ∼10 M was obtained. Approximately 1
of acetic acid was added to the solution, which was then infused into the APCI source at a flow rate of 180 µL/h, nebulised with N
at 2 bar and 180
C and vaporised at 410
C. The evaporating droplets travel towards the corona discharge needle where chemical ionisation occurs. The potential difference between the end plate and the capillary is 4500 V and the corona current is set to 6000 nA. The ions enter the vacuum of the mass spectrometer through the capillary and after passing through ion-transfer optics are trapped in the radio-frequency 3D ion trap. Ions of interest are accumulated for ∼20 ms and mass-isolated in the ion trap, where they are irradiated with the FELIX output, which consists of between one and six ∼10-µ s macropulses of 20–100 mJ (depending on the emission wavelength) at a 10 Hz repetition rate. When the laser frequency is resonant with a molecular vibration of the mass-selected ion, fragmentation occurs as a result of the absorption of multiple photons. In the present experiments, the m/z 172 isotopologue of protonated 4-bromoaniline is isolated. After IR irradiation, a mass spectrum is recorded and averaged six times for each IR wavelength. IRMPD generates product ions at m/z 93 and 67, as well as a minor peak at m/z 92. IRMPD spectra are obtained by determining the fractional fragmentation induced at each wavelength step (3 cm
), relating the precursor and fragment ion intensities
(1)
(1)
The fragment fluence S(λ) is then calculated as
(2)
(2)
The laser is calibrated with a grating spectrometer with an accuracy of ±0.01 µm and the fragment fluence is corrected linearly for variations in FELIX power over the spectral range [Citation45].
An ion of composition CH
NH
is obtained via collision-induced dissociation (CID) of the protonated precursor 4-bromoaniline (Figures S1–S2). Precursor ions are mass-selectively accelerated by excitation at their secular frequency in the quadrupole trap, leading to higher-energy collisions with the He buffer gas and the formation of the product ions. An ion accumulation time of 30 ms is used and the CID amplitude is optimised to maximise the m/z 93 product ion signal. Upon IRMPD, this ion mainly produces a fragment ion at m/z 67, which likely corresponds to the cyclopentenyl carbocation (C
H
), produced by expulsion of a CN radical.
2.1. Computational methods
To determine the molecular structure of the CH
NBr
and C
H
N
ions, their experimental IR spectra are contrasted against calculated IR spectra for different isomeric forms of the ions. Calculations are performed using density functional theory (DFT) at the B3LYP/6-31++G(d,p) level of theory employing the Gaussian16 software package [Citation46] as installed at the Snellius supercomputer at SURFsara, Amsterdam. Furthermore, geometry optimizations and frequency calculations were performed employing B3LYP/6-311++G(d,p) and MP2/ 6-31++G(d,p) to verify the relative energies of the different structures; single-point energy calculations at the B3LYP/6-311++G(d,p) geometry were performed using a larger basis as well as the CCSD(T) method. Geometry optimizations were performed with the standard convergence criteria and vibrational spectra were computed within the harmonic oscillator approximation. Harmonic frequencies were scaled by a factor of 0.975 [Citation47]. Anharmonic calculations were also performed using B3LYP/6-31++G(d,p) and the freq=anharm keyword in Gaussian16. Theoretical and experimental spectra were normalised along the intensity axis.
Transition state (TS) calculations were performed to elucidate the radical/proton migration pathways and the bond cleavage reaction. TS geometry optimisation and frequency calculations were performed following a quasi-Newton synchronous transit (QST3) calculation. All stationary points along the reaction pathways were verified through vibrational analysis to be local minima (zero imaginary frequencies) or first-order transition states (one imaginary frequency). Visualisation of the normal mode corresponding to the single imaginary frequency verified that the structures are indeed saddle points connecting the reactant and the product. The character of the normal mode associated with the imaginary frequency has been analysed to ensure it resembles the reaction coordinate under consideration. Zero-point corrected energies were used to calculate the activation energy barriers. In order to obtain the potential energy surface (PES) connecting the transition state structures and the minima, intrinsic reaction coordinate (IRC) calculations have been performed as implemented in Gaussian16.
3. Results and discussion
3.1. Precursor ion characterisation
The protonation site of gaseous aniline (and its halo-substituted derivatives) has been under long debate, as especially the para-position on the phenyl ring competes strongly with protonation on the amine nitrogen [Citation33–40]. Most computational studies place the ammonium isomer of anilinium marginally higher in energy than the phenylium isomer, although this is dependent on the level of theory [Citation37]. Experimentally, most studies identify the ammonium ion structure as the dominant contributor to the ion population, which is attributed to kinetic effects [Citation48]. Adding to the controversy, mixtures of structures have explicitly been reported and source conditions have been shown to influence the isomer population [Citation35,Citation39,Citation40]. Halo-substitution at the para-position on the ring influences the relative stability of the two protonation isomers, making the N-protonated form more favourable (see below and Refs. [Citation33,Citation34]), although margins remain narrow.
Given this ambiguity, we recorded the IRMPD spectrum of the precursor ion, protonated 4-bromoaniline, to firmly establish the starting ion structure. Figure compares the experimental IR spectrum to computed spectra for the four conceivable isomers that result from protonation at different sites in the molecule. Among the plausible alternatives, the best spectral match is obtained with the isomer protonated on the amino group, which is computed to possess the lowest energy (see Table ). The agreement in band positions and relative intensities between theoretical and experimental spectra suggests that protonation occurs exclusively on the NH group and not on the aromatic ring. Based on considerations of relative energies, the para-protonated structure is indeed a reasonable alternative at +13.8 kJ/mol (using the B3LYP energies), but the vibrational bands at 1083 cm
(inversion mode of the amino group) and 1460 cm
(umbrella mode) are not predicted by the calculations; moreover, the scissoring mode near 1580 cm
is shifted by 100 cm
and deviates in relative intensity. The clear mismatch of these signature IR features confirms that protonation of 4-bromoaniline occurs solely on the amino group in our APCI ion source. The experimental spectrum shows a hint of a band at 1670 cm
, which can be tentatively attributed to the minute presence isomer D contributing to the overall spectrum. Although relative energies of the isomers of protonated 4-bromoaniline differ between the B3LYP and MP2 calculations, the relative energy ordering of the isomers is retained; optimised geometries at the two levels of theory for structure A deviate in the torsional angle of the ammonium moiety relative to the aromatic plane, likely explaining the difference.
Figure 2. Experimental spectrum of protonated 4-bromoaniline (black in all panels) compared to computationally predicted spectra of its four different protonation isomers labelled A through D. The shaded coloured spectra represent the stick spectra convoluted with a 30 cm FWHM Gaussian profile. Harmonic frequencies are calculated at the B3LYP/6-31++G(d,p) level and are scaled by 0.975. Relative Gibbs energies (at 298 K) are shown for each isomer.
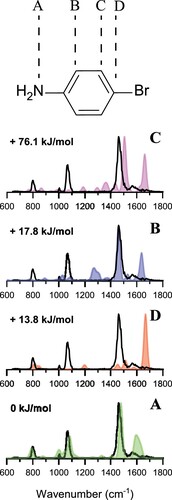
Table 1. Relative Gibbs energies (at 298 K) in kJ/mol of protonated 4-bromoaniline and radical cationic aniline, calculated at the different levels of theory.
3.2. Product ion (m/z 93) characterisation
Collisional activation induces facile homolytic cleavage of the C-Br bond, producing an ion at m/z 93 with elemental composition CH
N
. This ion could potentially correspond to the aniline π-radical cation or to a distonic radical cation, with the charge on the amino nitrogen and the radical on the C-atom in the para position [Citation34]. This phenylenyl-ammonium ion would derive from direct C-Br bond cleavage of protonated 4-bromoaniline in the amine-protonated structure established above. However, this distonic radical cation is higher in energy by a large margin (at least 180 kJ/mol) compared with the isomeric aniline radical cation (see Table ). Nonetheless, its formation has been reported using collisional activation of protonated iodoaniline and ion-molecule reactions to probe the distonic nature of the product ion formed [Citation38,Citation41].
To establish the actual structure of the CH
N
ion at m/z 93 in our experiment, we mass-isolate this ion and record its IRMPD spectrum, shown in Figure as the black curve. The experimental spectrum is compared with the computed spectra of the two suggested isomeric structures of the m/z 93 ion, with the phenylenyl-ammonium distonic radical cation in orange and the aniline radical cation in blue and red.
Figure 3. Experimental spectrum of the m/z 93 ion generated by CID of protonated 4-bromoaniline (black in both panels) compared with computationally predicted spectra of two different CH
N
isomers, the phenylenyl-ammonium distonic radical cation (orange) and the aniline π-radical cation (blue). For the latter, the anharmonic frequency calculation is reported as the shaded red spectrum. Computed stick spectra were convoluted with a 30 cm
FWHM Gaussian lineshape. In the bottom panel the IR spectrum of the aniline-Ar radical cation recorded in a supersonically cooled molecular beam taken from Ref. [Citation49] (green) is shown.
![Figure 3. Experimental spectrum of the m/z 93 ion generated by CID of protonated 4-bromoaniline (black in both panels) compared with computationally predicted spectra of two different C6H7N+∙ isomers, the phenylenyl-ammonium distonic radical cation (orange) and the aniline π-radical cation (blue). For the latter, the anharmonic frequency calculation is reported as the shaded red spectrum. Computed stick spectra were convoluted with a 30 cm−1 FWHM Gaussian lineshape. In the bottom panel the IR spectrum of the aniline-Ar radical cation recorded in a supersonically cooled molecular beam taken from Ref. [Citation49] (green) is shown.](/cms/asset/4426357f-9289-457a-93ae-6ca894972961/tmph_a_2192307_f0003_oc.jpg)
At first glance, none of the computed harmonic IR spectra appear to match the experimental IRMPD spectrum particularly well, especially due to the absence of the very strong feature observed at 1339 cm. However, gas-phase experimental spectra of the aniline radical cation have been previously reported [Citation49–52], showing an unusually strong band at 1319 cm
that has no counterpart in the DFT-computed spectrum. In earlier spectroscopic studies of the aniline radical cation, it was assigned to the overtone of the inversion mode [Citation49,Citation53]. Figure shows a back-to-back comparison of our spectrum and the molecular beam spectrum of Piest et al. [Citation49] (green curve) and overall, the frequencies of the observed vibrational bands are reproduced well. However, a significant mismatch in the relative intensities of the bands is observed, especially in the low-frequency range of the spectrum. We attribute this discrepancy mainly to the multiple-photon dissociation detection method in our experiment, which becomes less efficient at lower photon energies. In contrast, published gas-phase spectra of the aniline radical cation were obtained as predissociation spectra of aniline
-rare gas complexes [Citation49,Citation50]. Table provides a summary of observed vibrational frequencies in our IRMPD spectrum and their theoretical predictions, in the molecular beam spectrum of Ref. [Citation49], in helium nanodroplets [Citation52], in Ar matrix [Citation51] and in the gas phase [Citation53,Citation54]. We conclude that the ion probed in our experiment corresponds to the aniline radical cation, and not to the distonic phenylenyl-ammonium isomer. The absence of computed bands between 600 and 700 cm
as well as the inability to explain the strong feature near 1340 cm
exclude the presence of a major fraction of this isomer in the ion population.
Table 2. Vibrational band frequencies (cm) of the m/z 93 ion compared with the experimental peak positions for the aniline radical cation and the band centres of the convoluted computed spectrum.
We can now analyse the experimental spectrum in more detail using the computed normal mode vibrations for the aniline radical cation. The band at 617 cm can be assigned as an out-of-plane C-H and N-H bending mode of the molecule, whilst the band at 660 cm
is due to the inversion mode of the NH
group, usually found at 656-658 cm
[Citation53,Citation54]. As in previous studies [Citation49,Citation50], this band is not predicted by calculations due to the fact that this vibrational mode is not appropriately described by a harmonic potential, likely causing the discrepancy between experiment and theory. The anharmonic frequency calculation slightly improves the match with experiment (see the red shaded spectrum in Figure ), as it predicts the band at 660 cm
and an additional feature at 1285 cm
, possibly corresponding to the band at 1339 cm
. However, the band at 770 cm
appears to be blue-shifted in the anharmonic spectrum and it is better predicted by the scaled harmonic calculations. The strong band observed at 1339 cm
is attributed to the overtone of the inversion mode, following previous experimental studies that report it around 1323–1327 cm
[Citation49,Citation50,Citation52–54], which fits well within the envelope observed in our spectrum. The band at 770 cm
can be assigned as the C-H ring wagging mode. At 1635 cm
we observe the NH
scissoring mode. A number of weaker bands is also observed and is in line with the low-temperature spectra reported in the literature.
3.3. Computational evaluation of the reaction pathway
The ion spectroscopy experiments establish the precursor ion as the amine-protonated 4-bromoaniline isomer and the m/z 93 product ion as the aniline π-radical cation. Hence, the reaction must involve the transfer of a hydrogen from the amine nitrogen to the carbon in the para-position. Different reaction mechanisms were explored computationally and their potential energy surfaces were mapped, as shown in Figure . We can distinguish between mechanisms in which C-Br bond cleavage precedes or follows hydrogen transfer: if C-Br bond cleavage occurs first, the subsequent hydrogen migration is radical-driven and two plausible mechanisms can be devised. Alternatively, a rearrangement in the closed-shell ion may occur first by proton transfer from the ammonium moiety to the para-position of the phenyl ring, which then induces the expulsion of the Br radical. The possible pathways are summarised in Figure .
Figure 4. Potential energy surface at the B3LYP/6-31++G level of theory of the proton transfer mechanism in 4-Bromoaniline (black) and the two possible mechanism for hydrogen migration in radical cationic aniline: mechanism I (red) and mechanism II (blue). Gibbs energies (at 298 K) in kJ/mol of transition states and intermediates are relative to protonated 4-Bromoaniline.
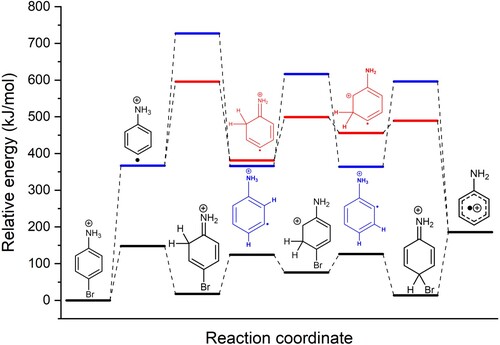
The two radical migration pathways are referred to as mechanisms I and II, whose intermediates and transition states are represented by red and blue lines, respectively, in Figure . The bond dissociation energy (BDE) for the Br-CH
NH
bond at the B3LYP/6-31++G
level of theory is 360 kJ/mol (Figure S3), which relates well with the value of 337 kJ/mol reported in literature for a Br−C
H
bond [Citation56]. Following C-Br bond cleavage, the first step in both mechanisms is rate limiting, although the barrier is significantly higher in mechanism II. All transition states (TSs) along pathway II are more than 100 kJ/mol higher in energy than their counterparts in mechanism I. In contrast, the intermediates along pathway II are more stable than along pathway I, which may reflect their retained aromaticity compared to mechanism I intermediates, where the sp
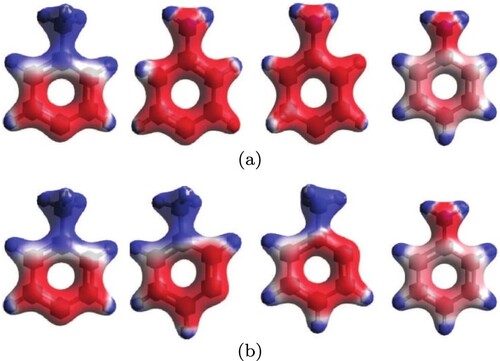
-hybridised carbon breaks the aromaticity. On the other hand, in mechanism I, the positive charge initially localised on the amino group becomes delocalised throughout the ring, providing an overall stabilising effect. In mechanism II, the positive charge remains localised on the ammonium moiety until charge delocalisation within the π-system occurs in the final product. Charge localisation/delocalisation in the intermediates along the two paths can be appreciated from the electrostatic potential maps in Figure . The computations suggest that the effect of charge delocalisation in mechanism I outweighs the retained ring aromaticity of mechanism II.
Figure 5. Total charge density maps of molecular structures for mechanism I (top row) and II (bottom row). Red regions represent high electron density, whereas blue regions are low electron density.
Careful inspection of the frontier molecular orbitals (see Figures S4–S5 in the Supplementary Information) and their energies provides further insight into the process. In the HOMO of the reactant, the radical is localised in a σ-orbital at the para-position and the LUMO reflects the positive charge localised on the ammonium group. In the product, both HOMO and LUMO are delocalised π-orbitals and consequently their MO energies are lowered. The frontier MOs of the intermediates display interesting differences between mechanisms I and II: in the SOMOs of the first intermediates, the radical is still localised in a σ orbital. However, the LUMO in mechanism I is now a delocalised π-orbital, having a significantly lower orbital energy and reduced SOMO-LUMO gap as compared with mechanism II. In the second intermediate, the SOMO delocalises the radical over the π-system including the N-atom in mechanism I, whereas in mechanism II the radical is delocalised over the phenyl ring but not onto the N-atom. Moreover, the SOMO-LUMO gap is smaller in mechanism I. Overall, these arguments suggest that mechanism I is favoured over II [Citation57,Citation58].
In contrast to the radical migration pathways described above, in the proton transfer pathway, homolytic C-Br bond cleavage occurs after proton migration from the nitrogen atom to the para-carbon atom. The potential energy surface for this mechanism is indicated by the black line in Figure . Apart from the expulsion of the bromine radical, the reaction path computed here is quantitatively close to that reported for unsubstituted protonated aniline [Citation38]. The main discrepancy compared to the values reported by Flammang et al. is the relative energy of the first transition state, which was found to be the rate limiting step for unsubstituted protonated aniline, with an energy about 100 kJ/mol higher than our value for the para-bromo derivative. This difference in transition state energy may be justified by the bromine atom in the para position acting as a π-donor to delocalise the charge and consequently stabilise the first transition state. From here on, the two subsequent energy barriers leading to the regio-isomer bearing the proton in the para-position are submerged. Finally, cleavage of the C-Br bond is the rate-limiting step in this mechanism; a relaxed potential energy scan shows that there is no significant barrier in this bond cleavage reaction.
Comparing the energetics of all reaction pathways evaluated above, the latter – proton transfer followed by C-Br bond homolysis – represents the lowest-energy path from reactants to products. The rate limiting step is the C-Br bond cleavage; the proton transfer steps, including the N-to-C migration, are lower in energy and may proceed prior to the ion acquiring enough energy to surmount the final threshold during the slow CID heating process. This scenario agrees with that put forward by Julian and coworkers describing the C-I bond cleavage reaction in iodotyrosine [Citation21]. Collisional activation does not allow regio-selective radical formation as it mobilises the proton before C-I bond homolysis occurs (in contrast to a UV-induced dissociation reaction).
4. Conclusions
IRMPD ion spectroscopy was used to probe reactant and product ion structures of protonated para-bromoaniline undergoing collision-induced homolytic C-Br bond cleavage. Protonation occurs on the amine moiety under our experimental conditions and the CID product ion corresponds to the π-radical cation of aniline. With these structures established, we reconstructed the PES describing the hydrogen transfer from the ammonium moiety to the para-position of the ring using transition-state calculations at the DFT level. This suggests that the reaction occurs through a series of proton transfers in the closed-shell ion, followed by expulsion of the bromine radical, generating the product ion in the global minimum-energy aniline radical cation structure.
Supplemental Material
Download Zip (661.3 KB)Acknowledgments
This contribution is dedicated to the memory of Prof. Dieter Gerlich and the vivid discussions with him on ion traps, ion chemistry and numerous other topics. We gratefully acknowledge the expert support by the FELIX staff. We thank Prof. P.B. Armentrout for providing the computational results at higher levels of theory and for his helpful comments.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- R.I. Kaiser, D.S. Parker and A.M. Mebel, Annu. Rev. Phys. Chem. 66 (1), 43–67 (2015). doi:10.1146/annurev-physchem-040214-121502
- K.H. Cheeseman and T.F. Slater, Br. Med. Bull. 49 (3), 481–493 (1993). doi:10.1093/oxfordjournals.bmb.a072625
- J. Roithová, Chem. Soc. Rev. 41 (2), 547–559 (2012). doi:10.1039/C1CS15133A
- F. Tureček and R.R. Julian, Chem. Rev. 113 (8), 6691–6733 (2013). doi:10.1021/cr400043s
- K.M. Stirk, L.K.M. Kiminkinen and H.I. Kenttamaa, Chem. Rev. 92 (7), 1649–1665 (1992). doi:10.1021/cr00015a008
- P.E. Williams, B.J. Jankiewicz, L. Yang and H.I. Kenttämaa, Chem. Rev. 113 (9), 6949–6985 (2013). doi:10.1021/cr400121wPMID: 23987564.
- B.F. Yates, W.J. Bouma and L. Radom, Tetrahedron 42 (22), 6225–6234 (1986). doi:10.1016/S0040-4020(01)88084-4
- T. Bjoernholm, S. Hammerum and D. Kuck, J. Am. Chem. Soc. 110 (12), 3862–3869 (1988). doi:10.1021/ja00220a023
- R.K. Sinha, P. Maître, S. Piccirillo, B. Chiavarino, M.E. Crestoni and S. Fornarini, Phys. Chem. Chem. Phys. 12 (33), 9794–9800 (2010). doi:10.1039/c003576a
- R. Zubarev, N. Kelleher and F. McLafferty, J. Am. Chem. Soc. 120 (13), 3265–3266 (1998). doi:10.1021/ja973478k
- R. Zubarev, Mass. Spectrom. Rev. 22 (1), 57–77 (2003). doi:10.1002/(ISSN)1098-2787
- J. Syka, J. Coon, M. Schroeder, J. Shabanowitz and D.F. Hunt, Proc. Natl. Acad. Sci. USA 101 (26), 9528–9533 (2004). doi:10.1073/pnas.0402700101
- N. Riley and J. Coon, Anal. Chem. 90 (1), 40–64 (2018). doi:10.1021/acs.analchem.7b04810
- G. Tsaprailis, H. Nair, A. Somogyi, V. Wysocki, W. Zhong, J. Futrell, S. Summerfield and S. Gaskell, J. Am. Chem. Soc. 121 (22), 5142–5154 (1999). doi:10.1021/ja982980h
- V. Wysocki, G. Tsaprailis, L. Smith and L. Breci, J. Mass. Spectrom. 35, 1399–1406 (2000). doi:10.1002/(ISSN)1096-9888
- B. Paizs and S. Suhai, Mass. Spectrom. Rev. 24 (4), 508–548 (2005). doi:10.1002/(ISSN)1098-2787
- R. Boyd and Á. Somogyi, J. Am. Soc. Mass. Spectrom. 21 (8), 1275–1278 (2010). doi:10.1016/j.jasms.2010.04.017
- F. Tureček, J. Phys. Chem. B. 125 (26), 7090–7100 (2021). doi:10.1021/acs.jpcb.1c03674
- J. Roithová, A. Gray, E. Andris, J. Jašík and D. Gerlich, Acc. Chem. Res. 49 (2), 223–230 (2016). doi:10.1021/acs.accounts.5b00489
- J. Jašík, J. Žabka, J. Roithová and D. Gerlich, Int. J. Mass. Spectrom. 354-355, 204–210 (2013). doi:10.1016/j.ijms.2013.06.007
- B.N. Moore, S.J. Blanksby and R.R. Julian, Chem. Commun. 33 (33), 5015–5017 (2009). doi:10.1039/b907833a
- A.P. Cismesia, G.R. Nicholls and N.C. Polfer, J. Mol. Spectrosc. 332, 79–85 (2017). doi:10.1016/j.jms.2016.10.020
- S. Yoon, J. Chamot-Rooke, B. Perkins, A. Hilderbrand, J. Poutsma and V. Wysocki, J. Am. Chem. Soc. 130 (52), 17644–17645 (2008). doi:10.1021/ja8067929
- N. Polfer, J. Oomens, S. Suhai and B. Paizs, J. Am. Chem. Soc. 129 (18), 5887–5897 (2007). doi:10.1021/ja068014d
- W. Tang, X. Mu, M. Li, J. Martens, G. Berden, J. Oomens, I. Chu and C.K. Siu, Phys. Chem. Chem. Phys. 22 (37), 21393–21402 (2020). doi:10.1039/D0CP00533A
- C. Shaffer, J. Martens, A. Marek, J. Oomens and F. Turecek, J. Am. Soc. Mass. Spectrom. 27 (7), 1176–1185 (2016). doi:10.1007/s13361-016-1390-4
- G. Frison, G. van der Rest, F. Turecek, T. Besson, J. Lemaire, P. Maitre and J. Chamot-Rooke, J. Am. Chem. Soc. 130 (45), 14916–14917 (2008). doi:10.1021/ja805257v
- S. Osburn, G. Berden, J. Oomens, R. O'Hair and V. Ryzhov, J. Am. Soc. Mass. Spectrom. 22 (10), 1794–1803 (2011). doi:10.1007/s13361-011-0198-5
- J. Martens, J. Grzetic, G. Berden and J. Oomens, Nat. Commun. 7 (1), 11754 (2016). doi:10.1038/ncomms11754
- J. Oomens, J.D. Steill and B. Redlich, J. Am. Chem. Soc. 131 (12), 4310–4319 (2009). doi:10.1021/ja807615vPMID: 19267428.
- J.J. Valle, J.R. Eyler, J. Oomens, D.T. Moore, A.F.G. van der Meer, G. von Helden, G. Meijer, C.L. Hendrickson, A.G. Marshall and G.T. Blakney, Rev. Sci. Instrum. 76 (2), 023103 (2005). doi:10.1063/1.1841953
- R.K. Roy, F. de Proft and P. Geerlings, J. Phys. Chem. A. 102 (35), 7035–7040 (1998). doi:10.1021/jp9815661
- H.T. Le, R. Flammang, M. Barbieux-Flammang, P. Gerbaux and M.T. Nguyen, Int. J. Mass. Spectrom. 217 (1-3), 45–54 (2002). doi:10.1016/S1387-3806(02)00530-4
- Z. Karpas, Z. Berant and R.M. Stimac, Struct. Chem. 1 (2-3), 201–204 (1990). doi:10.1007/BF00674262
- R.L. Smith, L.J. Chyall, B.J. Beasley and H.I. Kenttamaa, J. Am. Chem. Soc. 117 (30), 7971–7973 (1995). doi:10.1021/ja00135a016
- N. Russo, M. Toscano, A. Grand and T. Mineva, J. Phys. Chem. A. 104 (17), 4017–4021 (2000). doi:10.1021/jp991949e
- R. Flammang, N. Dechamps, L. Pascal, Y. Haverbeke, P. Gerbaux, P.C. Nam and M. Nguyen, Lett. Org. Chem. 1 (1), 23–30 (2004). doi:10.2174/1570178043488725
- F.M. Pasker, N. Solcà and O. Dopfer, J. Phys. Chem. A. 110 (47), 12793–12804 (2006). doi:10.1021/jp064571a
- A.B. Attygalle, H. Xia and J. Pavlov, J. Am. Soc. Mass. Spectrom. 28 (8), 1575–1586 (2017). doi:10.1007/s13361-017-1640-0
- L.J. Chyall and H.I. Kenttamaa, J. Am. Chem. Soc. 116 (7), 3135–3136 (1994). doi:10.1021/ja00086a058
- D. Oepts, A. van der Meer and P. van Amersfoort, Infrared. Phys. Technol. 36 (1), 297–308 (1995). doi:10.1016/1350-4495(94)00074-U
- J. Martens, G. Berden, C.R. Gebhardt and J. Oomens, Rev. Sci. Instrum. 87 (10), 103108 (2016). doi:10.1063/1.4964703
- F.J. Andrade, J.T. Shelley, W.C. Wetzel, M.R. Webb, G. Gamez, S.J. Ray and G.M. Hieftje, Anal. Chem. 80 (8), 2646–2653 (2008). doi:10.1021/ac800156y
- G. Berden, M. Derksen, K.J. Houthuijs, J. Martens and J. Oomens, Int. J. Mass. Spectrom. 443, 1–8 (2019). doi:10.1016/j.ijms.2019.05.013
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, G.A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A.V. Marenich, J. Bloino, B.G. Janesko, R. Gomperts, B. Mennucci, H.P. Hratchian, J.V. Ortiz, A.F. Izmaylov, J.L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V.G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J.A. Montgomery, Jr. J.E. Peralta, F. Ogliaro, M.J. Bearpark, J.J. Heyd, E.N. Brothers, K.N. Kudin, V.N. Staroverov, T.A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A.P. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, J.M. Millam, M. Klene, C. Adamo, R. Cammi, J.W. Ochterski, R.L. Martin, K. Morokuma, O. Farkas, J.B. Foresman and D.J. Fox, Gaussian 16 Revision C.01 (Gaussian Inc, Wallingford CT, 2016).
- A.P. Scott and L. Radom, J. Phys. Chem. 100 (41), 16502–16513 (1996). doi:10.1021/jp960976r
- T.M. Chang, J.S. Prell, E.R. Warrick and E.R. Williams, J. Am. Chem. Soc. 134 (38), 15805–15813 (2012). doi:10.1021/ja304929h
- H. Piest, G. von Helden and G. Meijer, J. Chem. Phys. 110 (4), 2010–2015 (1999). doi:10.1063/1.477866
- D. Zeh, M. Bast, D.B. Rap, P. Schmid, S. Thorwirth, S. Brünken, S. Schlemmer and M. Schäfer, J. Mol. Spectrosc. 378, 111453 (2021). doi:10.1016/j.jms.2021.111453
- S.Y. Lin, S.L. Chou, C.M. Tseng and Y.J. Wu, Spectrochim. Acta A. Mol. Biomol. Spectrosc. 276, 121233 (2022). doi:10.1016/j.saa.2022.121233
- X. Zhang, N.B. Brauer, G. Berden, A.M. Rijs and M. Drabbels, J. Chem. Phys. 136 (4), 044305 (2012). doi:10.1063/1.3678011
- X. Song, M. Yang, E.R. Davidson and J.P. Reilly, J. Chem. Phys. 99 (5), 3224–3233 (1993). doi:10.1063/1.465131
- X. Zhang, J.M. Smith and J.L. Knee, J. Chem. Phys. 97 (5), 2843–2860 (1992). doi:10.1063/1.463027
- M. Takahashi, H. Ozeki and K. Kimura, J. Chem. Phys. 96 (9), 6399–6406 (1992). doi:10.1063/1.462634
- D.F. McMillen and D.M. Golden, Annu. Rev. Phys. Chem. 33 (1), 493–532 (1982). doi:10.1146/annurev.pc.33.100182.002425
- J.i. Aihara, J. Phys. Chem. A. 103 (37), 7487–7495 (1999). doi:10.1021/jp990092i
- Y. Ruiz-Morales, J. Phys. Chem. A. 106 (46), 11283–11308 (2002). doi:10.1021/jp021152e