
Abstract
Aims. Low-dose acetylsalicylic acid (ASA; aspirin) for secondary prevention reduces cardiovascular disease mortality risk. ASA acetylates cyclooxygenase in the portal circulation and is rapidly (half-life, 20 min) hydrolyzed. Certain patients with cardiovascular disease may exhibit high on-therapy platelet reactivity as a result of high platelet turnover, a process whereby platelets are produced and are active beyond the duration of antiplatelet coverage provided by once-daily immediate-release (IR) ASA. A once-daily, extended-release (ER) ASA formulation using ER microcapsule technology was developed to release ASA over the 24-h dosing interval and reduce maximal plasma concentrations to spare peripheral endogenous endothelial prostacyclin production. Methods. Healthy adults (n = 50) were randomized in a crossover study to receive two different ER-ASA single doses (up to 325 mg) and two different IR-ASA single doses (up to 81 mg) in four periods, each separated by ≥14 days. Pharmacodynamics was assessed by measuring serum thromboxane B2 (TXB2), urine 11-dehydro-TXB2, and arachidonic acid-induced platelet aggregation. Pharmacokinetics was determined for ASA and salicylic acid (SA). Results: Both formulations produced dose-dependent inhibition on all pharmacodynamic parameters. Marked inhibition of TXB2 and 11-dehydro-TXB2 was maintained over the 24-h dosing interval after a dose of ≥81 mg ER-ASA or ≥40 mg IR-ASA. The dose required to achieve 50% of maximum TXB2 inhibition with ER-ASA was 49.9 mg versus 29.6 mg for IR-ASA, for a similar maximum pharmacodynamic effect (98.9% TXB2 inhibition). This suggests that an approximately twofold greater ER-ASA dose (162.5 mg) is necessary to obtain the same response as that of IR-ASA 81 mg. Peak ASA concentrations were lower and Tmax was longer with ER-ASA versus IR-ASA. Administration of IR-ASA resulted in a dose-normalized mean Cmax of ASA that was approximately sixfold higher than that for ER-ASA and a Cmax of SA approximately two- to threefold higher than that for ER-ASA. Conclusion. Both ASA formulations showed dose-dependent antiplatelet activity. Compared with the IR-ASA, ER-ASA released active drug more slowly, resulting in prolonged absorption and lower systemic drug concentrations, which is expected for an ER (24-h) formulation.
Introduction
Acetylsalicylic acid (ASA; aspirin) is a potent antiplatelet agent that is administered at low doses (≤325 mg/day) as secondary prevention to reduce the risk of cardiovascular events (e.g. myocardial infarction) and associated mortality [Citation1,2]. The antiplatelet effect of ASA is the result of irreversible inhibition of platelet cyclooxygenase-1 (COX-1), thereby preventing synthesis of thromboxane A2 (TXA2), a potent, short-lived platelet agonist and vasoconstrictor, and its inactive metabolite thromboxane B2 (TXB2) [Citation3].
However, COX-1 is constitutively expressed in other tissues, such as gastrointestinal (GI) mucosa, and its role in prostaglandin production (e.g. prostacyclin) is important for various physiologic processes (e.g. protection of the GI mucosa) [Citation4,5]. Some concerns with long-term ASA treatment have been raised due to its inhibition of prostacyclin synthesis in the systemic circulation (e.g. vascular endothelial cells and mucosal epithelial cells) and potential localized, direct effects on the GI tract [Citation6-8]. The risk of developing upper GI bleeding, although modest, is generally similar with enteric-coated, buffered, or immediate-release (IR) formulations, suggesting that the potential effects of ASA on the gastric mucosa are related to prostaglandin production inhibition to some degree, rather than localized irritation or injury to the stomach [Citation7,9].
A further concern with low-dose, once-daily ASA is that the antiplatelet effect is not consistent over the standard dosing interval. Although the exact prevalence is unclear and varies widely due to differences in assays used, 6 – 60% of patients may have an incomplete response to traditional low-dose ASA regimens [Citation10,11]. Data suggest that this phenomenon may occur because of physiologic differences among patient populations [Citation12]. Under normal situations, platelets are replenished in systemic circulation at a rate of approximately 10 – 15% per day, but in some high-risk individuals (e.g. patients with type 2 diabetes or coronary artery disease), this rate can be higher, resulting in new COX-1 activity in newly synthesized platelets and downstream TXA2 production beyond the ASA residence time in plasma and prior to the next dose of ASA [Citation13-16]. Therefore, given that the ASA molecule has a short half-life of 20 min in systemic circulation and only platelets exposed to nonhydrolyzed ASA are inactivated, other delivery methods for ASA have been developed to provide coverage during a full 24-h interval of ASA dosing [Citation8,13-15,17].
An extended-release (ER) formulation of ASA has been developed using ER microcapsule technology and is designed to release ASA over the entire 24-h (once-daily) dosing interval that is commonplace for current low-dose aspirin regimens. Additionally, whereas the formulation was designed to inhibit circulating thromboxane levels, the resulting low plasma concentration results in maintenance or “sparing” of endogenous peripheral endothelial prostacyclin levels. A previously published study in patients with atherosclerosis reported that this formulation of ER-ASA produced effective inhibition of TXB2 production and platelet aggregation at a dose of 162.5 mg, and that these antiplatelet effects were significantly greater than those of enteric-coated ASA 75 mg [Citation18]. The aim of the current study was to perform a bioequivalence analysis, based on pharmacodynamic (PD) measurements, as well as characterize the pharmacokinetic (PK) profile of ER-ASA compared with IR-ASA in healthy volunteers.
Materials and methods
This phase I, open-label, 4-way, randomized, crossover, dose-response study was conducted at a single center in the United States during April through July, 2013. It was conducted according to the principles of the Declaration of Helsinki and was approved by the Institutional Review Board. All participants provided written informed consent.
Participants
Healthy male and female volunteers, aged 18–55 years with a body mass index (BMI) between 18 and 30 kg/m2 were eligible for the study. Participants were excluded from participation if they had a history of clinically significant and relevant medical disorders, including metabolic, renal, and hepatic disorders, hemorrhagic stroke, GI bleeding, peptic ulcer and other chronic bleeding disorders, cardiovascular disease, or central nervous system disorders. Other exclusion criteria included pregnancy and lactation, smoking, use of aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs) within 14 days of study initiation, and use of prescription or over-the-counter medications within 7 days of study initiation. Concomitant administration of agents known to affect the absorption of ASA, such as antacids or proton pump inhibitors, was not permitted during the study.
Treatment and assessments
Participants were randomly assigned (1 of 50 sequences) to receive two single doses each of either a proprietary investigational ER-ASA capsule (NHP-554C) or IR-ASA capsule (aspirin powder USP; Letco Medical, Decatur, AL). Patients received two of five possible doses of ER-ASA (20, 40, 81, 162.5, or 325 mg) and two of five possible doses of IR-ASA (5, 10, 20, 40, or 81 mg) in a total of four periods, each separated by a ≥14-day washout period. Patients were fed a standard breakfast that was completed ≥1 h before drug administration. No food was permitted during the first 4 h postdose.
Serial blood samples (approximately 9 mL) for PD assessments were collected within 1 h before dosing and at 0.5, 1, 2, 4, 8, 16, and 24 h postdose. Urine samples were collected within 2 h before dosing and during 0–8 h, 8–16 h, and 16–24 h postdose. PD assessments included measurement of serum TXB2 levels, urine 11-dehydro-TXB2 levels, and percentage of platelet aggregation. Serum TXB2 levels were determined using a validated TXB2 enzyme immunoassay kit (Cayman Chemical Company, Ann Arbor, MI, USA) and urine 11-dehydro-TXB2 levels were determined using a validated 11-dehydro-TXB2 enzyme immunoassay kit (Cayman Chemical Company) and normalized to urine creatinine levels. For TXB2 measurements, accuracy and precision ranged from 99.6% to 108.1% and 5.1% to 7.1%, respectively, across a concentration range of 20–550 pg/mL, and the lower limit of quantification was 156 pg/mL. For 11-dehydro-TXB2 measurements, accuracy and precision were 88.7 – 97.8% and 9.4 – 14.9%, respectively, across a concentration range of 45 – 1100 pg/mL, and the lower limit of quantification was 93.3 pg/mL.
For platelet aggregation assays, platelet-rich plasma was prepared by centrifugation using a PDQ® platelet function centrifuge (Bio/Data Corporation, Horsham, PA). Platelet aggregation of the 0.25 mL of platelet-rich plasma was induced by arachidonic acid (final assay concentration, 500 µg/mL) or low-concentration collagen (final assay concentration, 2.0 ×10−5 M). Aggregation was allowed to generate for 5 min and then measured by light transmission aggregometry using a Platelet Aggregation Profiler PAP 8E (Bio/Data Corporation) [Citation19].
The primary PD endpoint was the ID50 (dose producing 50% of maximum inhibition of TXB2 production or platelet aggregation), which was determined by examining the relationship between the dose and the inhibition of TXB2 production or platelet aggregation at 24 h postdose, and the inhibition of urine 11-dehydro-TXB2 production at 16–24 h postdose. Secondary PD endpoints included the area under the effect curve (AUEC) for the inhibition of TXB2 production or platelet aggregation, and the maximum observed inhibition (Emax) of TXB2 production or platelet aggregation for each dose; the AUEC was calculated using the trapezoidal rule from 0 to 24 h postdose. PD parameters were analyzed using Phoenix WinNonlin® version 6.3 (Pharsight Corp., St. Louis, MO, USA).
Serial blood samples (∼4 mL) for PK analysis were collected within 1 h of dosing and at 0.25, 0.5, 1, 1.5, 2, 4, 8, 16, and 24 h postdose. Concentrations of ASA and salicylic acid (SA) were measured at NorthEast Bioanalytical Laboratories (Hamden, CT) and analyzed using LC/MS/MS (PE Sciex API-3000 triple quad mass spectrometer equipped with a TurboIonSpray). ASA and SA were separated by reverse-phase high pressure liquid chromatography (Prodigy™ 3 μm ODS-3 150 mm x 3.0 mm column [Phenomenex, Torrance, CA]) and eluted with retention times of 3.3 min for ASA and its internal standard ASA-D4 (Toronto Research Chemicals, Toronto, Canada) and 4.5 min for SA and its internal standard SA-D4 (Toronto Research Chemicals). Analytes were detected by MS/MS in the negative ion mode. The multiple reaction monitoring channels for ASA and its internal standard were a mass-to-charge ratio (m/z) 179→137 and 183→141, respectively. The multiple reaction monitoring channels for ASA and its internal standard were m/z 137→93 and 141→97, respectively. For ASA, accuracy and precision were 97.0 – 101.9% and 3.9 – 7.6%, respectively, across a concentration range of 30.50 – 4299.89 ng/mL. Accuracy and precision for SA were 95.0 – 101.3% and 2.1 – 5.0%, respectively, across a concentration range of 119.02 – 8450.08 ng/mL. The lower limits of quantification for ASA and SA were 10.21 ng/mL and 40.37 ng/mL, respectively. PK parameters were derived by noncompartmental methods, using Phoenix WinNonlin version 6.3 software. The following parameters were calculated: peak plasma concentration (Cmax); time to peak plasma concentration (Tmax); the area under the plasma concentration-time curve (AUC), calculated by the trapezoidal rule from time 0 to 8 h after dosing for ASA and from 0 to 24 h after dosing for SA; and the AUClast, which was defined as the AUC from time 0 to the last measurable ASA or SA concentration.
Safety
Safety was monitored throughout the study and included incidence of adverse events (AEs), physical examinations, vital sign measurements, electrocardiograms (ECG), and clinical laboratory analyses (i.e. hematology, coagulation, blood chemistry, and urinalysis).
Statistical analysis
The sample size was determined from AUEC data from previous unpublished studies, which showed that a sample size of 15 participants per group would permit the characterization of the mean AUEC over the 24 h following administration of ER-ASA with a relative standard error of 20%. Thus, a total population of 50 participants was planned, in order to ensure that ∼18 participants would complete each treatment. The PD population included all participants randomized who received ≥1 dose of study medication and had ≥1 postdose PD assessment. The PK dataset included all participants randomized who received ≥1 dose of study medication and had any plasma concentration data.
A model for determining the maximum PD effect (Emax) was used to analyze inhibition of TXB2 production and inhibition of urinary 11-dehydro-TXB2 excretion during the 16–24 h postdose with each formulation. Similarly, an inhibitory effect model was used to analyze effects on platelet aggregation 24 h postdose. The secondary PD analysis investigated the AUEC for inhibition of serum TXB2 production or inhibition of platelet aggregation. Demographics, PK parameters, and safety parameters were summarized using descriptive statistics. Data are mean ± standard deviation unless otherwise indicated.
Results
Participants
A total of 50 healthy volunteers were enrolled, of whom 47 completed the study (). Of the three participants who did not complete the study, one was withdrawn from participation because of an adverse event and two were withdrawn because of protocol violations (consumption of prohibited medications). The majority of the 50 participants (mean age, 36.9 ± 7.9 years; range, 19–55 years) were female (n = 38; 76.0%), white (n = 49; 98.0%), and had a mean BMI of 25.8 ± 2.7 (range, 20.7–29.9).
Figure 1. Study design.
Pharmacodynamic evaluation
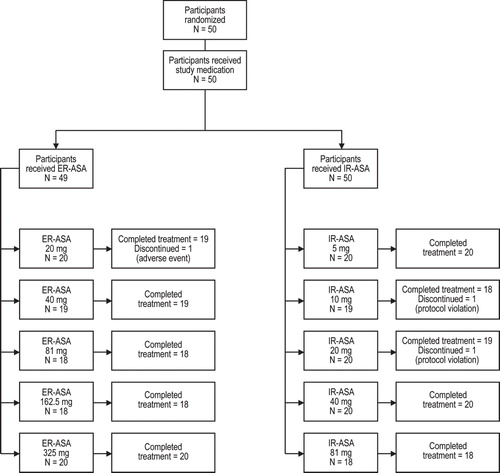
Although a single-dose study may not fully capture maximal PD effect of an ER product, both formulations produced dose-dependent decreases in serum TXB2 levels, urine 11-dehydro-TXB2 levels, and arachidonic acid-induced platelet aggregation across a dose range of 20 – 325 mg for ER-ASA and 5 – 81 mg for IR-ASA. Inhibition of serum TXB2 production was maximal at 2–4 h postdose for both formulations () and was essentially complete following single doses of ER-ASA 325 mg and IR-ASA 81 mg. Marked inhibition of TXB2 production () and mean inhibition of urinary 11-dehydro-TXB2 excretion () were both maintained through 24 h postdose with 81–325 mg ER-ASA and 40–81 mg IR-ASA dose ranges. Inhibition of arachidonic acid-induced platelet aggregation was sustained through 24 h postdose following single doses of 162.5 mg ER-ASA (at 24 h, 28.0% reduction), 325 mg ER-ASA (at 24 h, 90.1% reduction), and 81 mg IR-ASA (at 24 h, 60.0% reduction). Lower doses of both formulations produced only transient inhibition of platelet aggregation, which returned to baseline levels within 24 h. Neither aspirin formulation had an effect on collagen-induced platelet aggregation.
Figure 2. Inhibition of serum TXB2 production after single doses of: (a) ER-ASA 20 – 325 mg or (b) IR-ASA 5 – 81 mg.
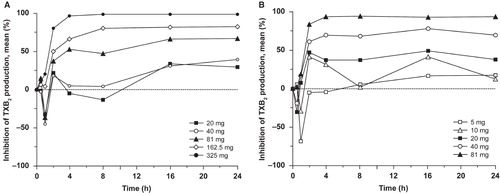
Figure 3. Inhibition of 11-dehydro-TXB2 production after single doses of: (a) ER-ASA 20–325 mg or (b) IR-ASA 5–81 mg. Assessed in urine and corrected for the urine creatinine level.
Analysis of the dose–response relationship for the two ASA formulations showed that the ID50 values for inhibition of serum TXB2 at 24 h postdose and urinary 11-dehydro-TXB2 excretion at 16–24 h postdose were 1.7-fold and 2.4-fold higher, respectively, with ER-ASA than with IR-ASA (). Similarly, the ID50 for inhibition of arachidonic acid-induced platelet aggregation was 2.6-fold greater with ER-ASA than with IR-ASA. The mean AUEC values for both inhibition of serum TXB2 and arachidonic acid-induced platelet aggregation increased with increasing dose for both ASA formulations ().
Table 1. Pharmacodynamic parameters of extended-release acetylsalicylic acid and immediate-release acetylsalicylic acida.
Table 2. AUEC analysis of inhibition of TXB2 production and arachidonic acid-induced platelet aggregationa.
Pharmacokinetic evaluation
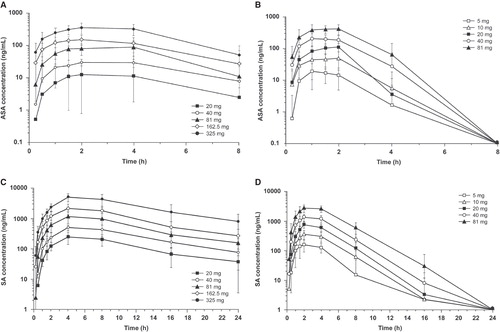
A dose-proportional increase in mean Cmax and AUClast for ASA and SA was observed for both formulations (). However, higher serum ASA and SA levels and shorter time to maximum plasma concentration were observed with IR-ASA (; ). With IR-ASA, Tmax for ASA was attained 1–2 h postdose, and ASA concentrations were only measurable up to 6 h postdose. However with ER-ASA, Tmax occurred at 2–4 h postdose, and ASA concentrations were measured throughout the 8 h evaluated in the study (complete sampling interval). Serum concentrations of SA showed similar trends (; ). With IR-ASA, dose-normalized mean Cmax and AUClast of ASA were approximately sixfold higher and Cmax of SA was approximately two to threefold higher compared with ER-ASA. However, the dose-normalized AUClast for SA was similar with both formulations (), indicating that for a given dose, both formulations released similar concentrations of active drug overall.
Table 3. Pharmacokinetics of acetylsalicylic acid and salicylic acid.
Figure 4. Single-dose mean concentration-time profiles of acetylsalicylic acid for: (a) ER-ASA and (b) IR-ASA; and single-dose mean concentration-time profiles of SA for: (c) ER-ASA and (d) IR-ASA.
Safety
A total of 23 AEs were reported by 8 of 49 participants (16.3%) receiving ER-ASA, and 20 AEs were reported in 8 of 50 participants (16.0%) receiving IR-ASA. There was no apparent dose-related increase in AEs for either ASA formulation. The most commonly reported AE was headache (five events in four participants receiving ER-ASA and three events in three participants receiving IR-ASA). Overall, 18 of the 23 (78.3%) AEs with ER-ASA and 18 of 20 AEs (90.0%) with IR-ASA were considered by investigators to be mild in intensity, and no severe or serious AEs were reported. One individual was withdrawn from the study due to elevated liver enzymes levels (alanine transaminase, aspartate aminotransferase, gamma-glutamyl transpeptidase) after dosing with ER-ASA 20 mg. There were no apparent trends in mean changes from the baseline in clinical laboratory parameters and vital sign and ECG measurements.
Discussion
The current study characterizes the pharmacodynamics, pharmacokinetics, and dose–response relationship of ER-ASA compared with an IR formulation in healthy volunteers. The benefits of aspirin as secondary prevention for cardiovascular disease are related to inhibition of the production of TXA2, a potent, locally acting vasoconstrictor and platelet activator [Citation3]. TXA2 is produced in platelets from arachidonic acid by the sequential action of the enzymes COX-1 and thromboxane synthase [Citation20], and is rapidly degraded to the stable metabolite TXB2. Aspirin (ASA) is a potent irreversible inhibitor of COX-1, preventing the synthesis of TXA2, hence reducing the production of TXB2 [Citation20]. The current study showed that serum TXB2 and urine 11-dehydro-TXB2 levels were inhibited in a dose-dependent manner following single doses of both ASA formulations. The inhibitory effects of ER-ASA 162.5 and 325 mg were maintained during the entire 24-h observation period and were similar to those of IR-ASA 81 mg. The safety profile of single-dose ER-ASA 20–325 mg was also similar to that of single-dose IR-ASA 5–81 mg and consistent with the previous data for ER-ASA [Citation18].
Platelets have a fairly short lifespan of ∼7–10 days, and under normal circumstances daily treatment with low-dose aspirin results in almost complete blockade of TXA2 formation and platelet aggregation [Citation17]. The platelet function is restored by the formation of new platelets from megakaryocytes [Citation17]: under healthy conditions ∼10 – 15% of circulating platelets are replaced each day [Citation13,17]. However, platelet production or reactivity may be increased in certain patient populations, including those with type 2 diabetes or coronary artery disease [Citation13-15,21,22]. Due to rapid hydrolysis of ASA into an inactive metabolite, the short duration of active metabolite (i.e. ASA) availability achieved with current ASA formulations may not provide sufficient exposure of new and highly reactive platelets to the drug throughout the entire 24-hour dosing interval, particularly in individuals with enhanced platelet turnover or activity. This would be consistent with the clinical finding that individuals with increased platelet turnover may be less responsive to antiplatelet drugs [Citation13,15,17]. In the current single-dose study, arachidonic acid-induced platelet aggregation was dose-dependent with both ASA formulations. However, the study also showed that an approximately twofold difference in dose between the two ASA formulations was required to achieve 50% of the maximum TXB2 inhibition for a similar maximum PD effect with single-dose administration, suggesting an approximately twofold greater ER-ASA dose (162.5 mg), still considered a low dose, is necessary to obtain the same steady-state PD response bioequivalent to that of IR-ASA 81 mg.
The ER formulation of aspirin in the current study was designed to deliver small amounts of ASA in the GI tract during a 24-h dosing interval. It was hypothesized that a more consistent, prolonged release of ASA over a 24-dosing interval would allow for inhibition of COX-1 in platelets in portal circulation (i.e. presystemic) [Citation23], without saturating hepatic metabolic mechanisms, thereby limiting the amount of ASA reaching systemic circulation. Thus, this new microcapsule formulation of ER-ASA was expected to exhibit a PK profile that differed from the PK profile of IR-ASA. The PK analysis in the current study confirmed that ER-ASA provided a more constant, prolonged release of ASA, compared with IR-ASA. In addition, although a delayed immediate-release (e.g. enteric coated) ASA formulation was not evaluated, the PK profile of the ER-ASA formulation evaluated in this study differs from that reported previously for an enteric-coated formulation [Citation23], thus supporting a PK profile that is unique versus other ASA formulations.
The PK analysis profile of ER-ASA indicating a more constant, prolonged release of ASA, compared with IR-ASA, supports the hypothesis that prolonged availability and extended coverage with ER-ASA that inhibits platelet COX-1 in the portal circulation might minimize the potential risk for deleterious effects on prostacyclin production related to reduced ASA availability in the systemic circulation. Furthermore, other controlled-release aspirin preparations administered at low doses support the achievement of cumulative maximum PD effects—TXB2 suppression in this case—as taking place after several daily doses (e.g. ∼4 days) [Citation20].
There are some limitations to this study, including that only single-dose administration was evaluated; thus steady-state PD and time to reach maximum PD effects were not analyzed. As noted previously, PD effects are cumulative at the initiation of lower dosages and require several doses (e.g. ∼4 days) to achieve a maximum PD (TXB2 cumulative suppression) effect. In addition, ASA serum concentrations were only assessed for up to 8 h, and only healthy volunteers were included.
Conclusion
Compared with IR-ASA, ER-ASA produced dose-dependent inhibition of platelet aggregation and reduced systemic ASA concentrations for a given dose. These single-dose data suggest that a higher dose (162.5 mg) of ER-ASA is necessary to achieve the same PD response as IR-ASA 81 mg in steady state. Overall, the differences in PK of ER-ASA versus IR-ASA, such as slower absorption and prolonged ASA release with ER-ASA, were expected for an ER (24-h) formulation.
Declaration of interest
This study was supported by New Haven Pharmaceuticals, Inc. Technical editorial and medical writing support, under direction of the authors, was provided by Mary Beth Moncrief, PhD, and Michael Shaw, PhD, Synchrony Medical Communications, LLC, West Chester, PA, USA, with support from New Haven Pharmaceuticals Inc. J Patrick and L Dillaha are employees of New Haven Pharmaceuticals. D Armas is an employee of Celerion, a company that received support from New Haven Pharmaceuticals for conducting this study. WC Sessa oversees a scientific advisory board for New Haven Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Notes
References
- Vandvik PO, Lincoff AM, Gore JM, Gutterman DD, Sonnenberg FA, Alonso-Coello P, et al. Primary and secondary prevention of cardiovascular disease: antithrombotic therapy and prevention of thrombosis, 9th Ed: American College Of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141:E637s–68s
- Furie KL, Kasner SE, Adams RJ, Albers GW, Bush RL, Fagan SC, et al. American Heart Association Stroke Council, Council On Cardiovascular Nursing, Council On Clinical Cardiology, And Interdisciplinary Council On Quality Of Care And Outcomes Research. Guidelines For The Prevention Of Stroke In Patients With Stroke Or Transient Ischemic Attack: A Guideline For Healthcare Professionals From The American Heart Association/American Stroke Association. Stroke 2011;42:227–76
- Patrono C, García Rodríguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med 2005;353:2373–83
- Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 2011;31:986–1000
- Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn’t the stomach digest itself? Physiol Rev 2008;88:1547–65
- Graham DY, Smith JL. Aspirin and the stomach. Ann Intern Med 1986;104:390–8
- Kelly JP, Kaufman DW, Jurgelon JM, Sheehan J, Koff RS, Shapiro S. Risk of aspirin-associated major upper-gastrointestinal bleeding with enteric-coated or buffered product. Lancet 1996;348:1413–16
- Awtry EH, Loscalzo J. Aspirin. Circulation 2000;101:1206–18
- Sørensen HT, Mellemkjaer L, Blot WJ, Nielsen GL, Steffensen FH, McLaughlin JK, Olsen JH. Risk of upper gastrointestinal bleeding associated with use of low-dose aspirin. Am J Gastroenterol 2000;95:2218–24
- Rocca B, Petrucci G. Variability in the responsiveness to low-dose aspirin: pharmacological and disease-related mechanisms. Thrombosis 2012;2012:376721
- Gasparyan AY, Watson T, Lip GY. The role of aspirin in cardiovascular prevention: implications of aspirin resistance. J Am Coll Cardiol 2008;51:1829–43
- Wurtz M, Grove EL. Interindividual variability in the efficacy of oral antiplatelet drugs: definitions, mechanisms and clinical importance. Curr Pharm Des 2012;18:5344–61
- Capodanno D, Patel A, Dharmashankar K, Ferreiro JL, Ueno M, Kodali M, et al. Pharmacodynamic effects of different aspirin dosing regimens in type 2 diabetes mellitus patients with coronary artery disease. Circ Cardiovasc Interv 2011;4:180–7
- Rocca B, Santilli F, Pitocco D, Mucci L, Petrucci G, Vitacolonna E, et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost 2012;10:1220–30
- Grove EL, Hvas AM, Mortensen SB, Larsen SB, Kristensen SD. Effect of platelet turnover on whole blood platelet aggregation in patients with coronary artery disease. J Thromb Haemost 2011;9:185–91
- Grove EL, Hvas AM, Kristensen SD. Immature platelets in patients with acute coronary syndromes. Thromb Haemost 2009;101:151–6
- Schnell O, Erbach M, Hummel M. Primary and secondary prevention of cardiovascular disease in diabetes with aspirin. Diab Vasc Dis Res 2012;9:245–55
- Brown N, May JA, Wilcox RG, Allan LM, Wilson AM, Kiff PS, Heptinstall S. Comparison of antiplatelet activity of microencapsulated aspirin 162.5 Mg (Caspac Xl), with enteric coated aspirin 75 Mg And 150 Mg in patients with atherosclerosis. Br J Clin Pharmacol 1999;48:57–62
- Frontroth JP. Light transmission aggregometry. In Monagle P, editor. Haemostasis methods and protocols. New York, NY: Humana Press; 2013. pp 227–40
- Clarke RJ, Mayo G, Price P, FitzGerald GA. Suppression of thromboxane A2 but not of systemic prostacyclin by controlled-release aspirin. N Engl J Med 1991;325:1137–41
- Simpson SH, Abdelmoneim AS, Omran D, Featherstone TR. Prevalence of high on-treatment platelet reactivity in diabetic patients treated with aspirin. Am J Med 2014;127:95.E1–9
- Martin JF, Kristensen SD, Mathur A, Grove EL, Choudry FA. The causal role of megakaryocyte-platelet hyperactivity in acute coronary syndromes. Nat Rev Cardiol 2012;9:658–70
- Patrignani P, Tacconelli S, Piazuelo E, Di Francesco L, Dovizio M, Sostres C, et al. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J Thromb Haemost 2014;12:1320–30
Notice of correction
Please note that in Pharmacokinetic evaluation section, “Figures 3a and 3b” in the sentence “Serum concentrations of SA showed similar trends (Table 3; Figures 3a and 3b).” has been changed to “Figures 4c and 4d” after initial online publication of this article (22nd May 2015).