
ABSTRACT
Gout is a progressive, painful, debilitating form of inflammatory arthritis. It is caused by factors that elevate the concentration of serum uric acid (sUA), leading to hyperuricemia (sUA >6.8 mg/dL). Continued elevated sUA can result in monosodium urate (MSU) crystal deposition in joints and soft tissues, and can cause acute and chronic inflammation. The prevalence of hyperuricemia and gout has increased over the last few decades, likely due to an aging population, changes in lifestyles and diet, and an increase in gout-associated comorbidities. Untreated or improperly treated gout can lead to chronic manifestation of the disease, including persistent inflammation, increased number of flares, development of tophi, and structural joint damage. Data show that even when patients are asymptomatic, ongoing inflammation and subsequent damage occurs locally at the joint and systemically. The aim of long-term treatment of gout is to reduce sUA levels to <6 mg/dL, which is below the saturation point of MSU (6.8 mg/dL), to inhibit formation of new crystals and to promote dissolution of existing crystals. Gout treatment should improve disease outcomes by eliminating gout flares, inducing long-term resolution of tophi, and more effectively managing comorbidities, many of which are associated with hyperuricemia. A number of studies have demonstrated that treating to the target of <6 mg/dL, by using effective therapies to lower sUA, results in reduction in the incidence of gout flares as well as shrinkage and eventual disappearance of tophi. Gout is often poorly managed due to a number of factors including lack of physician and patient adherence to treatment guidelines. Patients need to be educated about their diagnosis and management of the disease, such as the influence of diet and the importance of compliance with long-term treatment. With treatment, regular sUA monitoring, and patient adherence, gout is a curable disease.
Methods
PubMed and Google Scholar databases were searched to identify clinical trials and review articles on the epidemiology, disease mechanism, history, clinical presentation, and treatment and management of gout. The search also identified previously recommended guidelines which were incorporated into the review. Search terms included, but were not limited to, gout, hyperuricemia, treatment guidelines, American College of Rheumatology (ACR), European League Against Rheumatism (EULAR), comorbidity, flare, adherence, epidemiology, serum uric acid (sUA), monosodium urate (MSU), tophi, joint damage, joint erosion, <6 mg/dL, MSU crystal formation, and individual drug names and classes of treatments of interest. Both authors actively participated in the drafting and editing of the paper and offered comprehensive discussions regarding the content.
Gout background and epidemiology
Gout is a progressive, painful, debilitating form of inflammatory arthritis that has been affecting man since antiquity [Citation1–Citation3]. It is caused by factors that elevate the concentration of sUA, leading to hyperuricemia [Citation2,Citation4]. Continued elevated sUA can result in MSU crystal deposition throughout the body, including joints, soft tissues, cartilage, and organs (e.g. kidneys) [Citation5]. MSU crystals trigger the periodic occurrence of painful, acute, inflammatory flares [Citation1,Citation2]. If hyperuricemia persists, the acute gout flares may be superseded by long-term, chronic, tophaceous gout with bone and cartilage destruction [Citation1,Citation2].
Gout is one of the most common forms of inflammatory arthritis among men. In the USA, about 8.3 million adults (4%) are affected by gout and 43.3 million (21%) by hyperuricemia [Citation6]. Both occur with greater frequency in adults than other well-known, chronic, joint- or pain-related debilitating diseases such as fibromyalgia (5.0 million) [Citation7], rheumatoid arthritis (1.5 million) [Citation8], and psoriatic arthritis (0.5 million) [Citation9].
Gout and other crystal arthropathies accounted for 1.5% of the 1.2 million nonfederal, short-stay hospitalizations in 2007 [Citation1]. All-cause medical expenditures in persons with gout are estimated to total about $11,663 per person per year, and the direct burden for new gout in the USA may be greater than $27 million per year [Citation1,Citation10]. In addition, gout is a source of significant personal burden for patients. Among those with inadequately controlled gout, the condition is associated with significant functional disability, loss of work productivity, reduction in social activity, and poor health-related quality of life [Citation11–Citation13]. In fact, the work loss observed with gout is similar to that seen with rheumatoid arthritis [Citation8,Citation11].
The prevalence of gout and hyperuricemia has increased considerably over the last 25 years [Citation6,Citation14]. This increase may be due to an aging population, changes in lifestyle and diet (e.g. higher intake of fructose-containing soft drinks and alcoholic beverages, such as beer), and an increasing prevalence of comorbidities associated with hyperuricemia and gout, such as obesity, hypertension, widespread use of thiazide diuretics, and chronic kidney disease (CKD) [Citation14–Citation17]. Other factors associated with hyperuricemia and gout include metabolic syndrome, congestive heart failure, and organ transplantation [Citation18]. In fact, evidence suggests that increased sUA levels may play a role in the development of metabolic syndrome, type 2 diabetes, and hypertension [Citation19]. A history of gout is also an independent risk factor for cardiovascular disease, possibly due to increased systemic inflammation [Citation20]. Use of sUA-lowering therapy improved blood pressure and had a cardiovascular protective effect [Citation19]. Hyperuricemia has also been shown to be predictive of CKD; however, it is unclear whether elevated sUA levels have a causal role in the development of kidney disease [Citation20]. Similarly, the precise role of uric acid in the development of cardiovascular disease is not clear [Citation19]. Furthermore, age and genetic factors can predispose an individual to developing gout. Genetic variation in solute carrier family 2 member 9 (SLC2A9) and 11 (SLC22A11); solute carrier family member 17, member 1 (SLC17A1); coding PDZ domain-containing 1 (PDK-Z1); and ATP-binding cassette, subfamily G, member 2 (ABCG2) have been implicated in increasing the risk of gout [Citation21,Citation22]. Asymptomatic hyperuricemia can begin during puberty in boys, but is usually delayed until menopause in women [Citation3]. Estrogen has a uricosuric effect, making the disease less common among premenopausal women [Citation14,Citation23]. Gout commonly develops in men between 40 and 60 years of age after many (approximately 20–30) years of hyperuricemia [Citation3]. Before the age of 65 years, men have a fourfold greater incidence of gout than women (about 4 per 1000 persons in men compared with 1 per 1000 persons in women) [Citation23,Citation24]. After 65 years of age, prevalence increases for both men and women at a more comparable rate, with incidence about threefold higher in men [Citation24].
Presentation and characterization of gout
Classically, gout has been described as occurring in three stages: asymptomatic hyperuricemia, acute intermittent gout (flares), and advanced or chronic gout () [Citation25,Citation26]. A recent modification of this staging, however, has been proposed by Dalbeth and Stamp (2014) that includes four stages, with the period of asymptomatic hyperuricemia subdivided into two separate stages: Stage A (hyperuricemia without detectable MSU crystal deposition); Stage B (hyperuricemia with MSU crystal deposition but no signs or symptoms of gout); Stage C (MSU crystal deposition with prior or current symptoms of a gout flare); and Stage D (advanced gout requiring a specialist’s intervention) [Citation27]. Hyperuricemia is defined as sUA levels >6.8 mg/dL (404 µmol/L) and no symptoms from crystal deposition [Citation28,Citation29]. Of note, each laboratory calculates its own sUA threshold for hyperuricemia, often defined as the mean sUA plus 2 standard deviations for their local healthy population, and calculated separately according to sex [Citation30,Citation31]. Therefore, an sUA level in what an individual laboratory considers the ‘normal range’ may still reflect levels in joint tissues that are above the point for MSU crystal formation [Citation31]. The rate at which a person progresses from asymptomatic hyperuricemia to chronic advanced gout varies by individual and is dependent upon numerous factors, the most important of which is the degree of increase in the concentration of sUA () [Citation3,Citation32,Citation33].
Figure 1. Diagram showing the path from hyperuricemia to structural joint damage. Long-standing hyperuricemia may lead to monosodium urate (MSU) deposits. Hyperuricemia is typically asymptomatic and is often associated with subclinical inflammation and bone erosion. Crystal shedding into the joint can cause Intermittent, acute inflammation and a gout flare-up. Continued crystal deposition can result in chronic manifestation of the disease, including persistent inflammation, increased number of flares, development of tophi, and structural joint damage. Adapted from Adv Ther, ‘A review of uric acid, crystal deposition disease, and gout’, volume 32, 2015, Perez-Ruiz F, et al, Figure 1: ‘Diagram showing the path from hyperuricemia to structural joint damage’ on page 32, with permission of Springer (© 2014) [Citation26].
![Figure 1. Diagram showing the path from hyperuricemia to structural joint damage. Long-standing hyperuricemia may lead to monosodium urate (MSU) deposits. Hyperuricemia is typically asymptomatic and is often associated with subclinical inflammation and bone erosion. Crystal shedding into the joint can cause Intermittent, acute inflammation and a gout flare-up. Continued crystal deposition can result in chronic manifestation of the disease, including persistent inflammation, increased number of flares, development of tophi, and structural joint damage. Adapted from Adv Ther, ‘A review of uric acid, crystal deposition disease, and gout’, volume 32, 2015, Perez-Ruiz F, et al, Figure 1: ‘Diagram showing the path from hyperuricemia to structural joint damage’ on page 32, with permission of Springer (© 2014) [Citation26].](/cms/asset/dc0baeec-d4e0-48b9-8ff6-447adf87dc73/ipgm_a_1221732_f0001_c.jpg)
Figure 2. Five-year cumulative incidence of gout according to sUA level in men in the Normative Aging Study [Citation33]. Reproduced with permission from Roddy E and Doherty M: Arthritis Res Ther 2010;12(6):223. [Citation32].
![Figure 2. Five-year cumulative incidence of gout according to sUA level in men in the Normative Aging Study [Citation33]. Reproduced with permission from Roddy E and Doherty M: Arthritis Res Ther 2010;12(6):223. [Citation32].](/cms/asset/340e4741-73c1-426a-81f0-005786cb20ed/ipgm_a_1221732_f0002_c.jpg)
Of all patients with asymptomatic hyperuricemia, only about 15–20% will develop gout [Citation3]. In these patients, MSU crystal deposition, subclinical inflammation, and bone erosion can occur [Citation3,Citation18,Citation26,Citation33,Citation34]. The MSU crystals form small lattice structures on the surface of synovial lining and cartilage [Citation3]. The fact that not all patients with hyperuricemia develop gout suggests that other factors may contribute to the formation of crystals and the ability of formed crystals to trigger flares [Citation26]. MSU deposition is a precursor to gout, but it is unclear whether MSU crystals in patients with asymptomatic hyperuricemia predict development of clinically apparent gout [Citation26].
After a prolonged period of hyperuricemia, the shedding of asymptomatic, preformed crystals from the cartilage surface into the joint triggers inflammation characteristic of an acute flare [Citation26,Citation34]. The first manifestation of acute gout is an attack of synovitis typically affecting a single peripheral joint, most often the first metatarsophalangeal joint [Citation35]. Other joints commonly affected include the ankles, knees, mid-tarsal joints, fingers, elbows, and wrists [Citation35,Citation36]. Acute attacks are characterized by the sudden onset of extreme pain, which typically takes less than 24 h to peak in intensity () [Citation35,Citation36]. Flares are also associated with swelling, heat, tenderness, and erythema [Citation35,Citation36]. Acute gouty flares are self-limiting, with symptoms resolving in about 2 weeks [Citation26,Citation36]. Although acute flares appear episodic in nature, ongoing damage during intercritical asymptomatic periods occurs due to continuing MSU crystal deposition and inflammation [Citation26,Citation37,Citation38]. This continuing damage may not be apparent in normal plain radiographs; however, the destructive arthropathy can be detected by more advanced imaging procedures, such as dual-energy computed tomography (DECT), computed tomography (CT), magnetic resonance imaging (MRI), and ultrasound () [Citation39,Citation40].
Table 1. The ACR/EULAR gout classification criteria (reproduced with permission from Neogi et al. [Citation36].
Figure 3. Dual-energy computed tomography (DECT) images of ankle and foot (a, b) showing MSU crystal deposition in green color (arrows) and corresponding CT images (c, d) showing erosions (arrowheads). Reproduced with permission from Chowalloor PV, et al: Ther Adv Musculoskelet Dis 2014;6(4):131-43. [Citation40].
![Figure 3. Dual-energy computed tomography (DECT) images of ankle and foot (a, b) showing MSU crystal deposition in green color (arrows) and corresponding CT images (c, d) showing erosions (arrowheads). Reproduced with permission from Chowalloor PV, et al: Ther Adv Musculoskelet Dis 2014;6(4):131-43. [Citation40].](/cms/asset/3b63afd7-e837-4d80-b96e-a03d7d120635/ipgm_a_1221732_f0003_c.jpg)
Untreated or improperly treated crystal deposition can lead to chronic manifestation of the disease, including persistent inflammation, increased number of flares, development of tophi, and structural joint damage [Citation26]. Tophi can cause chronic inflammation and erosive arthritis [Citation16]. Chronic gout typically develops after 10 or more years of acute episodic gout, and is apparent when the pain-free intermittent periods disappear [Citation3].
Etiology
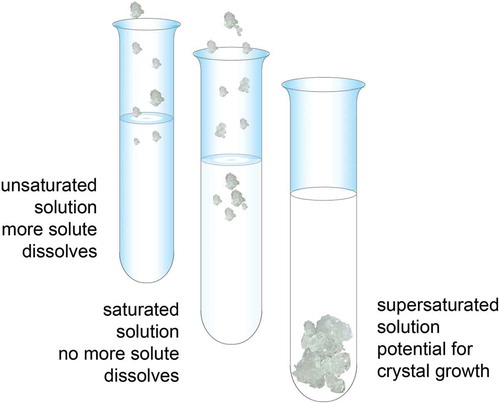
Hyperuricemia is the most important risk factor for gout [Citation33,Citation34]. In vitro, at 37°C and pH 7.4, levels of uric acid above 6.8 mg/dL result in crystal formation and aggregation [Citation41–Citation43]. Solubility of a substance (a solute) is a measure of how well one substance dissolves into another (). A solute can fully dissolve in a solution that contains less than the maximum amount of solute that can be dissolved. A saturated solution contains as much solute as it possibly can dissolve, and a supersaturated solution contains more solute than can be dissolved, resulting in crystal formation. As sUA levels rise, the blood and body fluids become supersaturated, causing crystals to form in synovial fluids, soft tissues, and organs [Citation5,Citation43,Citation44]. Crystal deposition is reversible, and crystals can dissolve when the concentration of sUA is reduced to levels <6 mg/dL [Citation43,Citation45]. The importance of maintaining sUA levels below 6.8 mg/dL is made evident by the observation that the incidence of gout increases with higher levels of sUA, starting at between 6.0 and 6.9 mg/dL () [Citation3,Citation32,Citation33]. One study found that the annual incidence of gouty arthritis for patients with sUA >9 mg/dL was 4.9%, compared with 0.5% for urate levels of 7.0–8.9 mg/dL and 0.1% for urate levels <7.0 mg/dL [Citation33]. Another study found that the 5-year incidence rate of gout was 0.6% in patients with sUA levels <7 mg/dL and about 30% for patients with sUA levels >10 mg/dL [Citation46].
Figure 4. Solubility of a substance (a solute) is the amount of that substance that will dissolve in a given amount of solvent; or it is a measure of how well one substance dissolves into another. A solute can fully dissolve in a solution that has less than the maximum amount of solute that can be dissolved. A saturated solution contains as much solute as it possibly can dissolve. A supersaturated solution contains more solute than can be dissolved, increasing the potential for crystal formation.
Uric acid levels are controlled by the rate of urate production and the elimination of urate via the kidneys and the gastrointestinal tract. In about 90% of patients with hyperuricemia, sUA levels are elevated mainly due to inefficient renal excretion rather than overproduction of uric acid [Citation3]. About one-third of uric acid comes from exogenous sources, such as diet (alcohol/beer, red meat, seafood) [Citation47,Citation48]. The other two-thirds derive from endogenous production via the degradation of purines, such as DNA and RNA [Citation48,Citation49]. In humans, urate is produced by the conversion of hypoxanthine to xanthine, which is subsequently converted to uric acid by an enzyme called xanthine oxidase [Citation3]. Most mammals possess an additional enzyme, uric acid oxidase, that converts uric acid into a soluble end product called allantoin that is readily eliminated in urine [Citation3]. The ability of humans and other higher primates to make uric acid oxidase was lost during evolution, and consequently, humans are prone to uric acid accumulation [Citation3]. The gastrointestinal tract eliminates about 20% of urate, while the rest is excreted by the kidney [Citation50]. The majority of uric acid (about 98–99%) circulates in the blood as ionized urate, and due to the high concentration of sodium in the extracellular compartment, urate is primarily present as MSU [Citation15,Citation51].
Guideline recommendations for sUA levels
The primary aim of treatment for gout is to lower sUA [Citation52]. Several guidelines, such as those published by the ACR [Citation53] and EULAR [Citation31,Citation54] (plus others, such as the American Society of Clinical Rheumatologists [ASCR] [Citation28], the 3Es [Evidence, Expertise, Exchange] Initiative [Citation55], Japanese Society of Gout and Nucleic Acid Metabolism [JSGNM] [Citation56], and the Dutch College of General Practitioners [DCGP] [Citation57]) recommend a target sUA level of <6 mg/dL (360 µmol/L). In some cases, a lower target (<5 mg/dL) may be recommended to more rapidly improve gout signs and symptoms in patients with more severe disease [Citation53,Citation54]. In contrast, the British Society of Rheumatology recommends the lower (<5 mg/dL) sUA target for all patients with gout [Citation58]. The idea behind treating to a target of <6 mg/dL and achieving lifelong maintenance of this level is to lower the concentration of sUA well below its saturation point and to provide some margin to accommodate the influence of natural fluctuations in uric acid levels. Lower sUA levels also increase the resolution rate of tophaceous deposits () [Citation59].
Figure 5. Mean serum urate levels versus velocity of tophi reduction. Reproduced with permission from Perez-Ruiz F, et al: Arthritis Rheum 2002;47(4):356-60. [Citation59].
![Figure 5. Mean serum urate levels versus velocity of tophi reduction. Reproduced with permission from Perez-Ruiz F, et al: Arthritis Rheum 2002;47(4):356-60. [Citation59].](/cms/asset/00a0fa4c-c208-435e-8932-aed7fe4048a4/ipgm_a_1221732_f0005_c.jpg)
Gout treatment should improve disease outcomes by eliminating gout flares, inducing long-term resolution of tophi, and more effectively managing comorbidities, many of which are associated with hyperuricemia [Citation31,Citation53]. There is growing evidence of the association of gout with several chronic diseases, including cardiovascular disease, and data exist that indicate gout treatment may improve some of these comorbidities [Citation60,Citation61]. The ACR recommends both nonpharmacologic and pharmacologic approaches to treatment after first establishing the diagnosis of gout () [Citation53]. Patient education is an important component of treatment and should include information on lifestyle adjustments, treatment objectives, and management of comorbidities [Citation52,Citation53]. Patients should be encouraged to lose weight and be cautioned about consumption of excessive amounts of foods with high purine content, such as organ meats, shell fish, and alcohol (especially beer), as well as foods containing high fructose corn syrup, all of which could trigger a flare [Citation14,Citation20]. However, diet change alone, although beneficial, is rarely sufficient to lower sUA levels to the degree necessary to prevent acute or chronic disease [Citation52,Citation62]. Physicians should consider all causes of hyperuricemia—both exogenous and endogenous—for all gout patients, and evaluate certain disorders that are associated with hyperuricemia and gout, such as obesity, metabolic syndrome, type 2 diabetes, hypertension, and CKD [Citation52,Citation53]. The ACR treatment guidelines also advocate consideration of the removal or replacement of nonessential prescription drugs (e.g. thiazide and loop diuretics, niacin, and calcineurin inhibitors), when appropriate, which are known to elevate sUA [Citation53].
Figure 6. Baseline ACR recommendations and overall strategy for treating and managing patients with gout. Data from Khanna D, et al: Arthritis Care Res (Hoboken) 2012;64(10):1431-46. [Citation53].
![Figure 6. Baseline ACR recommendations and overall strategy for treating and managing patients with gout. Data from Khanna D, et al: Arthritis Care Res (Hoboken) 2012;64(10):1431-46. [Citation53].](/cms/asset/104ceb70-c367-48fe-b4bd-8c57baa4f3fe/ipgm_a_1221732_f0006_c.jpg)
Following these initial steps, ACR recommends that a patient be evaluated for the use of urate-lowering therapy (ULT). A patient is considered a candidate if he/she has at least one of the following: tophus or tophi (verified by clinical examination or imaging study); ≥2 gout flares per year; stage 2 or worse CKD; or past urolithiases [Citation53]. If pharmacologic ULT is indicated, the patient should be treated to achieve an sUA target of <6 mg/dL [Citation53]. First-line pharmacologic therapies are xanthine oxidase inhibitors (e.g. allopurinol or febuxostat), or if necessary due to poor tolerability or contraindications, an alternative therapy (e.g. probenecid) can be used instead [Citation53]. If needed, ACR guidelines recommend the addition of probenecid to a xanthine oxidase inhibitor [Citation53]. Pegloticase can be considered if treatment goals are not achieved and there is continuing disease activity [Citation53]. If the target level of <6 mg/dL is not achieved, the dose of the ULT should be increased [Citation53]. When ULT is started, anti-inflammatory prophylactic treatment should also be initiated to reduce the increased risk of acute gouty attacks during the early phase (first 6 months) of treatment [Citation53]. Nonsteroidal anti-inflammatory drugs and low-dose colchicine are options for prophylactic therapy [Citation53]. Once the sUA target of <6 mg/dL is achieved continuously [Citation53], the dose of ULT should be maintained indefinitely with ongoing monitoring of sUA levels twice yearly [Citation53]. Anti-inflammatory prophylaxis should be maintained for at least 6 months after the target is achieved, or 3 months after achieving target sUA in patients without tophi, or 6 months in patients with tophi [Citation53]. It may be necessary to reduce sUA levels to <5 mg/dL for patients with advanced disease and tophaceous deposits [Citation53]. After all symptoms have resolved, ACR guidelines recommend continuing all measures and therapies needed to keep the patient’s sUA levels <6 mg/dL [Citation53]. In patients with more advanced disease, the primary care provider (PCP) may consider referral to a specialist, such as a rheumatologist, for cases of unclear etiology of hyperuricemia, refractory signs and symptoms of gout, lack of tolerability to standard therapy, or difficulty in reaching the sUA level target goal of <6 mg/dL [Citation53].
Reasoning behind the sUA target goal of <6 mg/dL
For patients whose sUA levels are not lowered sufficiently, gout will continue to be a chronic, progressive disease [Citation26]. Achieving a lower level of sUA can help prevent further MSU crystal deposition, inhibit tophi formation, and accelerate tophi dissolution [Citation59] (). Halting crystal formation and dissolving existing crystals is the only way to permanently stop the signs and symptoms of gout, and potentially ‘cure’ the disease [Citation59]. Undertreatment can have significant consequences for a patient, including increased number and frequency of flares, diminished quality of life and productivity, and increased use of systemic steroids and nonsteroidal anti-inflammatory drugs [Citation20]. Inadequate or delayed treatment and the failure to reduce sUA levels <6 mg/dL can result in increased crystal deposition and development of comorbidities, which can in turn result in more severe disease that is more difficult to treat and manage [Citation20,Citation26,Citation63]. Imaging studies have found a close relationship between features of structural joint damage and crystal deposition, with MSU crystals frequently observed within the areas of bone erosion [Citation40]. This close relationship between joint damage and MSU crystal formation emphasizes the need to reduce sUA levels before structural damage occurs [Citation26].
Table 2. Why keep serum uric acid levels <6 mg/dL?
A number of studies found that patients experienced fewer flares and better therapeutic outcomes when their sUA levels remained at <6 mg/dL [Citation45,Citation64–Citation68]. In one prospective study, sustained reduction of sUA levels <6 mg/dL over a 5-year period with long-term febuxostat therapy resulted in nearly complete elimination of gout flares in all patients (N = 116) and total dissolution of tophi in 69% of the patients who had palpable tophi at baseline () [Citation64]. A prospective study of patients with MSU crystal-confirmed gout (N = 18) found that lowering sUA to levels <5 mg/dL resulted in disappearance of MSU crystals [Citation66]. The time required for disappearance ranged from 3 to 33 months, and disappearance of crystals took longer in patients who had gout for a longer duration [Citation66]. A retrospective study of 267 patients found that the use of ULT and significant lowering of sUA levels reduced the risk of a gout attack over a 3-year period [Citation65]. Another retrospective study, which assessed gout outcomes over a 10-year period in patients with crystal-confirmed gout (N = 38), found that almost half of patients who maintained their sUA levels ≤6 mg/dL for more than 12 months had no gout attacks for 2 or more years [Citation45]. They also found that urate crystal deposition in knees and joint fluids were depleted in more than half (56%) of patients who maintained their sUA ≤6 mg/dL [Citation45].
Figure 7. Percentage of patients who required treatment for flares while receiving a maintenance dose of febuxostat to keep sUA levels <6 mg/dL. Reproduced with permission from Schumacher HR, Jr., et al: Rheumatology (Oxford) 2009;48(2):188-94. [Citation64].
![Figure 7. Percentage of patients who required treatment for flares while receiving a maintenance dose of febuxostat to keep sUA levels <6 mg/dL. Reproduced with permission from Schumacher HR, Jr., et al: Rheumatology (Oxford) 2009;48(2):188-94. [Citation64].](/cms/asset/1f2e3d60-4e0c-4eaa-ba07-5936d038f240/ipgm_a_1221732_f0007_c.jpg)
The effect of reducing sUA levels on radiographic changes is less clear. A retrospective study that assessed management of gout over a 10-year period found that tophaceous resorption correlated with serum levels ≤6 mg/dL [Citation67]. However, they found no correlation between sUA levels and radiographic changes [Citation67]. Similarly, another study in patients with early gout treated for 2 years with febuxostat found that treatment greatly reduced synovitis but did not alter joint erosion scores [Citation69]. In contrast, an exploratory study, which evaluated radiographic changes following intensive sUA-lowering therapy with pegloticase in patients with severe chronic gout, found that sustaining sUA levels <1 mg/dL for 1 year resulted in improvement in structural changes, particularly bone erosion, suggesting that some structural damage may be reversible [Citation68]. A key difference in these studies is the degree of uric acid suppression by different medical regimens, and this is the likely explanation for the different outcomes.
Although the disease is well understood, with good treatment options available, gout is often poorly managed [Citation28,Citation70]. Challenges of treating and managing gout include physician adherence to recommended guidelines when prescribing, and patient adherence to their prescribed treatment regimens [Citation71,Citation72]. In a retrospective study of 9482 patients with gout enrolled in managed care, only 18% of patients who filled at least two prescriptions were compliant with their allopurinol therapy throughout a 2-year study period [Citation73]. Another study found that of 4166 patients who initiated ULT for gout, more than 50% were nonadherent with their treatment, particularly younger patients (<50 years of age), patients with fewer comorbidities, and those with no visits to a healthcare provider prior to gout [Citation72]. These findings point out the importance of proactive education of patients about their disease [Citation74]. Patient education has been found to improve adherence, reduce the number of attacks, and improve the number of patients who reach sUA target levels [Citation74]. Rheumatologists and PCPs have also been found to have less than optimal overall adherence to treatment guidelines, particularly for first-line ULT and length of prophylactic treatment [Citation71,Citation75]. For example, a comprehensive quantitative survey that assessed adherence of US PCPs (n = 120) and rheumatologists (n = 71) to the ACR guidelines found that 53.7% of PCPs and 35.3% of rheumatologists had low adherence to treatment recommendations (defined as following ≤4 out of 8 ACR treatment recommendations), and even among highly adherent PCPs and rheumatologists, only 36.4% and 35.2%, respectively, were adherent to the initial ULT dose [Citation71].
Conclusions
Gout is a chronic, pervasive disease characterized by underlying hyperuricemia that results in recurrent flares and MSU crystal deposition. Even when patients are asymptomatic and free from flares, ongoing inflammation and subsequent damage occur locally at the joint and systemically [Citation26,Citation76]. Several guidelines recommend targets for sUA levels <6 mg/dL to prevent MSU crystal deposition, reduce the overall volume of crystals in the body, and dissolve tophi [Citation31,Citation53,Citation56–Citation58,Citation77]. Halting crystal formation and hastening dissolution of existing crystals are key to preventing the long-term negative effects of gout on a patient’s health and quality of life [Citation2,Citation52]. Importantly, only by keeping the sUA level <6 mg/dL can disease progression be halted and therapeutic outcomes improved [Citation13,Citation59,Citation64,Citation65,Citation78,Citation79]. While hyperuricemia is defined at sUA >6.8 mg/dL, the target goal of <6 mg/dL provides a necessary margin to mitigate the effects of fluctuations in factors that can influence the concentration and solubility of sUA. Physician and patient education is also key for properly managing the disease to ensure medication compliance and regular (6–12 months) monitoring of sUA levels. About two-thirds of gout patients in the USA are diagnosed and managed by PCPs [Citation80,Citation81]. Hence, these healthcare professionals can make a significant impact on treating this disease by ensuring that after gout diagnosis more patients receive optimal care [Citation28]. With proper titration and dosing of medication, regular sUA monitoring, and patient compliance, gout is a curable disease [Citation59,Citation82]. The old ‘treat-to-symptoms’ approach for managing gout is in large part responsible for the historically poor management of this disease. By contrast, the ‘treat-to-target’ recommendation of guidelines from multiple professional organizations [Citation28,Citation31,Citation53–Citation55,Citation58,Citation77] is based on clear and consistent demonstration that this approach improves long-term outcomes in this common disease.
Declaration of interest
Support for this paper was provided by AstraZeneca Pharmaceuticals, and editorial assistance was provided by Charlotte Singh, MD, CMPP, and Elizabeth Goodwin, PhD, of The Lockwood Group. NL Edwards has been a consultant for Takeda, AstraZeneca, Crealta/Horizon, and CymaBay Pharmaceuticals. G Ruoff has served on the advisory boards and speakers bureaus for Takeda, Endo, and Cephalon. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- CDC. Arthritis: Gout. 2015 [cited 2015 Aug 27]. Available from: http://www.cdc.gov/arthritis/basics/gout.html.
- Saccomano SJ, Ferrara LR. Treatment and prevention of gout. Nurse Pract. 2015;40(8):24–1.
- Edwards NL. Crystal deposition diseases. In: Goldman L, Schafer AI, editors. Goldman’s cecil medicine. 24th ed. Philadelphia, PA: Elsevier Saunders; 2012. p. 1737–1743.
- McQueen FM, Reeves Q, Dalbeth N. New insights into an old disease: advanced imaging in the diagnosis and management of gout. Postgrad Med J. 2013;89(1048):87–93.
- Scott JT. New knowledge of the pathogenesis of gout. J Clin Pathol Suppl (R Coll Pathol). 1978;12:205–213.
- Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141.
- CDC. Fibromyalgia. [ cited 2015 Sep 8]. Available from: http://www.cdc.gov/arthritis/basics/fibromyalgia.htm.
- Burton W, Morrison A, Maclean R, et al. Systematic review of studies of productivity loss due to rheumatoid arthritis. Occup Med (Lond). 2006;56(1):18–27.
- Liu JT, Yeh HM, Liu SY, et al. Psoriatic arthritis: Epidemiology, diagnosis, and treatment. World J Orthop. 2014;5(4):537–543.
- Kim KY, Ralph Schumacher H, Hunsche E, et al. A literature review of the epidemiology and treatment of acute gout. Clin Ther. 2003;25(6):1593–1617.
- Edwards NL, Sundy JS, Forsythe A, et al. Work productivity loss due to flares in patients with chronic gout refractory to conventional therapy. J Med Econ. 2011;14(1):10–15.
- Strand V, Khanna D, Singh JA, et al. Improved health-related quality of life and physical function in patients with refractory chronic gout following treatment with pegloticase: evidence from phase III randomized controlled trials. J Rheumatol. 2012;39(7):1450–1457.
- Becker MA, Schumacher HR, Benjamin KL, et al. Quality of life and disability in patients with treatment-failure gout. J Rheumatol. 2009;36(5):1041–1048.
- Doherty M. New insights into the epidemiology of gout. Rheumatology (Oxford). 2009;48(Suppl 2):ii2–ii8.
- Richette P, Bardin T. Gout. Lancet. 2010;375(9711):318–328.
- Terkeltaub R. Update on gout: new therapeutic strategies and options. Nat Rev Rheumatol. 2010;6(1):30–38.
- Choi HK, Liu S, Curhan G. Intake of purine-rich foods, protein, and dairy products and relationship to serum levels of uric acid: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2005;52(1):283–289.
- Neogi T. Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452.
- Soltani Z, Rasheed K, Kapusta DR, et al. Potential role of uric acid in metabolic syndrome, hypertension, kidney injury, and cardiovascular diseases: is it time for reappraisal? Curr Hypertens Rep. 2013;15(3):175–181.
- Baker JF, Schumacher HR. Update on gout and hyperuricemia. Int J Clin Pract. 2010;64(3):371–377.
- Li R, Miao L, Qin L, et al. A meta-analysis of the associations between the Q141K and Q126X ABCG2 gene variants and gout risk. Int J Clin Exp Pathol. 2015;8(9):9812–9823.
- MacFarlane LA, Kim SC. Gout: a review of nonmodifiable and modifiable risk factors. Rheum Dis Clin North Am. 2014;40(4):581–604.
- Saag KG, Choi H. Epidemiology, risk factors, and lifestyle modifications for gout. Arthritis Res Ther. 2006;8(Suppl 1):S2.
- Wallace KL, Riedel AA, Joseph-Ridge N, et al. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31(8):1582–1587.
- Hench PS. Diagnosis and treatment of gout and gouty arthritis. JAMA. 1941;116(6):453–459.
- Perez-Ruiz F, Dalbeth N, Bardin T. A review of uric acid, crystal deposition disease, and gout. Adv Ther. 2015;32(1):31–41.
- Dalbeth N, Stamp L. Hyperuricaemia and gout: time for a new staging system? Ann Rheum Dis. 2014;73(9):1598–1600.
- Hamburger M, Baraf HS, Adamson TC 3rd, et al. Recommendations for the diagnosis and management of gout and hyperuricemia. Postgrad Med. 2011;123(6Suppl 1):3–36.
- Becker MA. Arthritis and allied conditions. 14th ed. Philadelpia, PA: Lippincott Williams & Wlikins; 2001. p. 2281–2313.
- Zhang W, Doherty M, Pascual E, et al. EULAR evidence based recommendations for gout. Part I: diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65(10):1301–1311.
- Zhang W, Doherty M, Bardin T, et al. EULAR evidence based recommendations for gout. Part II: management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65(10):1312–1324.
- Roddy E, Doherty M. Epidemiology of gout. Arthritis Res Ther. 2010;12(6):223.
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med. 1987;82(3):421–426.
- Bardin T, Richette P. Definition of hyperuricemia and gouty conditions. Curr Opin Rheumatol. 2014;26(2):186–191.
- Roddy E. Revisiting the pathogenesis of podagra: why does gout target the foot? J Foot Ankle Res. 2011;4(1):13.
- Neogi T, Jansen TL, Dalbeth N, et al. Gout classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2015;74(10):1789–1798.
- Puig JG, de Miguel E, Castillo MC, et al. Asymptomatic hyperuricemia: impact of ultrasonography. Nucleosides Nucleotides Nucleic Acids. 2008;27(6):592–595.
- Pascual E, Batlle-Gualda E, Martínez A, et al. Synovial fluid analysis for diagnosis of intercritical gout. Ann Intern Med. 1999;131(10):756–759.
- Carter JD, Kedar RP, Anderson SR, et al. An analysis of MRI and ultrasound imaging in patients with gout who have normal plain radiographs. Rheumatology (Oxford). 2009;48(11):1442–1446.
- Chowalloor PV, Siew TK, Keen HI. Imaging in gout: A review of the recent developments. Ther Adv Musculoskelet Dis. 2014;6(4):131–143.
- Martillo MA, Nazzal L, Crittenden DB. The crystallization of monosodium urate. Curr Rheumatol Rep. 2014;16(2):400.
- Terkeltaub R, Bushinsky DA, Becker MA. Recent developments in our understanding of the renal basis of hyperuricemia and the development of novel antihyperuricemic therapeutics. Arthritis Res Ther. 2006;8(Suppl 1):S4.
- Fiddis RW, Vlachos N, Calvert PD. Studies of urate crystallisation in relation to gout. Ann Rheum Dis. 1983;42(Suppl 1):12–15.
- Sarawate CA, Patel PA, Schumacher HR, et al. Serum urate levels and gout flares: analysis from managed care data. J Clin Rheumatol. 2006;12(2):61–65.
- Li-Yu J, Clayburne G, Sieck M, et al. Treatment of chronic gout. Can we determine when urate stores are depleted enough to prevent attacks of gout? J Rheumatol. 2001;28(3):577–580.
- Agudelo CA, Wise CM. Crystal-associated arthritis. Clin Geriatr Med. 1998;14(3):495–513.
- Terkeltaub R, Edwards NL. Chapter 6. In: Terkeltaub R, Edwards NL, editors. Gout: diagnosis and management of gouty arthritis and hyperuricemia. 3rd ed. Durant (OK): Professional Communications; 2013.
- Fam AG. Gout, diet, and the insulin resistance syndrome. J Rheumatol. 2002;29(7):1350–1355.
- Keenan RT, Nowatzky J, Pillinger MH. Etiology and pathogenesis of hyperuricemia and gout. In Kelley’s textbook of rheumatology, 9th ed. 2013; Philadelphia, PA: Elsevier.
- So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120(6):1791–1799.
- Grassi D, Ferri L, Desideri G, et al. Chronic hyperuricemia, uric acid deposit and cardiovascular risk. Curr Pharm Des. 2013;19(13):2432–2438.
- Perez-Ruiz F. Treating to target: a strategy to cure gout. Rheumatology (Oxford). 2009;48(Suppl 2):ii9–ii14.
- Khanna D, Fitzgerald JD, Khanna PP, et al. American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431–1446.
- Richette P, Doherty M, Pascual E, et al. [SAT0531] Updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. 2014;73(Suppl2):783.
- Sivera F, Andres M, Carmona L, et al. Multinational evidence-based recommendations for the diagnosis and management of gout: integrating systematic literature review and expert opinion of a broad panel of rheumatologists in the 3e initiative. Ann Rheum Dis. 2014;73(2):328–335.
- Yamanaka H. Revised version of guideline for the management of hyperuricemia and gout. Nihon Rinsho. 2008;66(4):643–646.
- Romeijnders AC, Gorter KJ. Summary of the Dutch college of general practitioners’ “Gout” standard. Ned Tijdschr Geneeskd. 2002;146(7):309–313.
- Jordan KM, Cameron JS, Snaith M, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology (Oxford). 2007;46(8):1372–1374.
- Perez-Ruiz F, Calabozo M, Pijoan JI, et al. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum. 2002;47(4):356–360.
- Richette P, Frazier A, Bardin T. Impact of anti-inflammatory therapies, xanthine oxidase inhibitors and other urate-lowering therapies on cardiovascular diseases in gout. Curr Opin Rheumatol. 2015;27(2):170–174.
- Gasparyan AY, Ayvazyan L, Yessirkepov M, et al. Colchicine as an anti-inflammatory and cardioprotective agent. Expert Opin Drug Metab Toxicol. 2015;11(11):1781–1794.
- Holland R, McGill NW. Comprehensive dietary education in treated gout patients does not further improve serum urate. Intern Med J. 2015;45(2):189–194.
- Fels E, Sundy JS. Refractory gout: what is it and what to do about it? Curr Opin Rheumatol. 2008;20(2):198–202.
- Schumacher HR Jr., Becker MA, Lloyd E, et al. Febuxostat in the treatment of gout: 5-yr findings of the FOCUS efficacy and safety study. Rheumatology (Oxford). 2009;48(2):188–194.
- Shoji A, Yamanaka H, Kamatani N. A retrospective study of the relationship between serum urate level and recurrent attacks of gouty arthritis: evidence for reduction of recurrent gouty arthritis with antihyperuricemic therapy. Arthritis Rheum. 2004;51(3):321–325.
- Pascual E, Sivera F. Time required for disappearance of urate crystals from synovial fluid after successful hypouricaemic treatment relates to the duration of gout. Ann Rheum Dis. 2007;66(8):1056–1058.
- McCarthy GM, Barthelemy CR, Veum JA, et al. Influence of antihyperuricemic therapy on the clinical and radiographic progression of gout. Arthritis Rheum. 1991;34(12):1489–1494.
- Dalbeth N, Doyle A, McQueen FM, et al. Exploratory study of radiographic change in patients with tophaceous gout treated with intensive urate-lowering therapy. Arthritis Care & Research. 2012;66(1):82–85.
- Dalbeth N, Saag KG, Palmer W, et al. Imaging and safety assessments following treatment with febuxostat and placebo for 2 years in subjects with early gout [abstract]. Arthritis & Rheumatism. 2015;67(Suppl 10). Abstract no. 2110.
- Edwards NL. Quality of care in patients with gout: why is management suboptimal and what can be done about it? Curr Rheumatol Rep. 2011;13(2):154–159.
- Oderda GM, Shiozawa A, Walsh M, et al. Physician adherence to ACR gout treatment guidelines: perception versus practice. Postgrad Med. 2014;126(3):257–267.
- Edwards RR, Wasan AD, Bingham CO, et al. Enhanced reactivity to pain in patients with rheumatoid arthritis. Arthritis Res Ther. 2009;11(3):R46.
- Riedel AA, Nelson M, Joseph-Ridge N, et al. Compliance with allopurinol therapy among managed care enrollees with gout: a retrospective analysis of administrative claims. J Rheumatol. 2004;31(8):1575–1581.
- Rees F, Jenkins W, Doherty M. Patients with gout adhere to curative treatment if informed appropriately: proof-of-concept observational study. Ann Rheum Dis. 2013;72(6):826–830.
- Harrold LR, Mazor KM, Negron A, et al. Primary care providers’ knowledge, beliefs and treatment practices for gout: results of a physician questionnaire. Rheumatology (Oxford). 2013;52(9):1623–1629.
- Cronstein BN, Terkeltaub R. The inflammatory process of gout and its treatment. Arthritis Res Ther. 2006;8(Suppl 1):S3.
- Zhang W, Doherty M, Leeb BF, et al. EULAR evidence based recommendations for the management of hand osteoarthritis: report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2007;66(3):377–388.
- Perez-Ruiz F, Martin I, Canteli B. Ultrasonographic measurement of tophi as an outcome measure for chronic gout. J Rheumatol. 2007;34(9):1888–1893.
- Chandratre P, Roddy E, Clarson L, et al. Health-related quality of life in gout: a systematic review. Rheumatology (Oxford). 2013;52(11):2031–2040.
- Krishnan E, Lienesch D, Kwoh CK. Gout in ambulatory care settings in the United States. J Rheumatol. 2008;35(3):498–501.
- Singh JA, Sarkin A, Shieh M, et al. Health care utilization in patients with gout. Semin Arthritis Rheum. 2011;40(6):501–511.
- Wortmann RL. Effective management of gout: an analogy. Am J Med. 1998;105(6):513–514.