
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
We present a procedure, based on established principles, to accurately determine the ionisation energies of medium-sized to large polyatomic molecules in supersonic beams. The method combines single-photon excitation from the electronic ground state using tunable vacuum-ultraviolet laser radiation, the delayed pulsed-field ionisation of high Rydberg states with sequences of pulsed electric fields, and the modelling of the lineshapes from calculations of the field-ionisation dynamics of high Rydberg states and of the rotational contours of the photoelectron bands. The method is illustrated by a measurement of the adiabatic ionisation energy of p-diaminobenzene (p-DB), for which we obtain the value of 54,617.47(15) cm, which represents a more than 20-fold accuracy improvement over previous determinations.
GRAPHICAL ABSTRACT
Ionisation energies of atoms and molecules are important thermochemical quantities. Pulsed-field-ionisation zero-kinetic-energy photoelectron (PFI-ZEKE PE) spectroscopy [Citation1] and mass-analysed threshold ionisation (MATI) spectroscopy [Citation2] are the methods of choice to measure ionisation energies [Citation3]. With these methods, one monitors the delayed pulsed field ionisation of Rydberg states of high principal quantum numbers n () located just below molecular ionisation thresholds as the frequency of a tunable radiation source is scanned across these thresholds. To obtain accurate values of the field-free ionisation thresholds, the binding energies of the high Rydberg states need to be added to the observed transition energies [Citation4,Citation5].
In small molecules consisting of up to about 10 atoms, ionisation energies can routinely be determined with a precision of about 0.1–0.2 meV (or 0.8–1.6 cm, or 10–20 J/mol) or even better from rotationally resolved PFI-ZEKE-PE or MATI spectra (see, e.g. [Citation6–9]). In larger molecules, the rotational structure of the photoelectron spectra cannot be resolved and it is challenging to determine ionisation energies with an accuracy better than
meV. In many cases, the adiabatic ionisation energies of large molecules are determined using resonant two-photon excitation schemes via electronically excited intermediate states [Citation10–12]. These excitation schemes enable one to access the ionisation thresholds with commercial tunable visible or UV lasers and the choice of the intermediate state can be used to facilitate assignments, reach a broader range of ionisation thresholds, and select isomers or isotopologues in gas mixtures. At the same time, the resonant excitation typically introduces uncertainties in the values of adiabatic ionisation energies if the rotational structure of the intermediate state is not resolved spectrally. Numerous examples of the application of resonant two-photon excitation with PFI-ZEKE PE or MATI spectroscopy to large molecules and molecular complexes have been reported and selected examples over the past 20 years include studies of n-butylbenzene [Citation13], phenol-Ar
complexes [Citation14], p-diaminobenzene (p-DB) [Citation15,Citation16], mixed complexes of carboxylic acids and water [Citation17], 2-methoxybenzonitrile [Citation18] and m-chlorotoluene [Citation19].
We report here on the use of a narrow-band, intense vacuum-ultraviolet (VUV) laser system based on frequency upconversion in potassium-beryllium-fluoroborate (KBBF) crystals [Citation20] to determine the ionisation energies of large conjugated molecules with ionisation energies below 60,000 cm with an accuracy of better than 0.2 cm
. The procedure relies on established principles and involves the use of multipulse electric-field sequences to record PFI-ZEKE-PE spectra at high resolution, and calculations of the field-ionisation dynamics of high Rydberg states [Citation21,Citation22] and of the rotational structure of the photoelectron spectra [Citation23]. As an illustration, we present a measurement of the adiabatic ionisation energy of p-DB to clarify the discrepancy between the latest two values of this quantity reported in the literature (54,640(8) cm
[Citation15] and 54,624(5) cm
[Citation16]).
A pulsed supersonic expansion of p-DB was generated by loading approximately 15 mg of p-DB (Acros Organics, CAS 106-50-3) into a homemade pulsed valve, which was then heated to 85 C to produce a vapor pressure of 1.5 mbar (
Pa at 100
C [Citation24]). The vaporised p-DB was mixed with Ar and the pulsed valve released 80-μs-long pulses of the gas mixture from a reservoir held at a stagnation pressure of 6 bar into the vacuum chamber. The central part of the beam was selected by a 3.0-mm-diameter skimmer separating the gas-source chamber from the photoexcitation and detection chamber. Upon operation of the valve the background pressure rose from
mbar to
mbar (gas-source chamber) and
mbar (photoexcitation and detection chambers). The supersonic beam crossed a VUV laser beam at right angles on the axis of a 5.95-cm-long electrode stack consisting of 5 equidistant, resistively-coupled cylindrical electrodes.
To ionise p-DB with a single photon, VUV laser radiation around 180 nm was produced by quadrupling the frequency of a commercial dye laser pumped by an injection-seeded Nd:YAG laser (532 nm). The dye-laser output frequency with a bandwidth of 0.1 cm in the spectral range of 710–740 nm was doubled twice, first to the UV range 355–370 nm using a beta-barium-borate (BBO) crystal and then to the VUV range 178–185 nm using a KBBF crystal. The VUV radiation had a bandwidth of 0.2 cm
. To reduce absorption of the VUV radiation in air, the beam generated from the KBBF crystal was propagated in a nitrogen atmosphere. Typical VUV pulse energies at wavelengths near 180 nm were in the range between 90 and 120 μJ and constantly monitored with a homemade UV detector to compensate for shot-to-shot signal fluctuations by normalisation.
The VUV beam was directed into the photoexcitation region in a direction perpendicular to the electrode stack. An electric-field pre-pulse of V/cm was applied to the stack to sweep prompt electrons out of the photoexcitation volume, followed by pulses of
V/cm,
V/cm,
V/cm, and
V/cm used to field ionise high Rydberg states located just below the ionisation thresholds and to extract the electrons towards a microchannel-plate (MCP) detector located at the end of a short flight tube. Three PFI-ZEKE-PE spectra were recorded for each scan by monitoring the field-ionisation signal induced by each of the first three negative-field pulses of the sequence as a function of the VUV laser wavenumber.
The rotational contour of the ionisation transition of p-DB was predicted using the orbital-ionisation model originally developed for diatomic molecules [Citation25] and later extended to polyatomic molecules [Citation23]. In this model, the rotational structure of the photoionisation transition is expressed in terms of the angular-momentum composition of the orbital hole left in the molecule after photoionisation. The model assumes that the photon angular momentum is fully transferred to the photoelectron. Conservation of the total angular momentum implies that the rotational angular momentum of the neutral molecule must be the sum of the angular momentum of the ion and the electron hole
. p-DB is a near-prolate top (
), with the a axis corresponding to the axis passing through the two nitrogen nuclei. The highest occupied molecular orbital (HOMO) of p-DB has one nodal plane that includes the a axis and three nodal planes perpendicular to the a axis, two located between the phenyl ring and the two amino groups and one bisecting the phenyl ring. It can be approximated as an atomic
orbital centred in the middle of the ring, which results in a value of
but also contains higher contributions with even values of
to describe the outer lobes of the HOMO located on the amino groups. We have modelled the photoionisation transition assuming values of
and
. To calculate the rotational structure and predict relative intensities for the different rotational branches, we used Equation (Equation1
(1)
(1) ) [Citation23]
(1)
(1)
with
to account for the nodal plane of the HOMO containing the z axis (a axis), and
(2)
(2)
In these equations,
is the quantum number associated with the projection of the electron-hole orbital angular momentum
on the a axis,
is proportional to the populations of the initial rovibronic states of the neutral molecule, and
are radial electric-dipole transition integrals. In Equation (Equation2
(2)
(2) ), the sum over
and
comes from the expansions of the asymmetric top wavefunctions
and
of p-DB and p-DB
in the basis of symmetric-top functions
with expansion coefficients
[Citation23]. Rotational constants for the
B
ground state of neutral p-DB and the
B
ion ground state were estimated from those of aniline [Citation26], adding the appropriate contributions for the moments of inertia from the additional amino group. These approximate rotational constants, listed in Table , were sufficient for our purposes because our experimental resolution was not high enough to resolve individual rotational transitions.
Table 1. Estimated rigid-rotor rotational constants for the ground electronic states of neutral p-DB and its cation p-DB. Rotational constants were estimated from those of aniline (see text).
The rotational structure was calculated as a stick spectrum based on Equations (Equation1(1)
(1) ) and (Equation2
(2)
(2) ) and initially choosing the adiabatic ionisation threshold
, corresponding to the
B
(
0,0,0) –
B
(
0,0,0) transition, at a wavenumber of 0.0 cm
. The rotational temperature was then adjusted to best fit the contour of the experimental spectrum, from which we determined a value of
K. The final stick spectrum shown in Figure was obtained by weighting the
and
contributions by factors of 0.8 and 0.2, respectively, which gave best agreement with the measured intensity distribution.
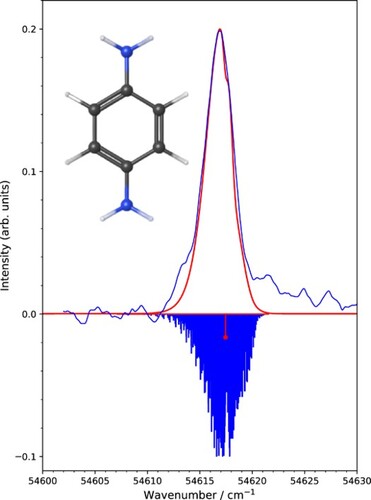
Figure 1. Comparison of experimental PFI-ZEKE-PE spectra of p-DB recorded by monitoring the pulsed-field-ionisation signals generated by the ,
, and
V/cm field pulses with calculated rotational contours. The spectra were shifted along the vertical axis so that their baselines correspond to the value of
. The corresponding positions of the band centres
B
(0,0,0) –
B
(0,0,0) are indicated with solid red circles. The dashed red line indicates the results of the extrapolation of these band centres to determine the field-free adiabatic ionisation energy, which corresponds to the position of the
B
(0,0,0) –
B
(0,0,0) transition highlighted in red in the inverted stick spectrum of the rotational structure. The summation of the field-corrected spectra (dark blue) is compared with the summation corresponding to the field-corrected calculated spectra at the bottom of the figure.
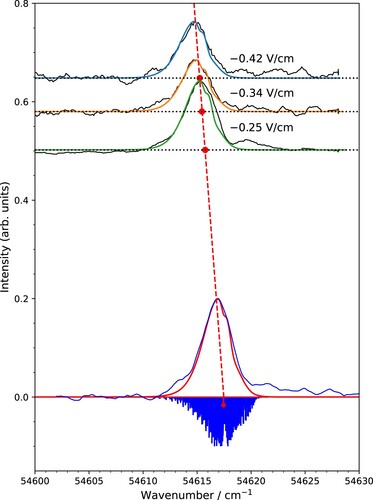
The ionisation energy of p-DB was determined from the experimental PFI-ZEKE-PE spectra in two related ways. First, lineshape functions for each of the electric-field pulses were generated following the procedure introduced by Hollenstein et al. [Citation21]. Asymmetric Gaussian functions were fitted to these lineshapes and the centre of gravity of each lineshape was determined. The calculated rotational stick spectrum was then convoluted with these Gaussian functions and shifted along the wave number axis to reach best agreement with the measured spectra. The procedure is illustrated in Figure , where the experimental PFI-ZEKE-PE spectra corresponding to the three negative pulses of the field ionisation sequence are shown in black. The calculated spectra, shifted to the optimal positions, are depicted in green, orange and blue for the ,
and
V/cm field pulses, respectively. In each case, the positions of the
B
(0,0,0) –
B
(0,0,0) transition are shown with solid red circles. The field-free ionisation energy
was determined by extrapolation to zero field using the fact that the field-induced shifts
are proportional to the square root of the field strength F [Citation27]:
(3)
(3)
where a is the proportionality constant. Values of a = 3.5(3) cm
and
were obtained from a linear regression (see dashed red line in Figure ).
In the second method to determine the ionisation energy, the experimental and calculated PFI-ZEKE-PE spectra were first corrected for the calculated field-induced shifts and then averaged to obtain the spectra in Figure displayed in blue and red, respectively, above the inverted calculated stick spectrum. The position of the
B
(0,0,0) –
B
(0,0,0) transition (red stick) was found to be 54,617.45(15) cm
, where the uncertainty corresponds to the range of values for which experimental and calculated spectra agree within the statistical limits. The ionisation energies derived from these two related methods agree within their uncertainties. In Table , we therefore report their average as our final value for the adiabatic ionisation energy of p-DB (
). Our new value for the adiabatic ionisation energy of p-DB is significantly smaller than earlier values and its uncertainty is reduced by a factor of more than 20.
Table 2. Values for the adiabatic ionisation energy of p-DB, corresponding to the
B
(0,0,0) –
B
(0,0,0) transition.
The difference between our new value of the adiabatic ionisation energy of p-DB and earlier values determined by resonant two-photon excitation is likely to be caused by uncertainties, in the earlier works, related to the correction of the field-induced shift of the ionisation thresholds and to the unresolved rotational structure. The multipulse electric-field sequence used in the present work offers the following advantages, both illustrated by Figure . First, it enables one to extrapolate the line positions of the spectra obtained from the different field pulses to the zero-field thresholds with an accuracy of about 0.1 cm. Second, the instrumental linewidth is guaranteed to be less than 0.5 cm
. Consequently, the observed linewidths are dominated by the unresolved rotational structure. Modelling the rotational structure makes it possible to locate the band origin with high precision. In this context, it is worth mentioning that a slowly growing electric-field ramp can be used instead of the multipulse sequence, as recently advocated by Harper et al. [Citation28]. The previous studies of Ozeki et al. [Citation15] and Colapietro et al. [Citation29] have established that gas-phase p-DB and p-DB
exhibit a trans-configuration of the two NH
planes with a high tunnelling barrier at the planar geometry. The adiabatic ionisation energy reported here is thus the energy of the transition between the rovibrational ground states of the trans-isomers of p-DB and p-DB
.
Acknowledgments
We thank Josef A. Agner for his help in optimising the supersonic beam of p-DB.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- G. Reiser, W. Habenicht, K. Müller-Dethlefs and E.W. Schlag, Chem. Phys. Lett. 152, 119–123 (1988). doi:10.1016/0009-2614(88)87340-8
- L. Zhu and P. Johnson, J. Chem. Phys. 94, 5769–5771 (1991). doi:10.1063/1.460460
- I. Fischer and S.T. Pratt, Phys. Chem. Chem. Phys. 24, 1944–1959 (2022). doi:10.1039/D1CP04984D
- W.A. Chupka, J. Chem. Phys. 98, 4520–4530 (1993). doi:10.1063/1.465011
- F. Merkt, S. Willitsch and U. Hollenstein, in Handbook of High-Resolution Spectroscopy, edited by M. Quack and F. Merkt, (John Wiley & Sons, Chichester, 2011), pp. 1617–1654, Vols. 3.
- R. Lindner, K. Müller-Dethlefs, E. Wedum, K. Haber and E.R. Grant, Science 271 (5256), 1698–1702 (1996). doi:10.1126/science.271.5256.1698
- R. Neuhauser and H.J. Neusser, Chem. Phys. Lett. 253, 151–157 (1996). doi:10.1016/0009-2614(96)00236-9
- H.J. Wörner and F. Merkt, J. Chem. Phys. 127, 034303 (2007). doi:10.1063/1.2748049
- U. Jacovella, C.J. Stein, M. Grütter, L. Freitag, C. Lauzin, M. Reiher and F. Merkt, Phys. Chem. Chem. Phys. 20, 1072–1081 (2018). doi:10.1039/C7CP06907C
- O. Dopfer, T.G. Wright, E. Cordes and K. Müller-Dethlefs, J. Am. Chem. Soc. 116, 5880–5886 (1994). doi:10.1021/ja00092a044
- A. Held, H.L. Selzle and E.W. Schlag, J. Phys. Chem. A 102, 9625–9630 (1998). doi:10.1021/jp982353e
- C.E.H. Dessent and K. Müller-Dethlefs, Chem. Rev. 100, 3999–4022 (2000). doi:10.1021/cr990060r
- X. Tong, J. Černý, K. Müller-Dethlefs and C.E.H. Dessent, J. Phys. Chem. A 112, 5866–5871 (2008). doi:10.1021/jp710997q
- A. Armentano, X. Tong, M. Riese, S.M. Pimblott, K. Müller-Dethlefs, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys. 13, 6071–6076 (2011). doi:10.1039/C004497K
- H. Ozeki, K. Okuyama, M. Takahashi and K. Kimura, J. Phys. Chem. 95, 4308–4313 (1991). doi:10.1021/j100164a025
- V. Shivatare, C.H. Wu and W.B. Tzeng, J. Photochem. Photobiol. A 251, 94–99 (2013). doi:10.1016/j.jphotochem.2012.10.020
- Q. Gu, Z. Tang, P. Su, W. Wu, Z. Yang, C.O. Trindle and J.L. Knee, J. Chem. Phys. 145, 051101 (2016). doi:10.1063/1.4959970
- Y. Zhao, Y. Jin, C. Li and S. Jia, J. Mol. Spectrosc. 363, 111182 (2019). doi:10.1016/j.jms.2019.111182
- D.J. Kemp, L.G. Warner and T.G. Wright, J. Chem. Phys. 152, 064303 (2020). doi:10.1063/1.5142992
- H. Herburger, U. Hollenstein, J.A. Agner and F. Merkt, Chem. Phys. 151 (14), 144302 (2019). doi:10.1063/1.5124477
- U. Hollenstein, R. Seiler, H. Schmutz, M. Andrist and F. Merkt, J. Chem. Phys. 115, 5461–5469 (2001). doi:10.1063/1.1396856
- S. Mollet and F. Merkt, Chem. Phys. 139 (3), 034302 (2013). doi:10.1063/1.4812376
- S. Willitsch and F. Merkt, Int. J. Mass Spectrom. 245, 14–25 (2005). doi:10.1016/j.ijms.2005.06.004
- World Health Organization, p-Phenylenediamine, (CAS:106-50-3). https://www.ilo.org/dyn/icsc (Accessed Apr, 20, 2023).
- A.D. Buckingham, B.J. Orr and J.M. Sichel, Philos. Trans. R. Soc. Lond. Ser. A 268, 147–157 (1970). doi:10.1098/rsta.1970.0068
- G. Roussy and A. Nonat, J. Mol. Spectrosc. 118, 180–188 (1986). doi:10.1016/0022-2852(86)90234-1
- T.F. Gallagher, Rydberg Atoms, (Cambridge University Press, Cambridge, 1994).
- O.J. Harper, N.L. Chen, S. Boyé-Péronne and B. Gans, Phys. Chem. Chem. Phys. 24, 2777–2784 (2022). doi:10.1039/D1CP04569E
- M. Colapietro, A. Domenicano, G. Portalone, G. Schultz and I. Hargittai, J. Phys. Chem. 91, 1728–1737 (1987). doi:10.1021/j100291a012