
Abstract
The transport of phosphorus (P) and bromide (Br) in an alluvial gravel aquifer was investigated in two tracing experiments: (1) injecting pulses of orthophosphate and (2) 10% diluted municipal effluent spiked with orthophosphate designed to simulate the leaching of P via preferential flow after the application of fertiliser or effluent on to shallow stony soils. Results indicated that compared with Br, filtered reactive P (FRP) transport was retarded, and its mass recovery relative to Br was only 6%–28%. Attenuation was probably a result of P sorption on iron (Fe) and manganese (Mn) oxides present in the aquifer media and filtration of colloidal-P. As a result, FRP could not be distinguished from background concentrations in the aquifer system when sampled 38 m from the injection site. At the concentrations/loadings used, surface water recharged from groundwater at this study site may receive P loading via the preferential flow of P into groundwater if the distance between source and recharge point is less than 38 m, and possibly at greater distances under higher loadings from intensive land use.
Introduction
Most phosphorus (P) loss to surface waterways originates as diffuse or non-point pollution from agricultural production systems, with smaller contributions from point sources associated with treated industrial or domestic wastewater discharges (Foy & Withers Citation1995; McDowell et al. Citation2003). Diffuse losses of phosphorus (P) from land to surface water are recognised worldwide as a major cause of accelerated eutrophication (Carpenter et al. Citation1998). In New Zealand for instance, surface water quality data collected from 35 major river systems between 1989 and 2009 indicated increasing P concentrations (Ballantine & Davies Colley Citation2014), with some sites exceeding national water quality trigger values. However, a more recent summary (2008–2012) of surface water total P (TP) concentrations from the Land, Air, Water Aotearoa website (www.lawa.org.nz) showed improving trends were detected at 30% of monitoring sites, although 10% of sites still showed a deteriorating trend across all land cover types (LAWA Citation2015). In response, there has been considerable research invested in quantifying the main sources of P (McDowell et al. Citation2007) and transport pathways by which P enters surface water bodies from land (Heathwaite & Dils Citation2000). This information is central to any attempt to identify how best to mitigate P losses and minimise or prevent surface water eutrophication.
One P loss pathway that has received comparatively little attention is transport through groundwater. This is due to the long-held assumption that P is not lost to groundwater because most soils have a moderate to high capacity to retain P (Correll Citation1998). However, an increasing number of studies investigating the state and trends of nutrient concentrations in groundwater have reported enrichment with P (Carlyle & Hill Citation2001; Burkart et al. Citation2004; Abraham & Hanson Citation2008; Holman et al. Citation2010; Heeren et al. Citation2011; Mittelstet et al. Citation2011).
Although these reports indicate that P can under certain conditions be transported from soils to groundwater, what still remains unclear is the impact this P-enriched groundwater may have on surface water quality. As base-flow supplied from groundwater often dominates inputs to surface waters during the warmer summer months when primary production is at its peak, even relatively low P concentrations delivered to surface water from groundwater at this time could potentially have a disproportionately large influence on algal growth (compared with storm flow events) (Biggs & Smith Citation2002).
To investigate this issue, several studies have explored the possibility that enriched concentrations of P in groundwater may influence surface-water quality (Holman et al. Citation2010; Rasiah et al. Citation2011). In New Zealand, a meta-analysis of three national databases was conducted to explore the potential link between soil and surface water and groundwater enrichment with P (McDowell et al. Citation2015). Data indicated that soil P concentrations were enriched (especially under dairying); median surface water P concentrations were enriched in locations receiving run-off from industrial and/or dairy land uses; and groundwater was enriched with P in locations used for dairying, especially in those aquifers with gravel or sand lithology. Furthermore, there was a good correlation between the enrichment of soil P under dairying, groundwater P concentrations and significant enrichment of surface water P concentrations. McDowell et al. (Citation2015) concluded that these data ‘raise the possibility’ that groundwater could contribute P to surface water if connectivity is good between surface and groundwater, intensive land use is coupled with soils prone to leaching, and P is mobile in aquifers.
This ‘possibility’ is particularly relevant given the recent large-scale expansion in parts of New Zealand of intensive dairying into areas traditionally farmed with sheep and beef (Carrick et al. Citation2013). A large proportion of this expansion has occurred on stony alluvial soils that are extensively irrigated (Carrick et al. Citation2013). It is recognised that many of these irrigated, stony soils have a high vulnerability to P leaching i.e. low anion storage capacity (ASC), rapid permeability and low water-holding capacity (McDowell et al. Citation2013). Furthermore, these soils are often over unconfined aquifers of alluvial sand and gravel lithology (White Citation2001). While recent evidence suggests that P enrichment of groundwater is occurring in some areas in New Zealand (Abraham & Hanson Citation2008; McDowell et al. Citation2015), there is a lack of understanding of the mobility of P once it gets into aquifer systems. This information is important to gauge the risk of P transport through certain aquifers into surface water, and therefore the impact of such transport on surface water quality.
The aim of this study was to determine the extent of mobility of P in a gravel aquifer that is typical of large parts of New Zealand. Two experiments were undertaken by injecting different sources of P into the aquifer i.e. orthophosphate (surrogate for mineral P fertiliser) and municipal effluent spiked with orthophosphate (an organic-based point source effluent). These P sources were designed to simulate the leaching of P via preferential downwards flow after the application of P fertiliser or effluent to stony soils. It was hypothesised that orthophosphate and P compounds in municipal effluent would move through groundwater at different rates.
Materials and methods
Experimental site and aquifer characteristics
The study site (), located at Burnham, Canterbury, New Zealand, consisted of an unconfined aquifer of medium to coarse alluvial sandy gravels with silt and clay bands (Pang & Close Citation1999a). The gravels are derived from indurated greywacke sandstone and they extend up to 125 m in depth, according to nearby well logs. Relatively impermeable fine-grained estuarine/marine sediments beneath the gravels separate the unconfined and confined aquifer system (Pang et al. Citation1998). The aquifer material had a low cation exchange capacity (0.2 cmolc kg−1 for gravels and 2.5 cmolc kg−1 for the fine material) and organic carbon concentration (0.4 g kg−1) (Hinton & Close Citation1998). Groundwaters were Na-Ca-HCO3 based, with a pH between 6.6 and 7.2, oxic (dissolved oxygen concentration) of 11.6 mg L−1 (Pang & Close Citation1999b) and electrical conductivities ranging between 6.4 and 15 mS m−1. The aquifer had previously been characterised using tracer and pump tests. Groundwater velocities at the site are high with a median of 63 m day−1 (range of 30–85 m day−1). The flow path at the site has previously been determined several times using different conservative tracers (Pang et al. Citation1998; Dann et al. Citation2008) and consistently showed a significant heterogeneity and curvature generally to the mid to left hand side of the well array ().
Figure 1 Location of the injection and sampling wells at the Burnham experimental site. Wells highlighted in black show sampling points for this experiment. The main flow line was inferred from earlier experiments (Pang et al. Citation1998; Dann et al. Citation2008).
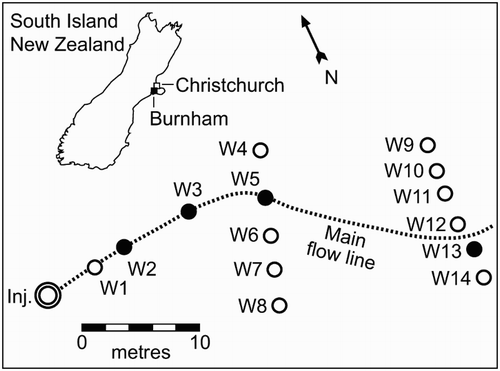
The experimental site was confined within a 50 × 100 m area with an up-gradient injection well and 21 down-gradient monitoring wells, all 100 mm in diameter and screened (screen porosity approximately 20%) between 12 and 18 m below ground level. The monitoring wells were arranged in four arrays approximately 20, 40, 65 and 90 m down-gradient of the injection well with the exception of the first three wells which were located 2, 6 and 12 m from the injection well ().
Experiment 1: injection of orthophosphate P
Before injection, groundwater levels were measured and monitoring wells sampled and analysed for background filtered reactive P (FRP) concentrations. An 80 L solution, containing P (as KH2PO4) at a concentration of 40 mg P L−1 and a non-reactive tracer, potassium bromide (KBr) at 5000 mg L−1, was injected simultaneously into the injection well at a depth of 13.5–15.5 m (1.8–3.8 m below the water table) over a period of 1 h at a rate of 1.3 L min−1. The injection depth was isolated with well-packers which remained inflated throughout the experiment. Groundwater samples were taken at a depth of 14.5 m from four of the wells along the flow line i.e. W2, W3, W5 and W13 (). These wells were 6.4, 12, 21 and 38 m down-gradient from the injection well. The sampling depth was based on preferential flow pathways through wells W5 and W13 determined in a previous experiment at the site (Pang et al. Citation1998).
Twenty-eight samples were collected from each monitoring well over 48 h after injection. Sampling intervals were initially 20 min, up to the maximum concentration of Br detected, and four-hourly after the Br concentration peak to give a well-defined breakthrough curve (BTC). Groundwater samples were taken from wells using either plastic or stainless steel bailers. Purging was not considered necessary because the aquifer formation in which the well is screened had a high hydraulic conductivity, resulting in a state of equilibrium existing between the water standing in the screened sampling interval and the formation water. Prior to the experiment, the samplers were confirmed as not sorbing P by adding a known P concentration and determining the degree of sorption after 24 h. Groundwater samples were field filtered through both 0.45 µm and 0.1 µm polyvinylidene membranes and kept cool (4 °C) until chemical analysis.
Experiment 2: Injection of municipal effluent spiked with orthophosphate P
A 7.2 L sample of treated municipal effluent was obtained from the Burnham wastewater treatment plant, spiked with orthophosphate (KH2PO4) and equilibrated for 48 h on a stirrer. The spiked effluent was then made up to 80 L in water to give a final total P concentration of 40 mg P L−1. The 80 L P-spiked effluent solution and Br at 5000 mg L−1 were injected simultaneously into the injection well at a depth of 13.5–15 m (0.5–2.5 m below the water table) over a period of 1 h. The injection depth was isolated with well-packers which remained inflated throughout the experiment. Samples were taken in the same manner and wells as for the orthophosphate experiment.
Sample analysis
The molybdate-blue method of Watanabe & Olsen (Citation1965) was used to measure FRP in <0.1 µm and <0.45 µm filtered samples. Total filtered P (TFP) was also measured using the molybdate-blue method after acid-persulphate digestion (Rowland & Haygarth Citation1997). Unreactive, largely organic P was taken as the difference between TFP and FRP. The detection limit for P was 0.007 mg L−1. Bromide samples were analysed using an ion selective electrode with a detection limit of 0.1 mg L−1.
Data analysis
The attenuation of Br and P in groundwater was evaluated using peak breakthrough attenuation (log10[cmax/c0] ; where cmax and c0 are the peak concentration and input solution concentration [mg L−1], respectively) and normalised mass recovery (RB) was estimated by integrating the area under the BTC of P normalised against that of Br:(1)
When RB is 100%, P would have the same mass recovery as that of the non-reactive tracer Br.
The transport speed of P relative to Br is described as the centre of mass retardation (Pang et al. Citation2003) which is determined from the normalised first moment (µ) of P and Br:(2)
Results and discussion
In both experiments, neither Br nor FRP could be detected at well 2 despite being closest to the site of injection. This indicated that well 2 was not in the main groundwater flow line () as previously determined (Pang et al. Citation1998) or that sampling was not at the correct depth. In addition, while the Br BTC for well 13 indicated that it was on the main flow line (data not shown), FRP concentrations in well 13 could not be differentiated from background concentrations measured at the site (). Therefore, Br and P data are reported only for wells 3 and 5.
Table 1 Average concentrations and standard deviation of P (mg P L−1) in the background groundwater (wells 2, 3, 5 and 13), the injection solution and in well 13 during the field experiments.
Analysis of the injection solution for FRP and TFP fractions indicated that in both experiments organic P was negligible. The very low dissolved organic carbon (DOC) concentration in the injection solution (i.e. 2.8 mg L−1 in the effluent injection solution) reinforced this observation, and was lower than the background concentration found in groundwater samples taken from wells before injection (i.e. 3.0 mg L−1). Therefore, only data for FRP in the <0.45 µm and <0.1 µm fractions are reported.
Transport and attenuation of the Br solute tracer
shows the attenuation of Br in both experiments using peak breakthrough attenuation, i.e. log10(cmax/c0) with distance. The Br peak was largely the same for both wells in each experiment, although Br concentrations were lower in the effluent experiment compared with the orthophosphate experiment. It is possible that this may be related to a change in the availability of some preferential pathways in the effluent experiment, with a drop of about 1.3 m in the water table level compared with the orthophosphate experiment undertaken a month earlier. However, previous experiments at the site which compared the depth of preferential flow pathways in different wells when the water level dropped by a similar amount to this study still showed a reasonable agreement in the preferential flow pathways between experiments (Pang et al. Citation1998).
Table 2 Peak concentration log10(cmax/c0) , centre of mass retardation, and relative mass (RB) of Br and FRP observed in wells 3 and 5 down-gradient of the injection.
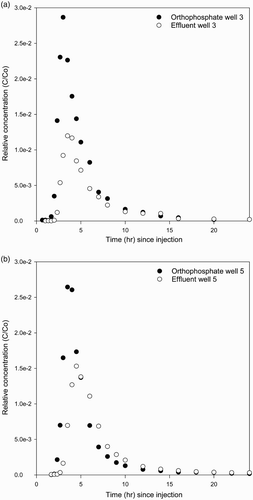
The observed Br BTCs for wells 3 and 5 for both experiments are given in . Bromide BTCs typically displayed early breakthrough and tail-off for both wells and both experiments (). The early breakthrough and tailing of the Br BTCs in the two experiments are consistent with previous observations at the Burnham site using other solute tracers such as rhodamine WT and chloride (Pang et al. Citation1998; Pang & Close Citation1999a). It has been suggested that the asymmetrical form of the BTC is caused by the presence of physical non-equilibrium conditions in the Burnham aquifer system and poor mixing caused by large portions of poorly permeable layers and lenses (silt and clay bands) within the aquifer (Pang et al. Citation1998). Put simply, when Br passes through the aquifer, a portion will preferentially move through highly permeable zones, while the remainder diffuses into poorly permeable zones. After moving through highly permeable zones, Br in the poorly permeable zones slowly diffuses back to the high-permeability zones driven by the concentration gradient. This water exchange between zones of contrasting permeability is considered to cause the observed tailing in the BTCs.
Figure 2 Observed concentration breakthrough curves for Br for well 3 (A) and well 5 (B).
Transport and attenuation of P
The attenuation of FRP, i.e. log10(cmax/c0), with distance overall was slightly greater in the <0.45 µm fraction than the <0.1 µm fraction (). There also appeared to be greater attenuation of FRP in the effluent experiment than the orthophosphate experiment. There were also differences in the time of centre of mass FRP breakthrough concentration (). While there did not appear to be any consistent difference between filtered pore size fractions, there appeared to be slightly less retardation in the effluent experiment than the orthophosphate experiment. While some of this may potentially have been an artefact of different flow paths between experiments as discussed already, this seems unlikely given the Br peaks were largely the same between experiments ().
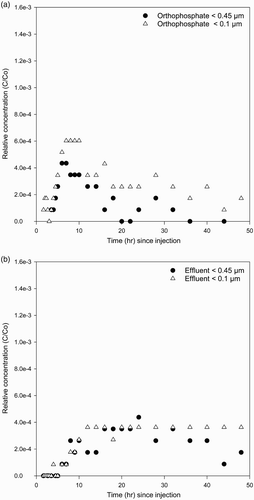
The observed FRP BTCs for wells 3 and 5 for both experiments are given in –. As for Br, FRP BTCs for the orthophosphate experiment typically displayed early breakthrough (especially for well 3) and notable tailing off slowly for both FRP pore size fractions. This is again indicative of non-equilibrium transport and has been found previously for both inorganic (e.g. cadmium ) and microbiological (bacteria) contaminants in field experiments undertaken at the Burnham site (Pang & Close Citation1999a; Sinton et al. Citation2000). In contrast, BTCs for FRP detected in <0.1 µm and <0.45 µm fractions were much less in the effluent experiment, although still with very notable tailing off. Filtered reactive P in the <0.1 µm fraction appears to be sustained for longer after injection than the <0.45 µm fraction (–), possibly as a result of breakdown of <0.45 µm filtrate in the effluent over time during aquifer transport.
Figure 3 Observed concentration breakthrough curves for <0.45 µm and <0.1 µm FRP in well 3 for the orthophosphate (A) and effluent (B) experiments.
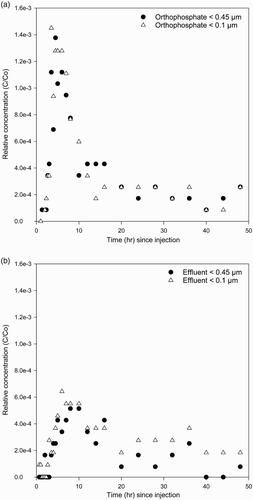
Figure 4 Observed concentration breakthrough curves for <0.45 µm and <0.1 µm FRP in well 5 for the orthophosphate (A) and effluent (B) experiments.
The normalised mass recovery of FRP with respect to Br (i.e. RB) measured in both experiments, wells and pore size fractions are given in . Recovery was less than 28%, indicating there was a substantial loss of FRP during transport from the injection site, but that there was still transport of potentially environmentally significant amounts of P (e.g. peak FRP concentration of 0.074 mg L−1 in well 3 in the orthophosphate experiment). The loss of FRP increased with distance. Recoveries were slightly greater in the effluent experiment than the orthophosphate experiment, perhaps caused by other organic constituents in the effluent known to impair sorption (McDowell et al. Citation2005). Recovery was also greater for the FRP <0.1 µm fraction than the FRP <0.45 µm fraction, probably due to a greater proportion of P in the effluent sample in the <0.1 µm fraction, although the reasons for this are unclear. It would appear therefore that potential P losses from effluent sources are at least the same as from orthophosphate, and that losses could potentially remain in groundwater for longer than for orthophosphate.
The reason for the relatively high retention of P in this study may be due to metal oxides present in the gravel aquifer material. Previous analyses of the gravels at the study site have revealed that surfaces were partially stained, 6 to 13 μm in thickness, and comprised predominantly of Fe oxide (>90% as hydrous ferric oxides) and Mn oxides (<10%) (Hinton & Close Citation1998). It is well recognised that Fe oxides and hydroxides are important components for P sorption in soils and sediments (Parfitt Citation1978; Hawke et al. Citation1989). Studies have also highlighted their role in retaining P within aquifers (Robertson Citation1995; Spiteri et al. Citation2007; Domagalski & Johnson Citation2011).
Filtered reactive P transport in both experiments and fractions appear to be restricted to less than 38 m from the contamination source in this aquifer system. Several studies have made estimates of P transport in aquifers from effluent or wastewater sources. Some have detected only limited transport away from the source (e.g. Robertson et al. Citation1998; Spiteri et al. Citation2007). For example, Fuchs et al. (Citation2009) used a trench to inject P into a groundwater flow system to study P transport. They found dissolved P was rapidly transported in preferential flow pathways with minimal attenuation, although in relatively short flow paths of 2–3 m. For example, at the same field site as this study, Close (Citation1989) found dissolved reactive P transport was limited to 43 m from a septic tank. In contrast, there are circumstances when P has been transported much greater distances in aquifers from their source (e.g. Walter et al. Citation1996). For instance, Corbett et al. (Citation2002) measured TP concentrations in groundwater in a sand aquifer downfield from three onsite sewage treatment and disposal systems in St George Island, Florida. Total P concentrations at all three sites were still significantly enriched compared with baseline (natural) concentrations at wells >50 m from wastewater disposal field. Close (Citation1984) showed for example that P was transported >900 m in an alluvial gravel aquifer from a flood-irrigated sewage system source in mid-Canterbury, New Zealand, which had been in use for about 50 years. It was speculated that sorption sites in soil and aquifer materials were saturated with P, and compared with the present study, groundwater velocity at the mid-Canterbury site was about twice as fast as those at the Burnham site.
Past studies at this site suggest that the aquifer was sampled under oxic conditions (Pang & Close Citation1999b). However, it is well known that the movement of P is much greater under anaerobic conditions where Fe and Mn oxides have little P-sorption capacity (Carlyle & Hill Citation2001). Leaching of P into anaerobic groundwater may substantially increase the distance P may be transported, as may saturation of P sorption sites with time following long-term inputs of P. This would expand the area at risk of P leaching from intensive land uses. Furthermore, various organic P compounds (e.g. diesters) are poorly sorbed compared with orthophosphate (Frossard et al. Citation1989). Such compounds are known to exist in effluent. Therefore, experiments are planned using the sustained application of farm dairy effluent, to explore the potential for greater transport of organic P.
Conclusions
The transport of FRP was retarded and showed significant losses of 72%–94% relative to Br during transport. As a result, FRP could not be distinguished from background concentrations in the aquifer system when sampled 38 m from the injection site. Attenuation during transport was considered likely to be a result of sorption of P with Fe and Mn oxides present in the aquifer media and filtration of colloidal-P. At the concentrations used, P could enter connected surface waters if the recharge occurred within 38 m of the P source, and possibly at greater distances under higher loadings from intensive land use such as winter grazing of forage crops.
Acknowledgements
Thanks to Phil Abraham and Bronwyn Humphries ESR Ltd for expertise and assistance in the trial set up and sampling, and Nick Reed (AgResearch) and ECAN staff for sampling.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Abraham P, Hanson C 2008. Annual groundwater quality survey, Spring 2008. Environment Canterbury technical report R09/46.
- Ballantine DJ, Davies-Colley RJ 2014. Water quality trends in New Zealand rivers: 1989–2009. Environmental Monitoring and Assessment 186: 1939–1950. doi: 10.1007/s10661-013-3508-5
- Biggs BJF, Smith RA 2002. Taxonomic richness of stream benthic algae: effects of flood disturbance and nutrients. Limnology and Oceanography 47: 1175–1186. doi: 10.4319/lo.2002.47.4.1175
- Burkart MR, Simpkins W, Morrow AJ, Gannon JM 2004. Occurrence of TDP in unconsolidated aquifers and aquitards in Iowa. Journal of the American Water Resources Association 40: 827–834. doi: 10.1111/j.1752-1688.2004.tb04461.x
- Carlyle GC, Hill AR 2001. Groundwater phosphate dynamics in a river riparian zone: effects of hydrologic flowpaths, lithology and redox chemistry. Journal of Hydrology 247: 151–168. doi: 10.1016/S0022-1694(01)00375-4
- Carpenter SR, Caraco NF, Correll DL, Howarth RW, Sharpley AN, Smith VH 1998. Nonpoint pollution of surface waters with phosphorus and nitrogen. Ecological Applications 8: 559–568. doi: 10.1890/1051-0761(1998)008[0559:NPOSWW]2.0.CO;2
- Carrick S, Palmer D, Webb T, Scott J, Lilburne L 2013. Stony soils are a major challenge for nutrient management under irrigation development. In: Currie LD, Christensen CL eds. Accurate and efficient use of nutrients on farms. Occasional Report No. 26. Palmerston North, Fertilizer and Lime Research Centre, Massey University. http://flrc.massey.ac.nz/publications.html (accessed 20 January 2015).
- Close ME 1984. Factors influencing the chemical quality of groundwater downstream of a sewage disposal area—Templeton, New Zealand. Water and Soil Miscellaneous Publication No 69: 47–61.
- Close M 1989. Effects of septic tank effluent on chemical quality of alluvial gravel aquifers. New Zealand Journal of Marine and Freshwater Research 23: 275–286. doi: 10.1080/00288330.1989.9516364
- Corbett DR, Dillon K, Burnett W, Schaefer G 2002. The spatial variability of nitrogen and phosphorus concentration in a sand aquifer influenced by onsite sewage treatment and disposal systems: a case study on St. George Island, Florida. Environmental Pollution 117: 337–345. doi: 10.1016/S0269-7491(01)00168-3
- Correll DL 1998. The role of phosphorus in the eutrophication of receiving waters: a review. Journal of Environmental Quality 27: 261–266. doi: 10.2134/jeq1998.00472425002700020004x
- Dann RL, Close ME, Pang L, Flintoft MJ, Hector RP 2008. Complementary use of tracer and pumping tests to characterize a heterogeneous channelized aquifer system in New Zealand. Hydrogeology Journal 16: 1177–1191. doi: 10.1007/s10040-008-0291-4
- Domagalski JL, Johnson HM 2011. Subsurface transport of orthophosphate in five agricultural watersheds, USA. Journal of Hydrology 409: 157–171. doi: 10.1016/j.jhydrol.2011.08.014
- Foy RH, Withers PJA 1995. The contribution of agricultural phosphorus to eutrophication. London, UK, The Fertiliser Society. 33 p.
- Frossard E, Stewart JWB, St Arnaud RJ 1989. Distribution and mobility of phosphorus in grassland and forest soils of Saskatchewan. Canadian Journal of Soil Science 69: 401–416. doi: 10.4141/cjss89-040
- Fuchs JW, Fox GA, Storm DE, Penn CJ, Brown GO 2009. Subsurface transport of phosphorus in Riparian Floodplains: influence of preferential flow paths. Journal of Environment Quality 38: 473–484. doi: 10.2134/jeq2008.0201
- Hawke D, Carpenter PD, Hunter KA 1989. Competitive adsorption of phosphate on goethite in marine electrolytes. Environmental Science & Technology 23: 187–191. doi: 10.1021/es00179a008
- Heathwaite AL, Dils RM 2000. Characterising phosphorus loss in surface and subsurface hydrological pathways. Science of the Total Environment 251–252: 523–538. doi: 10.1016/S0048-9697(00)00393-4
- Heeren DM, Fox GA, Miller RB, Storm DE, Fox AK, Penn CJ et al. 2011. Stage-dependent transient storage of phosphorus in alluvial floodplains. Hydrological Processes 25: 3230–3243. doi: 10.1002/hyp.8054
- Hinton C, Close ME 1998. Lead adsorption onto aquifer gravel using batch experiments and XPS. Water–rock interaction. In: Arehart GB, Hulston JR eds. Proceedings of the 9th International Symposium on Water–Rock Interaction—WRI-9. Taupo, New Zealand, Balkema, Rotterdam. Pp. 939–942.
- Holman IP, Howden NHK, Ballamy P, Willby N, Whelan MJ, Rivas-Casado M 2010. An assessment of the risk to surface water ecosystems of groundwater P in the UK and Ireland. Science of the Total Environment 408: 1847–1857. doi: 10.1016/j.scitotenv.2009.11.026
- LAWA 2015. Total phosphorus (TP). http://www.lawa.org.nz/explore-data/freshwater/national-picture/nutrients/total-phosphorus/ (accessed 16 July 2015).
- McDowell RW, Monaghan RM, Morton J 2003. Soil phosphorus concentrations to minimise potential P loss to surface waters in Southland. New Zealand Journal of Agricultural Research 46: 239–253. doi: 10.1080/00288233.2003.9513550
- McDowell RW, Condron L, Stewart I, Cave V 2005. Chemical nature and diversity of phosphorus in New Zealand pasture soils using 31P nuclear magnetic resonance spectroscopy and sequential fractionation. Nutrient Cycling in Agroecosystems 72: 241–254. doi: 10.1007/s10705-005-2921-8
- McDowell RW, Nash DM, Robertson F 2007. Sources of phosphorus lost from a grazed pasture receiving simulated rainfall. Journal of Environment Quality 36: 1281–1288. doi: 10.2134/jeq2006.0347
- McDowell RW, Dodd RJ, Cox N, Wheeler D 2013. Assessing and mitigating a potential legacy of phosphorus loss form land to surface waters: A national perspective. 2013 AWRA Spring Speciality Conference—Agriculture Hydrology and Water Quality II, March 25–27, St. Louis, MO.
- McDowell RW, Cox N, Daughney CJ, Wheeler D, Moreau M 2015. A national assessment of the potential linkage between soil and ground and surface water concentrations of phosphorus. Journal of the American Water Resources Association. In press.
- Mittelstet AR, Heeren DM, Fox GA, Storm DE, White MJ, Miller RB 2011. Comparison of subsurface and surface runoff phosphorus transport rates in alluvial floodplains. Agriculture, Ecosystems, and the Environment 141: 417–425. doi: 10.1016/j.agee.2011.04.006
- Pang L, Close ME 1999a. Non-equilibrium transport of Cd in alluvial gravels. Journal of Contaminant Hydrology 36: 185–206. doi: 10.1016/S0169-7722(98)00110-7
- Pang L, Close ME 1999b. Attenuation and transport of atrazine and picloram in an alluvial gravel aquifer: a tracer test and batch study. New Zealand Journal of Marine and Freshwater Research 33: 279–291. doi: 10.1080/00288330.1999.9516877
- Pang L, Close ME, Noonan M 1998. Rhodamine WT and Bacillus subtilis transport through an alluvial gravel aquifer. Ground Water 36: 112–122. doi: 10.1111/j.1745-6584.1998.tb01071.x
- Pang L, Goltz M, Close M 2003. Application of the method of temporal moments to interpret solute transport with sorption and degradation. Journal of Contaminant Hydrology 60: 123–134. doi: 10.1016/S0169-7722(02)00061-X
- Parfitt RL 1978. Anion adsorption by soils and soil materials. In: Brady NC ed. Advances in agronomy. Volume 30. New York, Academic Press. Pp. 1–50.
- Rasiah V, Moody PW, Armour JD 2011. Soluble phosphate in fluctuating groundwater under cropping in the north-eastern wet tropics of Australia. Soil Research 49: 329–342. doi: 10.1071/SR10167
- Robertson WD 1995. Development of steady-state phosphate concentrations in septic system plumes. Journal of Contaminant Hydrology 19: 289–305. doi: 10.1016/0169-7722(95)00022-N
- Robertson WD, Schiff SL, Ptacek CJ 1998. Review of phosphate mobility and persistence in 10 septic system plumes. Ground Water 36: 1000–1010. doi: 10.1111/j.1745-6584.1998.tb02107.x
- Rowland AP, Haygarth PM 1997. Determination of total dissolved phosphorus in soil solutions. Journal of Environment Quality 26: 410–415. doi: 10.2134/jeq1997.00472425002600020011x
- Sinton LW, Noonan M, Finlay RK, Pang L, Close ME 2000. Transport and attenuation of bacteria and bacteriophages in an alluvial gravel aquifer. New Zealand Journal of Marine and Freshwater Research 34: 175–186. doi: 10.1080/00288330.2000.9516924
- Spiteri C, Slomp CP, Regnier P, Meile C, Van Cappellen P 2007. Modelling the geochemical fate and transport of wastewater-derived phosphorus in contrasting groundwater systems. Journal of Contaminant Hydrology 92: 87–108. doi: 10.1016/j.jconhyd.2007.01.002
- Walter DA, Rea BA, Stollenwerk KG, Savoie J 1996. Geochemical and hydrological controls on phosphorus transport in a sewage-contaminated sand and gravel aquifer near Ashumut Pond, Cape Cod, Massachusetts. USGS Water-Supply paper 2463. Washington, USGS.
- Watanabe FS, Olsen SR 1965. Test of an ascorbic acid method for determining phosphorus in water and NaHCO3 extracts from soil. Soil Science Society of America Journal 29: 677–678. doi: 10.2136/sssaj1965.03615995002900060025x
- White PA 2001. Groundwater resources in New Zealand. In: Rosen MR, White PA eds. Groundwaters of New Zealand. Wellington, New Zealand Hydrological Society. Pp. 45–75.