
ABSTRACT
Species in Alternaria sections Infectoriae and Pseudoalternaria are commonly isolated from agricultural crops and a variety of other plant hosts. With the increasing appreciation that species from these two sections are often the dominant taxa recovered from important cereal crops, the need for improved understanding of their biodiversity and taxonomy has grown. Given that morphological characteristics and existing molecular markers are not sufficient for distinguishing among species, we expanded the genomic resources for these sections to support research in biosystematics and species diagnostics. Whole genome assemblies for 22 strains were generated, including the first genomes from section Infectoriae or Pseudoalternaria strains sampled from Canada, which significantly increases the number of publicly released genomes, particularly for section Pseudoalternaria. We performed comprehensive phylogenomic analyses of all available genomes (n = 39) and present the first robust phylogeny for these taxa. The segregation of the two sections was strongly supported by genomewide data, and multiple lineages were detected within each section. We then provide an overview of the biosystematics of these groups by analyzing two standard molecular markers from the largest sample of section Infectoriae and Pseudoalternaria strains studied to date. The patterns of relative diversity suggest that, in many cases, multiple species described based on minor morphological differences may actually represent different strains of the same species. A list of candidate loci for development into new informative molecular markers, which are diagnostic for sections and lineages, was created from analyses of phylogenetic signals from individual genes across the entire genome.
INTRODUCTION
Within the fungal genus Alternaria, sections Infectoriae and Pseudoalternaria are two closely related subgeneric groups composed of approximately 34 and 8 species, respectively (Gannibal and Lawrence Citation2016; Gannibal et al. Citation2022; Lawrence et al. Citation2016). Members of these sections can have broad distributions across temperate and tropical regions, with ecological roles ranging from harmless saprobes and endophytes to pathogens of plants and occasionally humans. Although species in sections Infectoriae and Pseudoalternaria (Inf/Pse hereafter) can infect a diversity of dicot and monocot hosts, their main pathological and economic impact is due to disease of important cereal crops such as wheat and barley (e.g., leaf spot, black point, black head mold) (Gannibal et al. Citation2022; Masiello Citation2020; Perelló et al. Citation2008; Poursafar et al. Citation2019, Citation2018; Somma et al. Citation2019; Turzhanova et al. Citation2020). Studies have found that the genus Alternaria can be the largest component of cereal mycobiota, and in many cases the most abundant Alternaria species is from sections Inf/Pse (Andersen et al. Citation1996; Barkat et al. Citation2016; Golosna Citation2022; Hafez et al. Citation2022; Kosiak et al. Citation2004). For example, a recent study by Hafez et al. (Citation2022), on wheat from western Canada, reported that 38.5% of all isolated fungi were from Alternaria species in sections Inf/Pse.
Species in sections Inf/Pse are considered “small-spored,” and their conidial morphology is very similar to that found in the more well-studied Alternaria section Alternaria (containing the species A. alternata; Simmons Citation2007). Despite this common morphology (i.e., darkly pigmented phaeodictyospores with tapering apical beaks), sections Inf/Pse are phylogenetically divergent from section Alternaria and are, in fact, more closely related to fungi that were originally described as separate genera such as Chalastospora (Lawrence et al. Citation2013; Woudenberg et al. Citation2013). While the shape and relatively small size (e.g., length under 60 um) of conidia are shared between sections Alternaria and Inf/Pse, the latter sections are characterized by longer and/or more highly branched secondary conidiophores, and the aggregation of conidiophores together, resulting in what is usually described as a complex, more three-dimensional sporulation pattern (Gannibal and Lawrence Citation2016; Lawrence et al. Citation2014). Under the traditional, morphological classification system for Alternaria, all strains or species with this sporulation morphology were placed into the “A. infectoria complex” or “A. infectoria species group” (Simmons Citation1994; Simmons and Roberts Citation1993) in reference to the representative species, A. infectoria. A series of taxonomic revisions supported by molecular information led to the synonymizing of all alternarioid hyphomycetes into the sole genus Alternaria and the elevation of species groups to the formal status of “sections” (Lawrence et al. Citation2013; Woudenberg et al. Citation2013). As a result, these species were all classified within section Infectoriae. Later, the genus Pseudoalternaria was converted into a new section named Pseudoalternaria (Lawrence et al. Citation2016), which is the sister group to section Infectoriae.
Historical use of the term “A. infectoria species group” for all strains that fall within sections Inf/Pse has created some challenges with interpreting past research. This naming convention is understandable given that reliable methods for identifying species are lacking, and that section Pseudoalternaria and many of its species were erected quite recently. Recognizing that sections Inf/Pse may in fact contain a significant amount of species diversity may explain some of the observed variation between strains in the “A. infectoria species group.” For example, several studies have clearly shown that section Inf/Pse strains can produce unique secondary metabolite profiles that are distinct from section Alternaria, and that substantial variation can occur between section Inf/Pse strains (Andersen et al. Citation2002, Citation2009; Andersen and Thrane Citation1996; Christensen et al. Citation2005; Patriarca et al. Citation2019). We hypothesize this variation is due, in part, to unrecognized species diversity within the “A. infectoria species group.” Being able to accurately identify these organisms is important for plant pathology and regulatory purposes, as well as for predicting mycotoxin production and its impacts on food/feed safety.
Cereal-associated Alternaria species are predominantly from sections Alternaria, Infectoriae, and Pseudoalternaria (Andersen et al. Citation2015; Golosna Citation2022; Kahl et al. Citation2015; Ramires et al. Citation2018; Somma et al. Citation2019; Orina et al. Citation2021; Castanares et al. Citation2021; Gannibal et al. Citation2022; Hafez et al. Citation2022). In a previous series of studies, we investigated the phylogenomic relationships, diagnostic markers, and species diversity of section Alternaria on Canadian cereal crops (Dettman and Eggertson Citation2021, Citation2022; Dettman et al. Citation2023). Here, we perform similar and complementary research but focus our attention on sections Inf/Pse. The included analyses and organization of this paper will be similar to our previous publication (Dettman and Eggertson Citation2021).
Given the limitations of morphological characteristics, and the lack of highly informative molecular markers, we aimed to expand the genomic resources for sections Inf/Pse and make them available for biosystematics and diversity research in this group. We sequenced the whole genomes of 22 strains from sections Inf/Pse, significantly increasing the number of publicly released genomes, particularly for section Pseudoalternaria. We provide the first genomes from section Inf/Pse strains sampled from Canada. All other available genomes were included in our comprehensive phylogenomic analyses to create the first robust phylogeny for these taxa, and to detect the main lineages within each section. To place our findings in context of other known species, we compiled and analyzed available data for two standard molecular markers, representing the largest sample of section Inf/Pse strains studied to date. Examination of existing, commonly employed molecular markers for phylogenetics in this group demonstrated that they are not sufficient, alone or together, to distinguish lineages. Analyses of phylogenetic patterns from individual genes across the entire genome identified candidate loci for development into new informative molecular markers.
MATERIALS AND METHODS
Fungal strains.—
Strains analyzed in this study are maintained by the Canadian Collection of Fungal Cultures (CCFC) or other mycological research collections held by Agriculture and Agri-Food Canada (AAFC) in Ottawa. Strains with DAOMC prefixes are from the CCFC, and strains with KAS or DET prefixes are from the AAFC collections of Keith Seifert or the Dettman laboratory, respectively.
Genome sequences.—
Fungal strains were grown on potato dextrose agar (potato extract 4 g/L, dextrose 20 g/L, agar 15 g/L) and incubated at 25 C for 10–14 days. For DET and KAS strains: Genomic DNA was extracted using previously described methods (Dettman and Eggertson Citation2021). After normalizing to 300 ng, genomic DNA was sheared to approximately 350 bp fragment size using a Covaris M220 sonicator (Covaris, Woburn, Massachusetts). The fragmented DNA was used as the template to construct polymerase chain reaction (PCR)-free libraries with a NxSeq AmpFREE Low DNA Library Kit (Lucigen, Middleton, Wisconsin) according to the manufacturer’s instructions. For DAOMC strains: Genomic DNA was extracted using a DNeasy Plant Mini Kit (Qiagen, Hilden, Germany). After normalizing to 100 ng, genomic DNA was used as the template to construct libraries with an Illumina DNA Prep kit (formerly Nextera DNA Flex; Illumina, San Diego, California) according to the manufacturer’s instructions. Single-indexed libraries were sequenced with paired-end reads (2 × 150 bp) on a NextSeq 500 instrument (Illumina) using a NextSeq High Output Reagent Kit (Illumina). For the 22 samples, an average of 21.0 million reads were generated for each (SUPPLEMENTARY TABLE 1). Eighteen existing genome assemblies were downloaded from the public repositories of the National Center for Biotechnology Information (NCBI) and Joint Genome Institute (JGI) MycoCosm (SUPPLEMENTARY TABLE 1).
Genome assembly and gene prediction.—
Genomic analyses methods were previously described in Dettman and Eggertson (Citation2021). Briefly, Illumina adapters were clipped from raw reads followed by quality trimming with Trimmomatic (0.38; Bolger et al. Citation2014). Assemblies were created with spades (3.15; Bankevich et al. Citation2012), and contigs were filtered out if they were less 500 bp in length or were potentially mitochondrial or bacterial in nature, as determined by BLASTn (2.10) searches. Assemblies then underwent four successive rounds of polishing (error correction) using the Illumina reads (Pilon 1.23; Walker et al. Citation2014). Assembly quality and completeness was assessed with QUAST (5.0.2; Mikheenko et al. Citation2018) and BUSCO (Benchmarking Universal Single-Copy Orthologs) (5.5; Simão et al. Citation2015), respectively. For BUSCO, genome assemblies were compared with the ascomycota_odb10 ortholog data set (accessed 5 Sep 2023) of 1706 conserved Ascomycete orthologs.
To generate a high-quality set of gene predictions, an A. rosae (Altro1 strain) genome assembly was subjected to training and gene prediction processes using funannotate (1.8.15; https://github.com/nextgenusfs/funannotate). The Altro1 (= DTO 242-I4) strain was chosen because it was the only strain for which a genome and RNASeq transcriptomic data were available (NCBI Sequence Read Archive [SRA] accession SRR7140916). Briefly, RNASeq data were trimmed with Trimmomatic and normalized using BBNorm (39.06; target = 50, min = 5; https://sourceforge.net/projects/bbmap) to reduce the number of redundant reads in areas of high coverage. The normalized RNASeq data were used as evidence to train gene models, which were then used for gene prediction.
Selection of gene data sets.—
BUSCO gene data set
BUSCO results were intersected to identify single-copy orthologs that were present in all included genomes. Nucleotide sequences of orthologs were aligned with MUSCLE (3.8; Edgar Citation2004), then alignments were filtered to exclude those with greater than 10% gaps or missing data, resulting in 1345 retained orthologs.
Custom gene data set
The gff3 (general feature format) coordinates from Altro1 gene prediction results were used by BEDTools (2.27; Quinlan and Hall Citation2010) to extract full gene sequences (including introns) from the Altro1 genome. Each of these 13 261 high-confidence genes was BLASTn-searched against a local, custom, 40-genome BLAST database. After filtering out sequences less than 25% of the query length, genes with single-copy hits in all 40 genomes were retained and aligned with MUSCLE. Alignments were filtered to exclude those with >10% gaps or missing data then filtered again to retain only those with ≥3% phylogenetically informative sites. The final custom data set included 5294 genes.
Standard molecular markers
To extract sequences of existing genes from genome assemblies, representative sequences from the A. infectoria ex-type strain (EGS 27-193, CBS 210.86) were used as queries against a local, custom, 40-genome BLAST database. Extracted sequences were aligned with MUSCLE.
Phylogenetic analyses.—
Model selection, data partitioning, and construction of maximum likelihood phylogenies were performed with IQ-TREE (2.0; Nguyen et al. Citation2015). Multispecies coalescent (MSC) analysis was implemented by ASTRAL-III (5.7.8; Zhang et al. Citation2018). A. panax (section Panax) was used for outgroup and tree rooting purposes because it’s the species with a genome sequence that is most closely related to the ingroup.
Selection of candidate diagnostic markers.—
Each of the 5294 genes in the Custom gene data set was scored for its support of the branches subtending the two sections and the five lineages (Inf1, Inf2, Inf3, Pse1, Pse2). If all seven of these branches were present in the maximum likelihood (ML) tree constructed from the single-gene alignment, that gene was flagged. As in Dettman and Eggertson (Citation2021), scaffold coordinates were used to identify regions of scaffolds containing multiple co-located genes that were consistent with the phylogenomic framework. Gene trees were manually examined to verify overall concordance, and eight candidate genes were chosen subjectively, attempting to include genes from as many scaffolds as possible.
See Supplementary Material for additional details on materials and methods.
RESULTS
Strain sampling.—
This study included 22 fungal strains in sections Infectoriae and Pseudoalternaria (). Seven strains are ex-type strains for described section Infectoriae species, isolated from four different hosts on five different continents. An additional 15 strains were isolated from barley, oats, wheat, or canola growing in Canada. These undescribed strains were chosen because examination of micromorphology, culture characteristics, and/or preliminary internal transcribed spacer (ITS) sequence data suggested that they were likely to belong to section Infectoriae (n = 10) or Pseudoalternaria (n = 5).
Table 1. Information on fungal strains and newly generated genome assemblies.
New genomic resources for sections Infectoriae and Pseudoalternaria.—
Summary statistics for our 22 newly generated genome assemblies (; SUPPLEMENTARY TABLE 1) were similar to previous assemblies for the genus Alternaria. New genomes had an average assembly length of 34.29 Mb distributed over an average of 120 contigs, with average sequence coverage of 89.6×. Genome contiguity was fairly high, with a mean N50 of 1.47 Mb, and BUSCO metrics also indicated high levels of genome completeness, with an average of 97.0% complete BUSCO genes being detected (Ascomycota ortholog set).
Seventeen other section Inf/Pse genomes were downloaded from public repositories (SUPPLEMENTARY TABLE 1). Six of the ex-type strains sequenced by us were also sequenced by Fulcher and Bergstrom (Citation2023).
Species phylogeny (combined gene analyses).—
Including 39 genomes from sections Inf/Pse and one outgroup (A. panax), we created two data sets for inferring the species (organismal) phylogeny:
BUSCO gene data set
The first data set was composed of conserved, single-copy BUSCO genes that were present in all 40 included genomes. The resulting data set contained 1345 BUSCO genes, with a cumulative alignment length of 2.88 Mb and 411 228 phylogenetically informative sites (14.3%).
Custom gene data set
The second data set was a curated selection of single-copy genes shared by all 40 genomes, customized for our specific sample of section Infectoriae and Pseudoalternaria diversity (Supplementary Material; Dettman and Eggertson Citation2021). Selected from a high-confidence gene set predicted with evidence from transcriptomic data, the resulting data set contained 5294 genes with a cumulative alignment length of 8.77 Mb and 1 371 562 phylogenetically informative sites (15.6%). Note that this data set represents 25.0% of the typical genome for these sections, which averages 34.9 Mb.
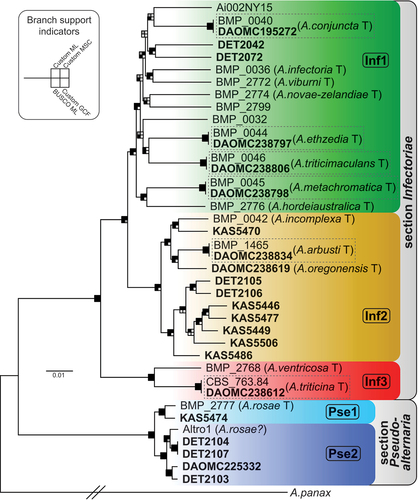
A maximum likelihood (ML) tree was constructed under the best-fit partition model for each of the combined gene data sets (; SUPPLEMENTARY FIG. 1). Owing to the large amount of sequence data being analyzed, the vast majority of branches were well supported. For example, 84% (31/37) of the branches received significant support in ML bootstrapping of the Custom gene data set ().
Figure 1. Maximum likelihood tree constructed from the Custom gene data set of 5294 genes combined. Bold taxon names indicate newly generated genome assemblies. Main lineages are color-coded and labeled within. Shading of squares at nodes represents the level of support each node received from four different methods of phylogenetic analysis (see inset). Support was considered significant where the analysis found ≥70% in ML bootstrapping for the Custom (Custom ML) and BUSCO (BUSCO ML) data sets or ≥0.95 posterior probability for the multispecies coalescent (Custom MSC) analysis. For shading, black indicates the branch was significantly supported, gray indicates the branch was present but not significantly supported, and white indicates the branch was absent. For shading of gene concordance factor (Custom GCF) analysis, black indicates the branch had a GCF ≥50, gray indicates GCF between 50 and 25, and white indicates GCF ≤25. The actual length of the branch leading to A. panax (outgroup) is 5× as long as shown. Previously existing species designations are shown in brackets. “T” indicates ex-type strains. Dashed-line boxes contain strains that should be the same ex-type strain.
Species phylogeny (individual gene analyses).—
As discussed in Dettman and Eggertson (Citation2021), combined gene analysis averages over the phylogenetic signal from all genes and may conceal incomplete lineage sorting that may occur at the species-population interface. We conducted two types of analyses on the Custom gene data set to investigate the concordance of signals from the 5294 individual genes.
Gene concordance factor (GCF)
A ML tree was constructed under the best-fit substitution model for each of the individual 5294 gene alignments. GCFs were calculated to determine which branches in the combined Custom ML tree were present in each of the 5294 individual gene trees. Only around one third (13/37) of the branches in combined ML analyses were present in the majority (>50%) of single-gene ML trees (; SUPPLEMENTARY FIG. 1).
Multispecies coalescent (MSC) approach
Ten nonparametric bootstrap ML trees were built from each of the 5294 genes, and these 52 940 trees were subjected to MSC analysis using ASTRAL-III, which accounts for incomplete lineage sorting and estimates the organismal tree from the distribution of individual gene trees. The resulting MSC consensus tree (SUPPLEMENTARY FIG. 1) was very similar to the combined analyses ML trees, and MSC branch support values are indicated in for comparison.
Comparison of genome sequences for different versions of the same strain.—
Six of the ex-type strains from section Infectoriae had two different genome assemblies, generated from different live versions of the strain, allowing for a test of consistency and strain purity between independent laboratories. As shown in , the paired versions of each strain appeared nearly identical at the sequence level, confirming their shared pedigree. We calculated the average genetic distance over 8.77 Mb of sequence in the Custom gene data set for each genome pair, which was only 1.14 × 10−5 (i.e., ~1 mismatch every 100 kb).
Lineages within section Infectoriae.—
The 32 (26 unique) strains in section Infectoriae were divided into three main lineages ():
Lineage Inf1 contained half (13) of the unique section Infectoriae strains, including eight ex-type strains of described species collected from around the world. There was little phylogenetic structure within the lineage; each strain was fairly distinct from the others, and the relationships among strains were poorly resolved. This lineage includes the type species of section Infectoriae, A. infectoria.
Lineage Inf2 contained 11 unique strains in total, including the three ex-types strains that were collected from western United States: A. arbusti from California, A. incomplexa from Idaho, and A. oregonensis from Oregon.
Lineage Inf3 was the earliest diverging lineage and had only two unique strains, the ex-types of A. ventricosa and A. triticina.
Lineages within section Pseudoalternaria.—
The seven section Pseudoalternaria strains consistently formed a distinct and well-supported clade that was clearly separate from section Infectoriae. Overall, the diversity within section Pseudoalternaria appeared to be lower than that observed in section Infectoriae. Our five newly sequenced strains were all collected in western Canada.
Even with our limited genome sample size for section Pseudoalternaria, two main lineages were apparent (Pse1 and Pse2). The only two strains with previously available genome sequences were both identified as A. rosae, but they did not group together in the phylogenomic analyses (). The ex-type strain (BMP 2777 or EGS 41-130) falls within lineage Pse1, but the Altro1 strain falls within Pse2.
Standard molecular phylogenetic markers.—
Next, we investigated the performance of standard molecular markers that have been employed for the systematics of sections Infectoriae and Pseudoalternaria (Gannibal et al. Citation2022; Iturrieta-González et al. Citation2020; Lawrence et al. Citation2014, Citation2013; Marin-Felix et al. Citation2019; Poursafar et al. Citation2019, Citation2018; Woudenberg et al. Citation2013). We scanned the literature and databases and identified five commonly used loci: ITS (rDNA internal transcribed spacer), ATPase (plasma membrane ATPase), gapdh (glyceraldehyde-3-phosphate dehydrogenase), rpb2 (RNA polymerase, second largest subunit), and tef1 (translation elongation factor 1-alpha). Locus-specific sequences were extracted from the 40 genomes, and the resulting single-locus phylogenies (SUPPLEMENTARY FIG. 2) were assessed for support of the sections and main lineages ().
Table 2. Performance of standard molecular markers.
Overall, the standard molecular markers did not reliably recover the main lineages identified by phylogenomic analyses. For example, section Infectoriae was a well-supported monophyletic group in only two single-locus trees (ATPase and ITS), and section Pseudoalternaria was well supported in three (ATPase, ITS, gapdh). Section monophyly was typically disrupted by the placement of the root (A. panax), but there was no intermingling of strains between sections (SUPPLEMENTARY FIG. 2). Members of different lineages within sections were often mixed together in the single-locus trees. When all five genes were combined and analyzed together (SUPPLEMENTARY FIG. 3), the two sections were recovered but only one of the five lineages (Inf3) was recovered ().
For placing an unknown strain into a section, we suggest that analyzing sequences for the ITS or ATPase markers should be adequate. For determining the finer-scale relationships among strains within sections, none of the standard molecular markers would be sufficient, but ATPase and gapdh should provide the most information.
Placing newly sequenced strains in phylogenetic context.—
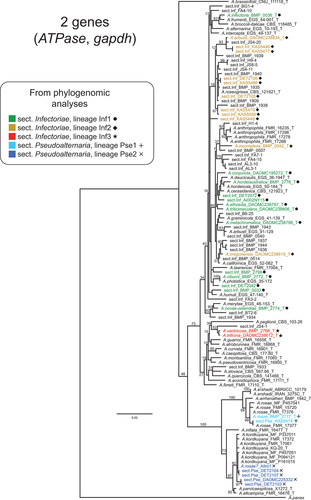
Although the genomes of multiple ex-type strains were incorporated into our phylogenomic analyses, not all described species were represented here. In particular, there are several other described species in section Pseudoalternaria whose relationships with these newly sequenced strains are unknown. In order to place our new and unidentified strains in the phylogenetic context of existing species, we gathered ATPase and gapdh sequences for the largest strain sample we could find (77 unique strains). Adding in just the unique strains in our genome sample, we compiled data for a total of 110 section Inf/Pse strains. shows the ML tree constructed from the two loci combined (see SUPPLEMENTARY FIG. 4 for single-locus trees). Acknowledging the limitations of these markers, we believe that they still provide a snapshot of relative genetic distances between strains.
Figure 2. Maximum likelihood tree constructed from the ATPase and gapdh loci combined, including as many unique strains as possible (total = 110 section Infectoriae and Pseudoalternaria strains). ML bootstrapping percentages (branch support) are shown on major branches. Sections and lineages are color-coded to match the phylogenomic analyses. “T” indicates ex-type strains. Previous species designations are shown in taxon labels. Strains not identified to species are indicated as “sect.Inf” and “sect.Pse,” for section Infectoriae and section Pseudoalternaria, respectively.
New candidates for diagnostic and informative molecular markers.—
Our phylogenomic analyses of genome-wide data sets have identified the Infectoriae and Pseudoalternaria sections, the main lineages within sections, and the relationships among them. Next, we examined the compatibility of single-gene trees with this phylogenomically informed biosystematics framework and searched for candidate genes that contained sequence polymorphisms that could discriminate between all sections and lineages. shows our selection for the top eight candidate genes for development into new diagnostic and informative molecular markers specific to sections Inf/Pse (SIP-01 to SIP-08; SIP = Sections Infectoriae/Pseudoalternaria).
Table 3. List of eight candidate genes that are diagnostic for the sections and lineages.
DISCUSSION
At the initiation of this project, sections Infectoriae and Pseudoalternaria were each represented by a single publicly available genome assembly (A. triticina and A. rosae, respectively). By sequencing and releasing the whole genomes of 22 strains, we have significantly increased the genomic resources for these two sections. During the course of our work, Fulcher and Bergstrom (Citation2023) reported the genomes of 14 section Infectoriae strains, of which six ex-type strains overlapped with our strain sample. Although this duplication of effort diminished the novelty of these six newly generated genomes, it also provided an opportunity to compare the results of sequencing different versions of the same strain, with different chains of custody. In all cases, the two versions of the same strain had nearly identical genomes as expected (), indicating that the independent laboratories have successfully maintained strain purity during handling and propagation. The very low, but nonzero, mismatch rate may represent Illumina sequencing errors or real mutations that can accumulate over many generations of growth.
Collectively, whole genome resources are now available for 26 and 7 unique strains in sections Infectoriae and Pseudoalternaria, respectively. We provide the first genomes for section Inf/Pse strains sampled within Canada, focusing mainly on cereal-associated species. Seventy percent (23 of 33) of the genomes represent North American biodiversity, creating a foundation for further work on the biosystematics, taxonomy, and molecular detection of section Inf/Pse strains in this region.
We present the first robust, genome-based phylogeny for members of sections Inf/Pse. The selection of the 5294 genes included in our custom data set was tailored to our specific sample of section Inf/Pse diversity. Our method maximizes the information content of the genes used for phylogenetic inference, which are distributed across the entire genome for a balanced sampling of genomic regions that may have variation in evolutionary history (Dettman and Eggertson Citation2021). Thus, we believe the phylogeny in is the best available estimate of the true phylogenetic relationships among included taxa. Our comprehensive phylogenomic analyses revealed the main lineages within sections. For clarity of discussion, we designated five lineages but recognize that further differentiation of clades may be present within them.
Early work based on metabolite profiles (Andersen et al. Citation2002; Andersen and Thrane Citation1996) hinted at the existence of subdivisions within the “A. infectoria species group.” Andersen et al. (Citation2009) explicitly tested for subdivisions and found that the groupings suggested by chemical data were not supported by morphological or sequence data. Subsequent studies that included larger multilocus sequence data sets showed mixed evidence for subdivisions within section Infectoriae. Some studies found no indication of distinct groups (Iturrieta-González et al. Citation2020; Marin-Felix et al. Citation2019; Poursafar et al. Citation2018), whereas others revealed an early diverging clade containing A. triticina and/or A. ventricosa (Lawrence et al. Citation2014, Citation2013; Masiello et al. Citation2020; Ramires et al. Citation2018; Somma et al. Citation2019). Although support for this clade was dependent upon which locus was analyzed, it appears to correspond to our well-supported section Infectoriae lineage Inf3. Our lineages Inf1 and Inf2 were well supported by phylogenomic data but have not been observed previously.
Although our ability to test for lineage-specific geographic patterns is limited, we did notice some potential trends. For example, all of the strains in lineage Inf2 were collected from western North America. The three ex-types strains of described morphospecies A. arbusti, A. incomplexa, and A. oregonensis were from western United States. Also, all eight of our new section Infectoriae strains that were collected from western Canada fell into lineage Inf2, whereas our two new strains from eastern Canada both fell into lineage Inf1. This pattern suggests that lineage Inf2 may be restricted to western North America; however, not all strains from western North America were in this lineage: two strains collected in California (BMP 0032 and BMP 2799) were within lineage Inf1.
Another potentially interesting trend is that strains recovered from surveys of wheat-associated section Infectoriae in Italy (Masiello et al. Citation2020; Ramires et al. Citation2018; Somma et al. Citation2019) tend to fall into lineage Inf3. Furthermore, many of the recently described species that appear to be within lineage Inf3 () were collected from Spain (Iturrieta-González et al. Citation2020; Marin-Felix et al. Citation2019). This is not a strict western Mediterranean trend, as the reference strains for A. triticina and A. ventricosa were collected from India and the United States, respectively. None of our strains from Canada fell within lineage Inf3, further suggesting nonuniform distribution of lineages across certain regions. During a survey of Japanese Alternaria, Nishikawa and Nakashima (Citation2020) reported no strains from sections Inf/Pse.
The two lineages within section Pseudoalternaria were well supported by all of our analyses of phylogenomic data. Previous studies relying on multilocus sequence data recovered two clades, despite fairly small sample sizes (Gannibal et al. Citation2022; Marin-Felix et al. Citation2019; Poursafar et al. Citation2019). Our lineages Pse1 and Pse2 correspond to “subclade I” and “subclade II,” respectively, from Poursafar et al. (Citation2019).
The genus Pseudoalternaria was established in 2014 (Lawrence et al. Citation2014) and was converted to Alternaria section Pseudoalternaria in 2016 (Lawrence et al. Citation2016). In studies prior to this taxonomic organization, strains that were actually within section Pseudoalternaria would likely have been referred to as “A. infectoria species group,” or even A. infectoria. Based on the close relationship between sections Infectoriae and Pseudoalternaria when inferred from some standard loci, the validity of section Pseudoalternaria as a separate section has been questioned by some authors (Ramires et al. Citation2018). However, our comprehensive analysis of thousands of genes provides strong support for the segregation of the two distinct sections.
With a genome-based phylogeny in hand, we were able to assess the informativeness and reliability of standard molecular marker that have been employed for the systematics of sections Inf/Pse. None of five investigated loci (ITS, ATPase, gapdh, rpb2, tef1) were able to recover or distinguish all of the five phylogenomically supported lineages from each other (; SUPPLEMENTARY FIG. 2). The most challenging lineages to distinguish were the sister groups Inf1 and Inf2. The small divergent lineage Inf3 was often recovered but was typically nested within Inf1 or Inf2. Surprisingly, the most conserved and slowest evolving locus, ITS, was able to segregate Inf1 from Inf2; however, Inf3 strains were intermixed within them (SUPPLEMENTARY FIG. 2).
Comparisons of standard marker performance for section Pseudoalternaria highlight how marker selection can heavily influence conclusions. Here, we demonstrated that the ATPase gene is the only standard marker that can recover the phylogenomically supported lineages Pse1 and Pse2 (; SUPPLEMENTARY FIG. 2); however, previous studies analyzing multiple strains in section Pseudoalternaria (Gannibal et al. Citation2022; Marin-Felix et al. Citation2019; Poursafar et al. Citation2019) consistently recovered these two groups. This pattern can be explained by the fact that the ATPase gene was included in the data set for all three of these studies, and owing to the particularly long length of the ATPase amplicon (~1200 bp), it contributed 52% or more of the sequence data. The ATPase gene was the main determinant of the resulting tree topologies, and the detection of two lineages was fully dependent upon ATPase inclusion. In this case, additional markers dilute the signal from ATPase; when analyzing five markers combined, with ATPase contributing only 36% of the data, the Pse1 and Pse2 lineages are not recovered (SUPPLEMENTARY FIG. 3). This simple example demonstrates the vagaries of locus selection and how, contrary to general expectations, more genes may not be better (i.e., sampling more genes did not bring the gene-based phylogeny closer to the genome-based phylogeny). This example also highlights the advantages of using phylogenomic data to select appropriate molecular markers that are proven to be consistent with the organismal phylogeny.
We have paid little attention to the morphological characters that are often considered relevant to the taxonomy of sections Inf/Pse, for multiple reasons. First, we reviewed the descriptions for section Inf/Pse morphospecies and found that important morphological characters (e.g., conidial dimensions, length of primary and secondary conidiophores) typically had highly overlapping ranges between species. Comparison of the specific measurements across described taxa (e.g., Dugan and Peever Citation2002; Poursafar et al. Citation2019, Citation2018) shows that these are continuous characters that may have limited ability to distinguish among taxa. Furthermore, there are many examples of strains that display intermediate morphologies and cannot be confidently segregated into described morphospecies (Andersen et al. Citation2002; Dugan and Peever Citation2002; Patriarca et al. Citation2019; Poursafar et al. Citation2018; Simmons and Roberts Citation1993). Second, even with standardized culture media and tightly controlled environmental conditions, individual strains in different developmental states can show variation in conidial size, shape, septation, etc. Third, most described species were erected based on single strains that showed minor morphological differences from other morphospecies, so no assessment of within-species variation can be made. The important question is how within-species variation relates to between-species variation? For example, if within-species variation in conidial length is equal to between-species variation, that morphological character has little taxonomic value. Fourth, morphological variation within sections Inf/Pse does not correlate with phylogenetic relationships. Lawrence et al. (Citation2014) showed good examples of how substantial morphological variation could occur in a clearly defined phylogenetic clade, confirming the plasticity of key morphological traits. Similarly, Poursafar et al. (Citation2019) concluded that there is no correlation between morphology and phylogeny in these sections.
At a higher taxonomic level, some aspects of morphology can distinguish sections Inf/Pse from other Alternaria sections. In addition to overall sporulation patterns, the morphology of the culture growing on specific medium (DRYES; Andersen and Thrane Citation1996) can differentiate strains belonging to sections Inf/Pse versus the other small-spored section Alternaria. Section Inf/Pse strains tend to produce white to off-white, floccose colonies on DRYES medium, versus the typically green to gray, velutinous colonies of section Alternaria strains. Although expanded sampling has shown that this character is not 100% consistent among sections (Dugan and Peever Citation2002; Poursafar et al. Citation2018), it performs fairly well for the majority of strains tested.
Preliminary examination of the conidial morphology of our newly characterized strains (data not shown) also found considerable variation between strains, but with overlapping ranges and no clear discontinuities between strains. To draw firm conclusions on the value of certain morphological characters, we need to perform a detailed analysis of character values and ranges, measured under specific and controlled conditions, for samples of phylogenetically characterized strains that allow within- and between-species comparisons.
Here, we focused on molecular data to characterize relationships among taxa in sections Inf/Pse. Our genomic and multilocus data sets contained a mix of ex-type strains, strains identified to species, and unidentified strains. For example, the tree of 110 strains in includes ex-type strains of 45 different species, but 47 strains (43%) were undescribed and not assigned to any existing species. Even the phylogenomic tree in contained 46% unidentified taxa. represents the largest sample, to date, of section Inf/Pse strains analyzed together. Previous studies characterizing sections Inf/Pse have focused on different sets of strains, using different sets of partially overlapping molecular markers. To provide an overview of known biodiversity beyond strains with whole genome sequences, we gathered together all strains for which sequence data were available for the two most informative existing markers for sections Inf/Pse ().
When considering the relative levels of genetic divergence between described morphospecies and nearest neighbors (), some important questions are raised regarding the current taxonomy of sections Inf/Pse. When the vast majority of species are known from single strains only, how do we distinguish strains from species? What is the significance of the morphological differences used to separate morphospecies? Based on genetic divergence alone, one could argue that many of the unidentified strains should be considered as novel and distinct species. Alternatively, one could argue that many of the previously described morphospecies collectively represent a single, larger species. Conceptually, we know that individuals from the same species are not expected to be morphologically or genetically identical. Therefore, finding a single strain with slight morphological or genetic differences from ex-type strains does not justify a new species; it could simply represent natural within-species variation.
Previous authors have raised the question of whether some section Infectoriae taxa represent multiple distinct species or a single morphologically diverse species (Andersen et al. Citation2009; Lawrence et al. Citation2014; Poursafar et al. Citation2018). Whereas morphology-based approaches suggest that each strain is a taxon in its own right, phylogenetic analysis favors the synonymization of multiple morphological species into one or a few phylogenetic species. Furthermore, when multiple strains that were morphologically identified to the same species are included, they often do not cluster together in phylogenetic trees (e.g., see A. arbusti, A. infectoria, and A. triticina in Lawrence et al. Citation2014; see A. arbusti in ; Poursafar et al. Citation2018). The lack of clustering of morphologically defined A. arbusti strains was also observed when analyzing metabolite data (Andersen et al. Citation2002). When A. arbusti was first described, it was placed by Simmons (Simmons and Roberts Citation1993) into “sporulation group 2,” which was represented by A. gaisen in section Alternaria. In other words, A. arbusti was not placed into “sporulation group 6,” which is associated with all other species in section Infectoriae. If strains from different sections can be more morphologically similar than strains within the same section, then sporulation morphology may not accurately reflect phylogenetic or evolutionary relationships.
In section Pseudoalternaria, the two A. rosae strains were placed in different lineages by phylogenomic analyses (). The ex-type strain clustered with three other A. rosae strains in the multigene phylogeny (), suggesting that the other strain, Altro1, has been misidentified. Strain Altro1 was identified via ITS sequence similarity (Mesny et al. Citation2021); however, all section Pseudoalternaria species have nearly identical ITS sequences (SUPPLEMENTARY FIG. 2), making this locus insufficient for species discrimination. It’s also possible that the ITS database used at the time of identification had limited representation of other section Pseudoalternaria species. Based on our analysis, Altro1 is most likely a strain of A. kordkuyana.
Our newly characterized Canadian strains that fall within lineages Inf1 and Inf2 are genetically distinct from the other included section Infectoriae species. Some of our strains that fall within section Pseudoalternaria lineages Pse1 and Pse2 are likely to be A. rosae and A. kordkuyana. Before making taxonomic and nomenclatural revisions, or erecting any new taxa, we want to expand our sampling of these lineages and/or potentially new species. Many of the taxonomic questions could be answered once detailed analyses of more strains, with sufficient levels of molecular data, are performed. Although identification to only the lineage or section may be unsatisfying, it allows for meaningful discussions while still accepting that taxonomic uncertainty exists.
Our study, based on genomic and molecular analyses of the largest data sets to date, leads to several important conclusions that may help guide future work. First, we clearly need greater genomic representation of the ex-types for species in section Pseudoalternaria. Second, our candidate diagnostic molecular markers () were chosen based on their congruence with the results of our comprehensive phylogenomic analyses and therefore should be appropriate for reliable and accurate tools for detection and identification. Future work should test how these proposed markers perform when confronted by a broader sample of section Inf/Pse strains, with the goal of developing markers that are diagnostic across all lineages within sections Inf/Pse. Third, our sampling of section Inf/Pse biodiversity is far from saturation, so we should continue to investigate and discover the important section Inf/Pse taxa associated with Canadian agriculture and the surrounding environments. Fourth, our analyses revealed that diversity among strains is similar to that for diversity among species that have been described based on minor morphological differences. This result suggests that, in many cases, the previously erected morphospecies may represent different strains from the same species. More detailed analyses of species boundaries, and implications on taxonomy and nomenclature, are clearly warranted. Finally, the increasing recognition that section Inf/Pse taxa may be common and sometimes dominant species in various agricultural crops highlights the need for further studies on biosystematics, species diagnostics, pathogenicity, and on the roles these fungi play in plant productivity, degradation of grain quality, and the production of harmful mycotoxins. With increasing scrutiny on Alternaria-derived toxins and their implications on international regulations, it’s critical to have a strong taxonomic framework and comprehensive genome resources available to support further research.
Supplementary_Material.pdf
Download PDF (793.2 KB)ACKNOWLEDGMENTS
We thank Allen Xue and Yuanhong Chen for providing access to grain samples for fungal isolation; Keith Seifert and staff at the Canadian Collection of Fungal Cultures for providing fungal strains; Quinn Eggertson and Natalie Kim for performing DNA extractions; and Kasia Dadej and staff at the Agriculture and Agri-Food Canada (AAFC) Molecular Technologies Laboratory for sequencing support.
DISCLOSURE STATEMENT
No potential conflict of interest was reported by the author(s).
SUPPLEMENTARY MATERIAL
Supplemental data for this article can be accessed online at https://doi.org/10.1080/00275514.2024.2354149.
Additional information
Funding
LITERATURE CITED
- Andersen B, Kroger E, Roberts RG. 2002. Chemical and morphological segregation of Alternaria arborescens, A. infectoria and A. tenuissima species-groups. Mycol Res. 106:170–182. doi:10.1017/S0953756201005263.
- Andersen B, Nielsen KF, Fernández Pinto V, Patriarca A. 2015. Characterization of Alternaria strains from Argentinean blueberry, tomato, walnut and wheat. Int J Food Microbiol. 196:1–10. doi:10.1016/j.ijfoodmicro.2014.11.029.
- Andersen B, Sørensen JL, Nielsen KF, van den Ende BG, de Hoog S. 2009. A polyphasic approach to the taxonomy of the Alternaria infectoria species-group. Fungal Genet Biol. 46:642–656. doi:10.1016/j.fgb.2009.05.005.
- Andersen B, Thrane U. 1996. Differentiation of Alternaria infectoria and Alternaria alternata based on morphology, metabolite profiles, and cultural characteristics. Can J Microbiol. 42:685–689. doi:10.1139/m96-093.
- Andersen B, Thrane U, Svendsen A, Rasmussen I. 1996. Associated field mycobiota on malt barley. Can J Bot. 74:854–858. doi:10.1139/b96-106.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477. doi:10.1089/cmb.2012.0021.
- Barkat EH, Hardy GESJ, Ren Y, Calver M, Bayliss KL. 2016. Fungal contaminants of stored wheat vary between Australian states. Australas Plant Pathol. 45:621–628. doi:10.1007/s13313-016-0449-9.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120. doi:10.1093/bioinformatics/btu170.
- Castañares E, da Cruz Cabral L, Dinolfo MI, Andersen B, Stenglein SA, Patriarca A. 2021. Alternaria in malting barley: characterization and distribution in relation with climatic conditions and barley cultivars. Int J Food Microbiol. 357:109367. doi:10.1016/j.ijfoodmicro.2021.109367.
- Christensen KB, Van Klink JW, Weavers RT, Larsen TO, Andersen B, Phipps RK. 2005. Novel chemotaxonomic markers of the Alternaria infectoria species-group. J Agr Food Chem. 53:9431–9435. doi:10.1021/jf0513213.
- Dettman JR, Eggertson Q. 2021. Phylogenomic analyses of Alternaria section Alternaria: a high-resolution, genome-wide study of lineage sorting and gene tree discordance. Mycologia. 113:1218–1232. doi:10.1080/00275514.2021.1950456.
- Dettman JR, Eggertson Q. 2022. New molecular markers for distinguishing the main phylogenetic lineages within Alternaria section Alternaria. Can J Plant Pathol. 44:754–766. doi:10.1080/07060661.2022.2061605.
- Dettman JR, Eggertson QA, Kim NE. 2023. Species diversity and molecular characterization of Alternaria section Alternaria isolates collected mainly from cereal crops in Canada. Front Microbiol. 14:1194911. doi:10.3389/fmicb.2023.1194911.
- Dugan FM, Peever TL. 2002. Morphological and cultural differentiation of described species of Alternaria from Poaceae. Mycotaxon. 83:229–264.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. doi:10.1093/nar/gkh340.
- Fulcher MR, Bergstrom GC. 2023. Draft genome sequences of 14 fungal species from Alternaria section Infectoriae. Microbiol Resour Announc. 12:e0108422. doi:10.1128/mra.01084-22.
- Gannibal PB, Lawrence DP. 2016. Distribution of Alternaria species among sections. 3. Sections Infectoriae and Pseudoalternaria. Mycotaxon. 131:781–790. doi:10.5248/131.781.
- Gannibal PB, Orina AS, Kononenko GP, Burkin AA. 2022. Distinction of Alternaria sect. Pseudoalternaria strains among other Alternaria fungi from cereals. J Fungi. 8:423. doi:10.3390/jof8050423.
- Golosna L. 2022. Mycobiota of wheat seeds with signs of “black point” under conditions of forest-steppe and forest zones of Ukraine. Chem Proc. 10:93.
- Hafez M, Gourlie R, Telfer M, Schatz N, Turkington TK, Beres B, Aboukhaddour R. 2022. Diversity of Fusarium spp. associated with wheat node and grain in representative sites across the Western Canadian Prairies. Phytopathology. 112:1003–1015. doi:10.1094/PHYTO-06-21-0241-R.
- Iturrieta-González I, Pujol I, Iftimie S, García D, Morente V, Queralt R, Guevara-Suarez M, Alastruey-Izquierdo A, Ballester F, Hernández-Restrepo M, et al. 2020. Polyphasic identification of three new species in Alternaria section Infectoriae causing human cutaneous infection. Mycoses. 63:212–224. doi:10.1111/myc.13026.
- Kahl SM, Ulrich A, Kirichenko AA, Müller MEH. 2015. Phenotypic and phylogenetic segregation of Alternaria infectoria from small-spored Alternaria species isolated from wheat in Germany and Russia. J Appl Microbiol. 119:1637–1650. doi:10.1111/jam.12951.
- Kosiak B, Torp M, Skjerve E, Andersen B. 2004. Alternaria and Fusarium in Norwegian grains of reduced quality - a matched pair sample study. Int J Food Microbiol. 93:51–62. doi:10.1016/j.ijfoodmicro.2003.10.006.
- Lawrence DP, Gannibal PB, Dugan FM, Pryor BM. 2014. Characterization of Alternaria isolates from the infectoria species-group and a new taxon from Arrhenatherum, Pseudoalternaria arrhenatheria sp. nov. Mycol Progr. 13:257–276. doi:10.1007/s11557-013-0910-x.
- Lawrence DP, Gannibal PB, Peever TL, Pryor BM. 2013. The sections of Alternaria: formalizing species-group concepts. Mycologia. 105:530–546. doi:10.3852/12-249.
- Lawrence DP, Rotondo F, Gannibal PB. 2016. Biodiversity and taxonomy of the pleomorphic genus Alternaria. Mycol Progr. 15:3. doi:10.1007/s11557-015-1144-x.
- Marin-Felix Y, Hernández-Restrepo M, Iturrieta-González I, García D, Gené J, Groenewald JZ, Cai L, Chen Q, Quaedvlieg W, Schumacher RK, et al. 2019. Genera of phytopathogenic fungi: GOPHY 3. Stud Mycol. 94:1–124. doi:10.1016/j.simyco.2019.05.001.
- Masiello M, Somma S, Susca A, Ghionna V, Logrieco AF, Franzoni M, Ravaglia S, Meca G, Moretti A. 2020. Molecular identification and mycotoxin production by Alternaria species occurring on durum wheat, showing black point symptoms. Toxins. 12:275. doi:10.3390/toxins12040275.
- Mesny F, Miyauchi S, Thiergart T, Pickel B, Atanasova L, Karlsson M, Hüttel B, Barry KW, Haridas S, Chen C, et al. 2021. Genetic determinants of endophytism in the Arabidopsis root mycobiome. Nat Commun. 12:7227. doi:10.1038/s41467-021-27479-y.
- Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. 2018. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics. 34:i142–i150. doi:10.1093/bioinformatics/bty266.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274. doi:10.1093/molbev/msu300.
- Nishikawa J, Nakashima C. 2020. Japanese species of Alternaria and their species boundaries based on host range. Fungal Syst Evol. 5:197–282. doi:10.3114/fuse.2020.05.13.
- Orina AS, Gavrilova OP, Gogina NN, Gannibal PB, Gagkaeva TY. 2021. Natural occurrence of Alternaria fungi and associated mycotoxins in small-grain cereals from the Urals and West Siberia Regions of Russia. Toxins. 13:681. doi:10.3390/toxins13100681.
- Patriarca A, da Cruz Cabral L, Pavicich MA, Nielsen KF, Andersen B. 2019. Secondary metabolite profiles of small-spored Alternaria support the new phylogenetic organization of the genus. Int J Food Microbiol. 291:135–143. doi:10.1016/j.ijfoodmicro.2018.11.022.
- Perelló A, Moreno M, Sisterna M. 2008. Alternaria infectoria species-group associated with black point of wheat in Argentina. Plant Pathol. 57:379. doi:10.1111/j.1365-3059.2007.01713.x.
- Poursafar A, Ghosta Y, Javan-Nikkhah M. 2019. Alternaria ershadii sp. nov. a new species isolated from wheat black head mold in Iran. Phytotaxa. 422:175–185. doi:10.11646/phytotaxa.422.2.4.
- Poursafar A, Ghosta Y, Orina AS, Gannibal PB, Javan-Nikkhah M, Lawrence DP. 2018. Taxonomic study on Alternaria sections Infectoriae and Pseudoalternaria associated with black (sooty) head mold of wheat and barley in Iran. Mycol Progr. 17:343–356. doi:10.1007/s11557-017-1358-1.
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26:841–842. doi:10.1093/bioinformatics/btq033.
- Ramires FA, Masiello M, Somma S, Villani A, Susca A, Logrieco AF, Luz C, Meca G, Moretti A. 2018. Phylogeny and mycotoxin characterization of Alternaria species isolated from wheat grown in Tuscany, Italy. Toxins. 10:472. doi:10.3390/toxins10110472.
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31:3210–3212. doi:10.1093/bioinformatics/btv351.
- Simmons E 2007. Alternaria. An identification manual. CBS biodiversity series 6. Utrecht (The Netherlands): CBS Fungal Biodiversity Centre.
- Simmons EG. 1994. Alternaria themes and variations (106-11). Mycotaxon. 50:409–427.
- Simmons EG, Roberts R. 1993. Alternaria themes and variations (73). Mycotaxon. 47:109–140.
- Somma S, Amatulli MT, Masiello M, Moretti A, Logrieco AF. 2019. Alternaria species associated to wheat black point identified through a multilocus sequence approach. Int J Food Microbiol. 293:34–43. doi:10.1016/j.ijfoodmicro.2019.01.001.
- Turzhanova A, Khapilina ON, Tumenbayeva A, Shevtsov V, Raiser O, Kalendar R. 2020. Genetic diversity of Alternaria species associated with black point in wheat grains. PeerJ. 8:e9097. doi:10.7717/peerj.9097.
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE. 9:e112963. doi:10.1371/journal.pone.0112963.
- Woudenberg JHC, Groenewald JZ, Binder M, Crous PW. 2013. Alternaria redefined. Stud Mycol. 75:171–212. doi:10.3114/sim0015.
- Zhang C, Rabiee M, Sayyari E, Mirarab S. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinf. 19(Suppl 6):153. doi:10.1186/s12859-018-2129-y.