
ABSTRACT
The genus Alternaria contains a diversity of saprobic and pathogenic species that can be found in a wide range of environments. Alternaria is currently divided into 26 subgeneric sections, and the “small-spored” Alternaria section Alternaria includes many species that are economically important agricultural pathogens. Recognizing that a stable framework for systematics and species identification is essential for management and regulation purposes, this section has experienced much taxonomic debate and systematic revision in recent years. Molecular phylogenetic studies have challenged the reliability of using morphological characteristics to differentiate Alternaria species but have also suggested that commonly used molecular markers for fungal phylogenetics may not be sufficiently informative at this taxonomic level. To allow the assessment of molecular variation and evolutionary history at a genome-wide scale, we present an overview and analysis of phylogenomic resources for Alternaria section Alternaria. We review the currently available genomic resources and report five newly sequenced genomes. We then perform multiple comparative genomic analyses, including macrosynteny assessment and inference of phylogenetic relationships using a variety of data sets and analysis methods. Fine-scale, genome-wide phylogenetic reconstruction revealed incomplete lineage sorting and the genomic distribution of gene/species tree discordance. Based on these patterns, we propose a list of candidate genes that may be developed into informative markers that are diagnostic for the main lineages. This overview identifies gaps in knowledge and can guide future genome sequencing efforts for this important group of plant pathogenic fungi.
INTRODUCTION
The genus Alternaria, characterized by chains of darkly pigmented, multicelled conidia with transverse and longitudinal septa (phaeodictyospores) and tapering apical beaks, was established by Nees in Citation1817 (Nees Citation1817). Over the next 150 years, other genera (e.g., Brachycladium, Macrosporium, Stemphylium, Ulocladium) were erected to accommodate variations in Alternaria-like phaeodictyospore morphologies (Lawrence et al. Citation2016). Decades of comprehensive revision of Alternaria and related genera based on morphological characteristics culminated in the recognition of a total of 275 Alternaria species. These species have been further divided into subgeneric groups based on broad patterns of morphological similarity (Simmons Citation1967, Citation2007).
Results of recent molecular phylogenetic studies have challenged the morphological basis of the accepted taxonomy of Alternaria and close relatives (Pryor and Gilbertson Citation2000; Peever et al. Citation2004; Hong et al. Citation2005; Andrew et al. Citation2009; Lawrence et al. Citation2013; Woudendberg et al. Citation2013, Citation2015; Armitage et al. Citation2015, Citation2020). In general, the morphologically distinct subgeneric species groups, now called sections, tend to be supported by molecular data. However, the phylogenetic relationships within species groups are often inconsistent with the subtle phenotypic characteristics used to delineate morphospecies. This pattern was also observed at the generic level: lack of phylogenetic differentiation led to the synonymization of 13 other genera into Alternaria (Woudenberg et al. Citation2013), thereby redefining and expanding the generic concept of Alternaria. Collectively, these studies suggest that morphological characteristics typically used to delineate species (e.g., conidium length, width, septation; chain structure; beak shape) may not reflect evolutionary relationships between taxa.
A subgeneric species group that has experienced much taxonomic debate and confusion is the “small-spored” Alternaria section Alternaria (Lawrence et al. Citation2013; Woudenberg et al. Citation2015). This section contains many species that cause disease of economically important crops, so consistent species delineation and identification are essential for management and quarantine purposes. Particular species are associated with specific host ranges and pathogenicity, due in part to the ability to produce host-specific toxins (Meena et al. Citation2017). Some species are included on regulated organism lists for international phytosanitary certification, further highlighting the need for stable taxonomy and nomenclature. Even in the hands of experienced mycologists, the morphological characteristics used to differentiate species in section Alternaria may overlap between species and be sensitive to variation in growth conditions. Different molecular studies have led to different systematics proposals for section Alternaria, potentially due to the use of different molecular markers for inferring phylogenetic relationships and the differentiation among taxa. When working with strains that are so closely related, the commonly used genes for fungal phylogenetics may not be informative enough at this taxonomic level. In such cases, adopting a phylogenomics approach and assessing variation at a genome-wide scale may provide the necessary discriminatory power.
Here, we explore a phylogenomics approach for Alternaria section Alternaria (also known as Alternaria alternata species group) to address questions of molecular systematics and evolutionary history. First, we provide a review of the genomic resources currently available for this section and release five newly sequenced genomes from isolates collected from cereal crops in Canada. We then perform a range of comparative genomic analyses, including macrosynteny assessment and inference of phylogenetic relationships using a variety of data sets and analysis methods. Fine-scale, genome-wide phylogenetic reconstruction allowed us to investigate incomplete lineage sorting and the genomic distribution of gene/species tree discordance. By viewing how patterns of concordance varied with genomic context, we determined the regions of the genome that supported the main lineages identified by whole-genome analyses. We then propose a list of candidate genes that can be developed into diagnostic markers for distinguishing the main lineages. This overview identifies gaps in knowledge and can guide future genome sequencing efforts for this important group of plant pathogenic fungi.
MATERIALS AND METHODS
Existing genomic resources.—
All genome assemblies for section Alternaria that were publically available on January 27, 2020, were downloaded. Assembly quality was assessed with QUAST (5.0.2; Mikheenko et al. Citation2018), and genomes with inconsistent genome sizes (>50 Mb) or low quality scores (N50 <10 kb) were omitted. For one additional isolate (EGS 90.391), raw short-read sequence data were obtained directly from the authors who published genome analyses previously (Woudenberg et al. Citation2015), resulting in 33 genomes (). Here, we present the original species names given to the strains at the time of genome submission but also indicate what their current names would be based on the revision of section Alternaria by Woudenberg et al. (Citation2015).
Table 1. Summary of information and statistics for the analyzed section Alternaria genomes
Genome sequencing of new isolates.—
For sequencing of new genomes, we chose five section Alternaria isolates collected from rye or wheat in Canada between 2013 and 2018 (). Fungal isolates were grown on potato dextrose agar (potato extract, 4 g/L, dextrose, 20 g/L, agar 15 g/L) plates for 14 days at 25 C. Genomic DNA was extracted following Möller et al. (Citation1992) with modifications (see SUPPLEMENTARY MATERIAL for details). The RNA polymerase second largest subunit (rpb2) gene was sequenced following polymerase chain reaction (PCR) amplification using primers and thermocycler conditions described in Woudenberg et al. (Citation2013). Sequences of new isolates (data now shown) were compared with reference sequences for preliminary identification to species or species complex (under the taxonomic framework of Woudenberg et al. [Citation2015]), later confirmed by genome sequencing described below.
Genomic DNA samples were normalized to 300 ng and sheared to approximately 350-bp fragment size using a Covaris M220 sonicator (Woburn, Massachusetts). The fragmented DNA was used as the template to construct PCR-free libraries with a NxSeq AmpFREE Low DNA Library Kit (Lucigen, Middleton, Wisconsin) according to the manufacturer’s instructions. Single-indexed libraries were sequenced with paired-end reads (2 × 150 bp) on a NextSeq 500 instrument (Illumina, San Diego, California) using a NextSeq High Output Reagent Kit (Illumina) according to the manufacturer’s recommendations. An average of 24.2 million reads were generated per sample (SUPPLEMENTARY TABLE 1).
Genome assembly.—
For new assemblies, raw reads were clipped of Illumina adapters and quality trimmed with Trimmomatic (0.38; Bolger et al. Citation2014). Assemblies were created with SPAdes (3.12; Bankevich et al. Citation2012), and contigs less than 500 bp in length were removed. See SUPPLEMENTARY MATERIAL for details. Contigs were also removed if BLASTn searches indicated that they were mitochondrial or bacterial in nature. The resulting assemblies had an average sequence coverage of 105× (SUPPLEMENTARY TABLE 1). The final versions of five new genome assemblies are available under National Center for Biotechnology Information (NCBI) accession numbers JAAOQT010000000 to JAAOQX010000000.
Gene prediction (and annotation).—
The previously published genomes are not all in the same state of gene prediction and annotation. Different software/pipelines, or even different versions of the same pipeline, can introduce technical variation that may obscure direct comparison across genomes. To remove this potential source of bias, all assemblies were subjected to new gene prediction using funannotate (1.7.2; https://github.com/nextgenusfs/funannotate, see SUPPLEMENTARY MATERIAL for programs and parameters). In brief, gene prediction parameters were obtained from training with high-quality genomic and transcriptomic data that were available for the same isolate (SRC1lrK2f; Zeiner et al. Citation2016). Pretrained SRC1lrK2f parameters were used to guide gene prediction in the other genomes.
Genome completeness and macrosynteny.—
The BUSCO (benchmarking universal single-copy orthologs) metric was applied to assess genome completeness using BUSCO (4.0.6; Simão et al. Citation2015). Genome assemblies (nucleotide level) were compared with the ascomycota_odb10 ortholog data set, which contains 1706 conserved ascomycete orthologs. Whole-genome macrosynteny comparisons were performed with the Nucmer module of MUMmer (3.1; Kurtz et al. Citation2004; SUPPLEMENTARY MATERIAL). To restrict comparisons to only near-complete genomes, genomes with more than 30 scaffolds were excluded.
Phylogeny.—
Sequence alignment was performed with MUSCLE (3.8; Edgar Citation2004), and model selection and construction of maximum likelihood phylogenies were performed with IQ-TREE (2.0; Nguyen et al. Citation2015). Tree construction was performed under best-fit substitution models and partitions. Multispecies coalescent (MSC) analysis was implemented by ASTRAL-III (5.7.3; Zhang et al. Citation2018). See SUPPLEMENTARY MATERIAL for full details of phylogenetic analyses and tree comparison methods. Previous studies (Woudenberg et al. Citation2015; Armitage et al. Citation2020) have shown that the gaisen clade is the earliest diverging of the clades studied here, so it was used for rooting displayed trees.
RESULTS
Genome assembly and gene prediction.—
Summary statistics for the 38 genomes are shown in . Our newly generated assemblies for Canadian section Alternaria isolates were very similar to previously published genomes in regard to GC content (mean = 51.0%) and genome length (mean = 34.11 Mb). Overall, the quality of 38 analyzed genome assemblies was quite high, with a mean of 531 contigs and an N50 of 904 kb. As expected, genomes sequenced using long-read technologies such as PacBio (Menlo Park, California) or Oxford Nanopore (Oxford, UK) tended to have higher qualities than short-read Illumina-based genomes. For example, long-read assemblies had ~14× fewer contigs, and ~5× longer N50 values, than short-read assemblies.
To standardize our downstream comparisons, we subjected each assembly to ab initio gene prediction using the funannotate pipeline. An average of 12 788 gene models per genome were predicted (SUPPLEMENTARY TABLE 2), with long-read genomes having an average of 212 more gene models than short-read genomes.
Assessment of genome completeness.—
The BUSCO metrics were quite high (; SUPPLEMENTARY TABLE 2), indicating that most of these Alternaria assemblies were not missing significant genomic content. Based on the Ascomycota ortholog set (1706 genes), the average percentage of complete genes detected was 98.4%, with an average of only 1.3% of genes missing. Interestingly, the genome with the highest percent missing genes was JS-1623, which was sequenced using long reads and had the highest assembly quality scores. When this outlier was excluded, the long-read genomes had greater average BUSCO scores than the short-read genomes (99.2% versus 98.4% complete, respectively). Investigation of BUSCO genes that were missing from all 38 genomes (SUPPLEMENTARY TABLE 3) did not reveal any shared functional roles.
Genome structure and macrosynteny.—
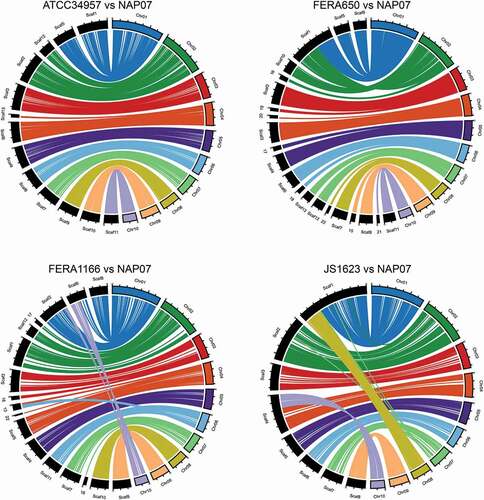
Patterns of large-scale synteny along chromosomes were compared for the five genomes with near-complete assemblies. The 10 core chromosomes of the NAP07 strain have been matched to the 10 A. solani chromosomes, so the NAP07 genome was used as the reference genome for these analyses. Based on the four pairwise comparisons (), the overall structural arrangement of the genomes was fairly well conserved across strains. For example, although three NAP07 chromosomes (Chr01, Chr02, Chr04) were each matched to two scaffolds in ATCC 34957, there were no large-scale conflicts. This situation likely represents a lack of centromere coverage and failure to join the two chromosome arms in the ATCC 34957 assembly. The opposite situation appears in the JS-1623 comparison: NAP07 Chr01 and Chr08 equals JS-1623 Scaffold 1, and NAP07 Chr04 and Chr10 equals JS-1623 Scaffold 4. Although it is possible that JS-1623 has only eight core chromosomes, the more parsimonious explanation is that chromosomes were simply misjoined in the assembly process due to similarity of telomeric sequences. Despite some potential discrepancies observed for FERA 1166, the general conclusion is that large-scale duplications or rearrangements of core chromosomes were not common during the evolutionary history of the section Alternaria clade.
Figure 1. Circular plots showing macrosynteny between section Alternaria genomes sequenced with long-read technology. Four genomes were compared with the NAP07 genome. NAP07 chromosomes are color-coded and shown on the right side of each plot. Scaffolds from other genomes are in black and shown on the left side of each plot. Tick marks along chromosomes/scaffolds indicate 1 Mb distances
Species phylogeny (concatenated analyses).—
We used three data sets for inferring the species (organismal) phylogeny:
Data set 1—Orthologous proteins. The first approach was to extract all protein sequences and determine single-copy orthogroups with OrthoFinder (2.3.11; Emms and Kelly Citation2019; SUPPLEMENTARY MATERIAL). Consistent with macrosynteny results, OrthoFinder analyses of section Alternaria revealed very few gene duplication events: 95.9% of orthogroups experienced no duplications across the entire phylogeny. In total, protein sequences from 8750 single-copy orthologs shared by all taxa were used to construct a consensus phylogeny (SUPPLEMENTARY FIG. 1).
Data set 2—BUSCO genes. The second data set was composed of conserved, shared genes identified during BUSCO analyses. Nucleotide sequences for BUSCO genes present in all 38 genomes were extracted, and 983 genes were chosen for the final data set (SUPPLEMENTARY MATERIAL; SUPPLEMENTARY TABLE 4), with a cumulative alignment length of 1.97 Mb and 101.7 × 103 (5.2%) variable sites. A maximum likelihood (ML) tree was constructed under the best-fit partition model (SUPPLEMENTARY FIG. 1).
Data set 3—Custom gene selection. The third data set was a custom selection of genes that were likely to be the most informative for phylogenetic reconstruction of our sample of strains from section Alternaria. Starting with a high-confidence gene set predicted with evidence from transcriptomic data, each of the 12 860 genes were searched for and extracted from the genomes. Only single-copy genes that were present in all 38 genomes, and contained sufficient phylogenetic signal (i.e., >3% phylogenetically informative sites; SUPPLEMENTARY MATERIAL), were retained. The final custom data set had 6209 genes, with a cumulative alignment length of 8.58 Mb and ~756 × 103 (8.8%) variable sites. The resulting ML tree constructed under the best-fit partition model is presented in .
Figure 2. Maximum likelihood tree constructed from the custom data set of 6209 genes concatenated. Shading of squares at nodes represents the level of support each node received from bootstrapping, but also from other data sets or analyses methods (see inset). For shading, black indicates that the branch was present and significantly supported, gray indicates that the branch was present but not significantly supported, and white indicates that the branch was absent. Significant support was considered >95% in ML ultrafast bootstrapping for the BUSCO and custom data sets, >95% in posterior probability for the custom MSC data set, and presence in consenses tree for OrthoFinder data set. Numbers on branches indicate the gene concordance factor (GCF) for the 6209 genes trees compared with the displayed species tree. Current species name is indicated in brackets in taxon labels, if different from original species name. T indicates ex-type strain
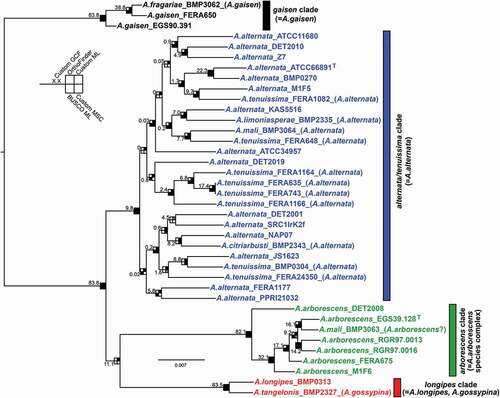
Comparison of branch support values from the three data sets () revealed that four main clades were identified by all analyses, hereafter referred to as the (i) “alternata/tenuissima” (alt/ten), (ii) “arborescens,” (iii) “longipes,” and (iv) “gaisen” clades. As predicted, the tree constructed from protein sequences (OrthoFinder) provided less resolution within clades, whereas the data set of 6209 custom genes provided the most resolution.
Species phylogeny (analyses of individual loci).—
Joint analyses of the 6209 concatenated genes included a very large amount of sequence data, so the resulting phylogeny was well resolved with nearly every branch receiving 100% bootstrap support. This approach averages over the signals from all genes but obscures the differences that may exist between the evolutionary histories of individual genes. When the sampled taxa are likely to span the species-population interface, one must accept that incomplete lineage sorting may cause discordance between gene trees (Pamilo and Nei Citation1988) and that gene trees may not match the species tree.
When gene tree discordance is likely to be high, we can apply multispecies coalescent (MSC) approaches that account for incomplete lineage sorting and estimate the species tree from the distribution of gene trees. To more comprehensively explore gene-tree space, and to account for gene-tree uncertainty, 10 nonparametric bootstrap ML trees were constructed for each of the 6209 genes under their respective best-fit models. MSC analysis of the resulting 62 090 gene trees with ASTRAL-III produced a species tree with topology similar to that from concatenated analyses (SUPPLEMENTARY FIG. 1).
Although all analyses support the existence of the four main clades (), the relationships among clades received mixed support. For example, the concatenated custom 6209-gene set suggests that the longipes clade is sister to the arborescens clade [longipes,arborescens], but most evidence supports a sister relationship between the longipes and alt/ten clades [longipes,alt/ten].
Gene concordance factor.—
To further explore gene/species tree concordance, we determined which branches in the species tree were present in each of the individual gene trees (gene concordance factor, GCF). As shown in , most of the main branches that received full support in the concatenated ML and MSC analyses were supported by a large proportion of the gene trees. The most notable exception was the branch for the alt/ten clade, which was supported by only 9.8% of individual gene trees. The placement of the longipes clade again received mixed support, with [longipes,alt/ten] and [longipes,arborescens] being supported by 12.4% and 11.1% of individual gene trees, respectively.
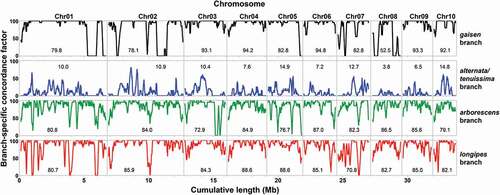
Genomic distribution of gene/species tree discordance.—
To visualize the relationship between gene/species tree concordance and genomic location, individual genes were scored for their support of the four main branches and the patterns were plotted against physical coordinates along chromosomes (). At the chromosome level, the proportion of analyzed genes that supported a particular main branch could vary quite drastically. For example, the gaisen clade was supported by 94.8% of genes from chromosome 6, but only 52.5% of genes from chromosome 8. When analyzed at a finer scale, gene/species tree discordance was not evenly distributed along individual chromosomes. Genomic clustering of discordance was most apparent for the gaisen clade where, for example, chromosomes 1 and 2 both had much of their gene/species discordance concentrated into regions of ~0.75 Mb in length (). Discordance for the arborescens and longipes clades was more evenly distributed, but they still had regions where continuous stretches of analyzed genes failed to support the clade branch. The distribution for the alt/ten branch was fundamentally different because gene/species discordance was the rule rather than the exception, but there were still peaks of genomically colocated concordance.
Figure 3. Plots of branch-specific concordance factor as a function of genomic location. Lines represent moving averages based on a sliding window of 20 genes. A concordance factor of 100 indicates that the branch was present in all included gene trees, whereas 0 indicates that the branch was absent in all included gene trees. The mean for the entire chromosome is indicated within each panel
We examined the gene trees located in some regions of high overall gene/species discordance to reveal the phylogenetic history of those regions (e.g., 34 genes in the ~154 kb region centered around the 5.9 Mb mark of the chromosome 1; 23 genes in the last ~185 kb at one end of chromosome 5). Although the topology of the trees varied quite drastically, a common observation was that the gaisen clade often intermingled with the alt/ten clade and the longipes clade often intermingled with the arborescens clade. ASTRAL MSC consensus trees produced from these discordant genomic regions are shown in SUPPLEMENTARY FIG. 2.
Putative diagnostic markers for detection and identification of main lineages.—
Now that the main lineages were circumscribed by genome-wide data, we searched for diagnostic molecular markers that were consistent with this framework. To support accurate identification, we chose molecular markers that contained sequence polymorphisms that could distinguish among all four of the main section Alternaria lineages (i.e., genes that had polymorphism arise, sort and fix between the divergence events giving rise to the lineages). We focused on regions of the genome that contained multiple consecutives genes that were fully concordant with the species phylogeny. lists our selection of top 20 candidate diagnostic genes. We were able to find appropriate candidate genes from all chromosomes except chromosome 8, which is consistent with chromosome 8 having the lowest mean concordance factor for two of the four main branches (). Genes were chosen irrespective of their function, and information on predicted gene products provided no evidence for any obvious trends of functional enrichment.
Table 2. List of 20 candidate genes that are diagnostic for the four analyzed clades in section Alternaria
For comparison, we also examined existing loci that previously have been applied to the molecular systematics of section Alternaria (Hong et al. Citation2005; Andrew et al. Citation2009; Woudenberg et al. Citation2015; Armitage et al. Citation2015). Gene trees built from extracted sequences for 15 of these loci () were scored for the presence of main clades. The rDNA region is typically informative at deeper phylogenetic depths, so as expected, the 28S nuc rDNA, internal transcribed spacer (ITS), and 18S nuc rDNA markers provided very little resolution and did not support any of the four main clades. Only one of the 15 gene trees had the alt/ten clade present, consistent with our previous finding that less than 10% of loci support that branch. Loci that were designed specifically for Alternaria provided greater resolution, but only one locus (Tma22) supported all four main clades. Therefore, most existing loci may be able to place an unknown strain into a section, but section-specific molecular markers are required to distinguish lineages within section Alternaria.
Table 3. Existing markers and their ability to discriminate the four analyzed clades in section Alternaria.
Based on genome coordinates alone (SUPPLEMENTARY TABLE 5), many of these loci are not predicted to support the species tree because they are located in genomic regions with low gene/species tree concordance (). The one locus (Tma22) that supported the species tree was within a genomic region of high concordance and, coincidently, was directly adjacent to one of our chosen candidate diagnostic genes (ASA-13).
Variation in genome content.—
The previous analyses were based on genes that were present in the genomes of all sampled taxa. OrthoFinder analyses revealed that 6004 (38.8%) of 15 479 orthogroups were missing from at least one strain, and 99.3% (5964) of these were shared between multiple strains. Although macrosynteny results indicate that large-scale variation in core chromosome structure may be rare in section Alternaria, smaller-scale variation in genome content (i.e., one to a few genes) appears to be more common.
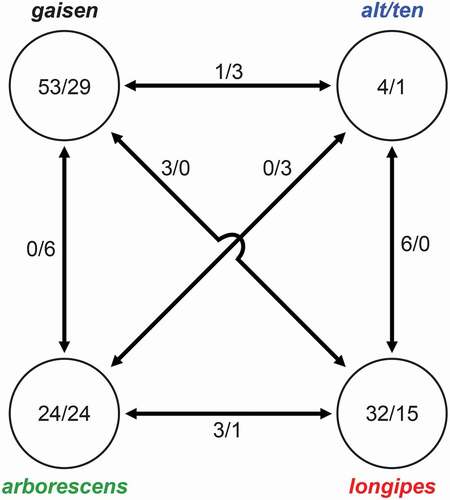
Although a detailed pan-genomic analysis is outside the scope of this study, we did search for genes or genomic regions that were potentially diagnostic for the major clades. Based on filtered OrthoFinder results, we identified 113 and 69 orthogroups whose presence or absence, respectively, was specific to one of the four major clades (i.e., present in all strains in one clade, and absent from all other strains; ). Consistent with the relative branch lengths in the species phylogeny, the gaisen clade possessed the highest number clade-specific orthogroups (53), whereas the alt/ten clade had the lowest (4). Patterns of clade-specific orthogroup absence were very similar to that of orthogroup presence. The functional annotations of clade-specific orthogroups were investigated for the two clades with the largest samples sizes (alt/ten and arborescens). We identified putative functions or conserved protein domains for 55% of these genes (SUPPLEMENTARY TABLE 6), but no clear trends in function were observed. When considering orthogroups that were shared exclusively by all members of two clades (), the alt/ten and longipes clades shared the highest number (6), supporting a [longipes,alt/ten] sister relationship.
Figure 4. Numbers of orthogroups that were clade specific (present/absent). Numbers within circles indicate orthogroup patterns for single clades (e.g., present in all taxa in clade but absent from all other taxa/absent from all taxa in clade but present in all other taxa). Numbers between circles indicate orthogroup patterns shared between two clades
When the genomic locations of clade-specific genes were examined, little evidence of physical clustering was found. In total, 163 of the 182 (89.6%) genes were singletons that were not physically adjacent to other clade-specific genes. The mean and maximum numbers of physically consecutive clade-specific genes were 1.12 and 4, respectively, indicating that clade-specific genomic regions are very limited in size.
DISCUSSION
Review of genomic resources for Alternaria section Alternaria.—
Our first goal was to provide a brief review of the genomic resources that are currently available for Alternaria section Alternaria. Over the past 9 years, Alternaria researchers have generated and released genomes for strains sampled from a variety of hosts, with a focus on pathotypes infecting citrus, apple, and pear (see for list of references). Although infection of cereals and contamination of harvested grains is a major problem caused by species in section Alternaria (Tralamazza et al. Citation2018; Tittlemier et al. Citation2019), there were no genomes available for strains sampled from major agricultural crops such as wheat, oat, barley, or rye. Here, we sequenced and assembled five new genomes from strains from wheat and rye, representing the first genomes produced for section Alternaria strains of Canadian origin.
The quality of the 38 analyzed genome assemblies was quite high, with some of the long-read-based genomes nearing the complete chromosome level, allowing for macrosynteny analyses and the detection of large-scale chromosomal rearrangements. Although we made only four pairwise comparisons, the structure of the genomes appeared to be fairly well conserved across section Alternaria. Note that we focused on core chromosomes and did not specifically analyze accessory chromosomes, also known as conditionally dispensable or supernumerary chromosomes. Although these accessory chromosomes are very interesting and often play important roles in conferring host-specific pathogenicity (Hu et al. Citation2012; Wang et al. Citation2019), a detailed analysis of their evolution and gene content will be performed in a future study. Recent work in other fungi have found that large-scale variation in genome content can be quite common and contribute to adaptive evolution (Möller and Stukenbrock Citation2017). A possible explanation for high macrosynteny observed here is that the depth of phylogenetic divergence within section Alternaria is relatively low, providing only a short time frame for major structural changes. When similar analyses are extended across additional sections of the Alternaria genus, more evidence of large-scale genome content variation may be found.
Phylogenomics of Alternaria section Alternaria.—
Another goal was to build a robust phylogeny for section Alternaria using a phylogenomics approach. Most previous phylogenetic work has relied on sequences from up to seven independent loci (Andrew et al. Citation2009; Lawrence et al. Citation2013; Armitage et al. Citation2015; Woudenberg et al. Citation2015), but a recent genome-based study included 500 BUSCO genes (Armitage et al. Citation2020). To fully explore the phylogenetic signals present in the genomic data, we compared multiple data sets and tree-building approaches.
An advantage of the OrthoFinder approach is that it allows sample-specific ortholog sets to be identified using established, user-friendly software packages. A disadvantage is that it relies heavily on reading frame and protein prediction, processes that may not be accurate or precise for the taxa under study. BUSCO genes are predesignated as single-copy orthologs across a large taxonomic group, in this case, all ascomycete fungi. Thus, the same set of genes can be used for molecular systematics of any ascomycete fungi, making comparisons across studies and taxa possible. However, BUSCO genes are by definition highly conserved, so their phylogenetic informativeness decreases as the taxonomic level decreases. Much like the OrthoFinder approach, BUSCO genes may not possess sufficient sequence variation to be useful at phylogenetic divergence levels found in section Alternaria (i.e., species-population interface). To maximize the phylogenetic informativeness of included loci, we also created a 6209-gene data set that was customized to section Alternaria. The advantages of this method are that it requires confident gene prediction for only one representative genome, which is often the case for many taxonomic groups, and it is well suited for large-scale studies that utilize mainly draft genomes without annotation.
One consistent pattern that was shared among all data sets and phylogenetic analyses was the well-supported existence of four main clades (): (i) alternata/tenuissima (alt/ten), (ii) arborescens, (iii) longipes, and (iv) gaisen clades. However, the relationships among the four distinct clades received mixed support. For example, the concatenated custom 6209-gene set suggests [longipes,arborescens], but most other analyses support [longipes,alt/ten]. The [longipes,alt/ten] grouping was recovered by Armitage et al. (Citation2020) with a genome sample that overlaps significantly with ours, albeit using a different gene set and analysis method.
Gene/species tree discordance.—
Visualization of gene/species tree discordance () revealed the dynamic nature of the evolutionary histories of specific regions along the length of chromosomes. Although the genomic regions displaying gene/species tree discordance could have arisen due to more complex evolutionary scenarios (e.g., horizontal gene transfer, genome duplication and loss), our findings of high macrosynteny and low rates of gene duplication provide strong evidence that our 6209 chosen loci are orthologous within our sample and therefore accurately reflect the evolutionary histories of the organisms. Since genes with low levels of informative polymorphism result in poorly resolved trees, we purposely excluded genes with low phylogenetic signal from our custom gene set. This screening criterion prevented low-signal genes from interfering with the high-signal genes during consensus and tree comparison analyses (i.e., a poorly supported polytomy should not detract from well-supported monophyly). Our approach is an extension of genealogical concordance concepts originally applied to species delineation using multilocus sequence data (Taylor et al. Citation2000; Dettman et al. Citation2003). Newly diverged species lose ancestral polymorphism via genetic drift, which may eventually lead to reciprocal monophyly. If effective population sizes are large, and time between divergence events is short, these random processes are expected to result in exclusive reciprocal monophyly for only a small proportion of genes (Pamilo and Nei Citation1988; Mehta and Rosenberg Citation2019).
The comparative levels of incomplete lineage sorting and resulting nonmonophyly can be estimated from the summed, genome-wide GCF of the section Alternaria clade-specific branches (). The alt/ten branch had the lowest GCF (9.8), and the internal branches within the alt/ten clade also had low GCF compared with other clades, suggesting that the alt/ten clade has had a larger historical effective population size. The alt/ten clade being large, widespread, and diverse is consistent with the frequency of occurrence of species in natural settings and surveys: the majority of small-spored Alternaria strains collected worldwide fall into the alt/ten clade (e.g., Zheng et al. Citation2015; da Cruz Cabral et al. Citation2017), although this may be due in part to difficulties with definitive taxonomic identification within section Alternaria. The other three clades are sampled less commonly and appear to have smaller effective population sizes, allowing for more rapid fixation of polymorphisms during divergence and higher probabilities of genealogical coalescence within clades. Although we included most publicly available genomes, the small sample size for some clades is a clear limitation of this study. The addition of new genomes may affect GCFs, but the magnitude of the effect, and which branches it would affect, would depend on the nature of the strains themselves. For example, adding a genome from a strain that is a first-generation hybrid between two clades would have a greater effect than a strain that falls cleanly into one clade. We look forward to expanding the genome sampling of underrepresented lineages and testing how well this framework holds up when challenged with more data.
Although no sexual state has been observed for species in section Alternaria, there is strong genetic evidence that recombination does occur in these taxa (Stewart et al. Citation2014; Meng et al. Citation2015). The high levels of gene tree discordance observed here are consistent with recombination within clades. In particular, the alt/ten clade displays high levels of within-clade diversity, and the within-clade branches typically have very low GCF values. Collectively, most evidence suggests that the alt/ten clade represents a single large, recombining species with wide geographic distribution. Further population-level studies are required to assess recombination in the other main clades.
Taxonomy and nomenclature of Alternaria section Alternaria.—
When erected by Lawrence et al. (Citation2013), section Alternaria contained ~60 small-spored catenulate species, including well-studied species such as A. alternata, A. tenuissima, A. arborescens, and A. gaisen. Later, a comprehensive revision by Woudenberg et al. (Citation2015) concluded that section Alternaria was composed of only 11 phylogenetic species and one species complex. Here, we describe the correspondence between named taxa and the four main clades identified in our phylogenomic analyses.
i) alternata/tenuissima (alt/ten) clade. Most of the 26 strains included in this clade were originally described as A. alternata or A. tenuissima (see ). High plasticity and overlap among morphological characters makes reliable and consistent differentiation of these morphospecies very difficult. Our phylogenomic analyses failed to support a distinction between the two taxa, in agreement with most molecular phylogenetic studies involving these two morphospecies (Andrew et al. Citation2009; Armitage et al. Citation2015, Citation2020; Woudenberg et al. Citation2015; da Cruz Cabral et al. Citation2017). Highlighting the lack of phylogenetic divergence, Woudenberg et al. (Citation2015) synonymized 35 morphospecies into A. alternata, including A. tenuissima. The name A. alternata was retained because it was the name of the type species for the genus and was well established in the research literature. Given the large body of supporting evidence, we also recommend that the name A. tenuissima be retired from use and be replaced by A. alternata instead.
As with many other plant pathogenic fungi, Alternaria strains that infect specific host plants or produce host-specific toxins have been described as formae speciales or pathotypes. In many cases, these strains were raised to the status of formally named species based on slight morphological differences (e.g., Simmons Citation1999a). Three examples of this are included in our alt/ten clade, all of which were named after their host associations: A. mali (apple), A. citriarbusti (citrus), and A. limoniasperae (lemon). Molecular studies of such named strains have shown that they are intermixed with other species and do not represent genetically distinct taxa (Peever et al. Citation2004; Rotondo et al. Citation2012). Accordingly, A. mali, A. citriarbusti, and A. limoniasperae were among the 35 morphospecies synonymized into A. alternata (Woudenberg et al. Citation2015). Our phylogenomic results support these large-scale synonymizations and the idea that apparent host-specific pathogenicity is a characteristic of individual strains rather than species.
For the sake of nomenclatural stability, we recommend that all strains in the alt/ten clade be referred to as A. alternata, with the recognition of host-specific pathotypes within the species.
ii) arborescens clade. A. arborescens is characterized morphologically by more frequent branching of the conidiophore and conidial chains, producing a three-dimensionally arborescent appearance (Simmons Citation1999b). This species was designated to contain strains causing stem canker of tomato (formerly known as the tomato pathotype, or A. alternata f. sp. lycopersici), but subsequent work has shown that host specificity is lacking and the species may be isolated from a wide range of hosts (Woudenberg et al. Citation2015). Phylogenomic analyses placed a strain named A. mali into this clade, likely representing an A. arborescens strain that was named A. mali simply because it was sampled from an apple host.
The strong phylogenomic support observed here for the distinct arborescens clade is consistent with previous molecular phylogenetic studies (e.g., the “arborescens clade” identified by Armitage et al. [Citation2020]). Woudenberg et al. (Citation2015) found that multilocus sequence data could not resolve the relationships between A. arborescens and ex-type strains from three other closely related morphospecies (A. cerealis, A. geophila, and A. senecionicola) and collectively considered them as the A. arborescens species complex. Support for an A. arborescens species complex is found when comparing the GCF values for the within-clade branches: values for the arborescens clade are greater than that for alt/ten clade, suggesting that the former clade has lower rates of recombination and higher levels of phylogenetic substructure. We note that uneven sampling across the main clades may introduce bias, so further sampling of the arborescens clade is required before meaningful comparisons of within-clade diversity can be made. Our newly sequenced strain DET2008 appears to be early diverging and may represent a previously undescribed species, pending increased sampling and analyses. Furthermore, better representation of the other species within the complex are needed to determine whether they represent multiple distinct species or one larger species. Until these issues are resolved, we follow Woudenberg et al. (Citation2015) and recommend using the name A. arborescens species complex.
iii) longipes clade. The two strains comprising this clade are from closely related, sister species. One strain was from A. longipes, representing the tobacco pathotype of small-spored Alternaria. The second strain was originally identified as A. tangelonis (tangerine pathotype), however, A. tangelonis was synonymized into A. gossypina based on comparative analyses of ex-type strains (Woudenberg et al. Citation2015). The placement of a tangerine pathotype in the longipes clade highlights the fact that citrus and apple pathotypes are typically polyphyletic and cannot be assumed to represent distinct species (Armitage et al. Citation2020).
iv) gaisen clade. A. gaisen encompasses strains that are specifically pathogenic on Japanese pear. This clade included a genome that was accessioned as A. fragariae, named after the fact the strain was isolated from strawberry. However, this is not a formally named species and should actually be A. alternata f. sp. fragariae (Simmons Citation1999b). Recent work has found that some strawberry and pear pathotypes are morphologically and phylogenetically indistinguishable and should both be included within A. gaisen (Nishikawa et al. Citation2019). Once again, the apparent misplacement of taxa can be explained by erroneous identifications based on host association.
Our study highlights how nomenclature at the point of submission to public sequence databases typically does not get updated when taxonomic revisions occur. Of the 32 genomes obtained from public databases, 14 (44%) of them are accessioned under species names that have been synonymized or changed (sensu Woudenberg et al. Citation2015). However, this is not just a problem for retroactive correction of names in existing database accessions. Revisions in nomenclature or taxonomy can take time to be incorporated into the research community. For example, even though evidence shows that A. tenuissima is not a distinct species, and was synonymized into A. alternata in 2015, the tenuissima epithet continues to be frequently used in publications even today.
Future directions.—
Our list of candidate diagnostic molecular markers was developed for distinguishing among the four main section Alternaria clades identified in our phylogenomic analyses. Appropriate marker choice is critical for accurate and reliable methods of detection and identification, and these tools are critical for dealing with quarantine and restricted species that impact international trade. Future work is required to test how well these newly proposed markers perform when challenged by a wider range of section Alternaria strains, including strains from other species not studied here. Ideally, we hope to identify markers that are consistent within clades but also diagnostic across the entire section Alternaria.
The five newly sequenced genomes were all from isolates sampled from cereal crops of Canadian origin, but they did not form a monophyletic clade that corresponded to host or geography. Given the observed diversity among these five isolates, it is clear that our sampling of section Alternaria biodiversity in Canada is far from complete, and we will continue to characterize the species of Alternaria infecting a wide range of agricultural hosts.
Our analyses included whole genome sequences for as many section Alternaria species as were available; however, section Alternaria contains at least seven other species for which genome sequences do not currently exist. To characterize the biodiversity of this important group of fungi, genome sampling should be expanded to encompass the full phylogenetic diversity of small-spored Alternaria. Increased sampling of the arborescens clade will help to determine whether it represents a species complex, and proper population samples will allow more comprehensive tests for recombination.
Supplemental Material
Download PDF (2 MB)ACKNOWLEDGMENTS
We thank Allen Xue and Yuanhong Chen for providing access to grain samples for fungal isolation; Keith Seifert for providing a fungal strain; Joyce Woudenberg and Ewald Groenewald for sharing sequence data; Kasia Dadej and staff at the Agriculture and Agri-Food Canada (AAFC) Molecular Technologies Laboratory for sequencing support; and Hai Nguyen for reviewing an early draft of the manuscript.
supplementary material
Supplemental data for this article can be accessed on the publisher’s Web site.
Additional information
Funding
Related Research Data
LITERATURE CITED
- Andrew M, Peever TL, Pryor BM. 2009. An expanded multilocus phylogeny does not resolve morphological species within the small-spored Alternaria species complex. Mycologia 101:95–109.
- Armitage AD, Barbara DJ, Harrison RJ, Lane CR, Sreenivasaprasad S, Woodhall JW, Clarkson JP. 2015. Discrete lineages within Alternaria alternata species group: identification using new highly variable loci and support from morphological characters. Fungal Biology 119:994–1006.
- Armitage AD, Cockerton HM, Sreenivasaprasad S, Woodhall J, Lane CR, Harrison RJ, Clarkson JP. 2020. Genomics evolutionary history and diagnostics of the Alternaria alternata species group including apple and Asian pear pathotypes. Frontiers in Microbiology 10:3124.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19:455–477.
- Bihon W, Cloete M, Gerrano AS, Oelofse D, Adebola P. 2016. Draft genome sequence of Alternaria alternata isolated from onion leaves in South Africa. Genome Announcements 4:301022–16.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120.
- da Cruz Cabral L, Rodriguero M, Stenglein S, Fog Nielsen K, Patriarca A. 2017. Characterization of small-spored Alternaria from Argentinean crops through a polyphasic approach. International Journal of Food Microbiology 257:206–215.
- Dang HX, Pryor B, Peever T, Lawrence CB. 2015. The Alternaria genomes database: a comprehensive resource for a fungal genus comprised of saprophytes, plant pathogens, and allergenic species. BMC Genomics 16:239.
- Dettman JR, Jacobson DJ, Taylor JW. 2003. A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote neurospora. Evolution 57:2703–2720.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32:1792–1797.
- Emms DM, Kelly S. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology 20:238.
- Gebru ST, Gangiredla J, Tournas VH, Tartera C, Mammel MK. 2020. Draft genome sequences of Alternaria strains isolated from grapes and apples. Microbiology Resource Announcements 9:e01491–19.
- Hong SG, Cramer RA, Lawrence CB, Pryor BM. 2005. Alt a 1 allergen homologs from Alternaria and related taxa: analysis of phylogenetic content and secondary structure. Fungal Genetics and Biology 42:119–129.
- Hu J, Chen C, Peever T, Dang H, Lawrence C, Mitchell T. 2012. Genomic characterization of the conditionally dispensable chromosome in Alternaria arborescens provides evidence for horizontal gene transfer. BMC Genomics 13:171.
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biology 5:R12.
- Lawrence DP, Gannibal PB, Peever TL, Pryor BM. 2013. The sections of Alternaria: formalizing species-group concepts. Mycologia 105:530–546.
- Lawrence DP, Rotondo F, Gannibal PB. 2016. Biodiversity and taxonomy of the pleomorphic genus Alternaria. Mycological Progress 15:3.
- Meena M, Gupta SK, Swapnil P, Zehra A, Dubey MK, Upadhyay RS. 2017. Alternaria toxins: potential virulence factors and genes related to pathogenesis. Frontiers in Microbiology 8:1451.
- Mehta RS, Rosenberg NA. 2019. The probability of reciprocal monophyly of gene lineages in three and four species. Theoretical Population Biology 129:133–147.
- Meng JW, Zhu W, He MH, Wu EJ, Duan GH, Xie YK, Jin YJ, Yang LN, Shang LP, Zhan J. 2015. Population genetic analysis reveals cryptic sex in the phytopathogenic fungus Alternaria alternata. Scientific Reports 5:18250.
- Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. 2018. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34:i142–i150.
- Möller EM, Bahnweg G, Sandermann H, Geiger HH. 1992. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Research 20:6115–6116.
- Möller M, Stukenbrock EH. 2017. Evolution and genome architecture in fungal plant pathogens. Nature Reviews Microbiology 15:756–771.
- Nees von Esenbeck C. 1817. Das System der Pilze und Schwämme. Wurzburg, Germany: Stahel. 474 p.
- Nguyen HDT, Lewis CT, Lévesque CA, Gräfenhan T. 2016. Draft genome sequence of Alternaria alternata ATCC 34957. Genome Announcements 4:e01554–15.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution 32:268–274.
- Nishikawa J, Nakashima C. 2019. Morphological and molecular characterization of the strawberry black leaf spot pathogen referred to as the strawberry pathotype of Alternaria alternata. Mycoscience 60:1–9.
- Pamilo P, Nei M. 1988. Relationships between gene trees and species trees. Molecular Biology and Evolution 5:568–583.
- Park SY, Jeon J, Kim JA, Jeon MJ, Jeong MH, Kim Y, Lee Y, Chung H, Lee YH, Kim S. 2020. Draft genome sequence of Alternaria alternata JS-1623, a fungal endophyte of Abies koreana. Mycobiology, doi:https://doi.org/10.1080/12298093.2020.1756134
- Peever TL, Su G, Carpenter-Boggs L, Timmer LW. 2004. Molecular systematics of citrus-associated Alternaria species. Mycologia 96:119–134.
- Pryor B, Gilberston R. 2000. Molecular phylogenetic relationships amongst Alternaria species and related fungi based upon analysis of nuclear ITS and mt SSU rDNA sequences. Mycological Research 104:1312–1312.
- Rotondo F, Collina M, Brunelli A, Pryor BM. 2012. Comparison of Alternaria spp. Collected in Italy from apple with A. mali and other AM-toxin producing strains. Phytopathology 102:1130–1142.
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva E V., Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212.
- Simmons E. 1999a. Alternaria themes and variations (236–243). Host-specific toxin producers. Mycotaxon 70:325–369.
- Simmons E. 1999b. Alternaria themes and variations (226–235). Classification of citrus pathogens. Mycotaxon 70:263–323.
- Simmons E. 2007. Alternaria: an identification manual. CBS Biodiversity Series 6. Utrecht, The Netherlands: CBS Fungal Biodiversity Centre. 780 p.
- Simmons EG. 1967. Typification of Alternaria, Stemphylium, and Ulocladium. Mycologia 59:67–92.
- Stewart JE, Timmer LW, Lawrence CB, Pryor BM, Peever TL. 2014. Discord between morphological and phylogenetic species boundaries: incomplete lineage sorting and recombination results in fuzzy species boundaries in an asexual fungal pathogen. BMC Evolutionary Biology 14:38.
- Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC. 2000. Phylogenetic species recognition and species concepts in fungi. Fungal Genetics and Biology 31:21–32.
- Tittlemier SA, Blagden R, Chan J, Gaba D, Mckendry T, Pleskach K, Roscoe M. 2019. Fusarium and Alternaria mycotoxins present in Canadian wheat and durum harvest samples. Canadian Journal of Plant Pathology 41:403–414.
- Tralamazza SM, Piacentini KC, Iwase CHT, Rocha Lde O. 2018. Toxigenic Alternaria species: impact in cereals worldwide. Current Opinion in Food Science 23:57–63.
- Ushijima K, Yamamoto M. 2019. A sequence resource of autosomes and additional chromosomes in the peach pathotype of Alternaria alternata. Molecular Plant-Microbe Interactions 32:1273–1276.
- Wang M, Fu H, Shen XX, Ruan R, Rokas A, Li H. 2019. Genomic features and evolution of the conditionally dispensable chromosome in the tangerine pathotype of Alternaria alternata. Molecular Plant Pathology 20:1425–1438.
- Wang M, Sun X, Yu D, Xu J, Chung K, Li H. 2016. Genomic and transcriptomic analyses of the tangerine pathotype of Alternaria alternata in response to oxidative stress. Scientific Reports 6:32437.
- Woudenberg JHC, Groenewald JZ, Binder M, Crous PW. 2013. Alternaria redefined. Studies in Mycology 75:171–212.
- Woudenberg JHC, Seidl MF, Groenewald JZ, de Vries M, Stielow JB, Thomma BPHJ, Crous PW. 2015. Alternaria section Alternaria: species, formae speciales or pathotypes? Studies in Mycology 82:1–21.
- Zeiner CA, Purvine SO, Zink EM, Paša-Tolić L, Chaput DL, Haridas S, Wu S, LaButti K, Grigoriev IV, Henrissat B, Santelli CM, Hansel CM. 2016. Comparative analysis of secretome profiles of manganese(II)-oxidizing ascomycete fungi. PLoS ONE 1:e0157844.
- Zhang C, Rabiee M, Sayyari E, Mirarab S. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19(Suppl 6):153.
- Zheng HH, Zhao J, Wang TY, Wu XH. 2015. Characterization of Alternaria species associated with potato foliar diseases in China. Plant Pathology 64:425–433.