
ABSTRACT
Objective: The concurrent use of an insulin sensitizer in type 2 diabetes mellitus (T2DM) patients with inadequate glycemic control on basal-bolus insulin may help improve glycemic control while limiting further insulin requirement. Bromocriptine-QR (B-QR), a quick release, sympatholytic, dopamine D2 receptor agonist therapy for T2DM, is a postprandial insulin sensitizer. This study evaluated the effect of B-QR on dysglycemia in T2DM subjects with suboptimal glycemic control on basal-bolus insulin plus metformin.
Methods: The effect of once-daily morning administration of B-QR on dysglycemia was evaluated in 60 T2DM subjects derived from the Cycloset Safety Trial, with HbA1c >7% on basal-bolus insulin plus metformin at baseline, randomized to B-QR (N = 44) versus placebo (N = 16) and completed 12 weeks of study drug treatment. The analyses also included a subset of subjects on high-dose insulin (total daily insulin dose (TDID) ≥70 units; N = 36: 27 B-QR; 9 placebo).
Results: Subjects were well matched at baseline. After 12 weeks of B-QR treatment, mean % HbA1c decreased by -0.73% relative to baseline (p < 0.001) and by -1.13 relative to placebo (p < 0.001). In the high-dose insulin subset, B-QR therapy resulted in % HbA1c reductions of -0.95 and -1.49 relative to baseline (p < 0.001) and placebo (p = 0.001) respectively. Secondary analyses of treatment effect at 24 and 52 weeks demonstrated similar influences of B-QR on HbA1c. The fasting plasma glucose (FPG) and TDID changes within each treatment group were not significant. More subjects achieved HbA1c ≤7 at 12 weeks with B-QR relative to placebo (36.4% B-QR vs 0% placebo, Fisher’s exact 2-sided p = 0.003 in the entire cohort and 37% vs 0%, 2-sided p = 0.039 in the high-dose insulin subset).
Conclusion: B-QR therapy improves glycemic control in T2DM subjects whose glycemia is poorly controlled on metformin plus basal-bolus insulin, including individuals on high-dose basal-bolus insulin. This glycemic impact occurred without significant change in FPG, suggesting a postprandial glucose lowering mechanism of action.
Cycloset Safety Trial registration: ClinicalTrials.gov Identifier: NCT00377676
1. Introduction
The natural progression of disease in type 2 diabetes mellitus (T2DM) is marked by progressive decline in pancreatic beta-cell function resulting in the need for exogenous insulin therapy in a large proportion of patients, particularly with increasing duration of the disease [Citation1]. The declining beta-cell function along with the presence of longstanding and often severe insulin resistance results in the need for exogenous basal as well as prandial insulin and often escalating doses of insulin to achieve adequate glycemic control. Chronic high-dose insulin therapy can be difficult to manage due to the potential risks of such therapy such as hypoglycemia and weight gain [Citation2–Citation7]. This sequence of disease progression with increasing insulin requirements and difficulty achieving adequate glycemic control occurs frequently even in patients on high-dose insulin therapy. The concurrent use of an insulin sensitizer in such patients whose glycemia is not adequately controlled on high-dose insulin therapy may offer a strategy to improve glycemic control while limiting further insulin requirement but safe, effective options for such agents are limited. The thiazolidinediones (TZDs) such as pioglitazone and rosiglitazone are effective insulin sensitizers but the use of these agents in insulin-treated patients can be associated with significantly increased risk of adverse effects such as congestive heart failure (CHF), weight gain, edema, and bone fractures [Citation8–Citation16]. Bromocriptine-QR (B-QR), a quick-release, high absorbing formulation of the potent dopamine D2 receptor agonist bromocriptine, approved in the United States for the treatment of T2DM, is a unique insulin sensitizer with a good safety profile [Citation17–Citation23]. Circadian-timed administration (within 2 h of waking) of this quick-release formulation of bromocriptine results in a discrete and brief daily interval of circulating bromocriptine, thereby providing a timed pulse of increased dopaminergic activity centrally at the time of day that studies suggest is the natural daily peak of central dopaminergic activity in healthy individuals [Citation24] which studies suggest is diminished in insulin-resistant individuals [Citation25,Citation26].
Based on preclinical study findings [Citation27–Citation33], it is postulated that appropriately circadian-timed B-QR administration restores the diminished circadian peak of dopaminergic activity present in insulin resistant states, to thereby improve central fuel-sensing mechanisms and insulin sensitivity [Citation34]. Available evidence from preclinical and clinical studies suggests that B-QR is a postprandial-weighted insulin sensitizer promoting glucose disposal following a meal [Citation21–Citation23,Citation35–Citation39]. Hyperinsulinemic–euglycemic clamp studies indicate that B-QR improves maximally insulin-stimulated glucose disposal [Citation21]. Moreover, consistent with B-QR’s meal-time insulin sensitizing properties, the HbA1c-lowering effect of B-QR has been shown to correlate with the postprandial insulin level [Citation26]. In a previous small open-label pilot study [Citation38], B-QR therapy in T2DM subjects with inadequate glycemic on metformin plus high-dose basal–bolus insulin resulted in a significant mean % HbA1c reduction of −1.76 with a concurrent 27% reduction in total daily insulin dose (TDID) and a 32% decrease in postprandial glucose response to a mixed-meal tolerance test with no significant change in fasting plasma glucose (FPG) level. Given the import of these findings highlighting a potential potent method of improving glycemic control in these very difficult to manage T2DM patients, in the present study, we sought to evaluate the reproducibility of the above pilot study within a double-blind, placebo-controlled study design utilizing a cohort of subjects derived from a randomized, placebo-controlled safety study of B-QR therapy in T2DM subjects (Cycloset Safety Trial [CST]) [Citation17].
2. Methods
2.1. Study subjects and design
Study subjects were derived from the CST. The total CST study population had included subjects whose T2DM was managed with either lifestyle interventions alone, oral antihyperglycemic agents (≤2), or insulin either alone or in combination with 1 oral antihyperglycemic agent. The present study is a post-hoc analysis limited to a subset (N = 60) of the CST study population that was not prespecified in the original CST study protocol and included only those CST subjects on metformin and basal plus prandial insulin therapy (including mixed insulin regimens) with HbA1c >7% at baseline, randomized to B-QR therapy (N = 44) versus placebo (N = 16) and completing 12 weeks on the study drug with a 12-week HbA1c measurement.
The study design and protocol for the CST have been previously described [Citation17]. Briefly, the CST was a 12-month, multicenter, randomized, placebo-controlled, double-blind, parallel-group safety and efficacy study in outpatient T2DM subjects between the ages of 30 and 80 years, with body mass index <43 kg/m2 and HbA1c ≤10.0%. Subjects were randomized 2:1 to B-QR therapy versus placebo added on to their baseline diabetes treatment regimen as described above. The study drug was titrated by adding 1 tablet (0.8 mg B-QR per tablet) per week until a maximum tolerated daily dose between 2 and 6 tablets (1.6–4.8 mg/day) was achieved. The study drug was taken once daily with the morning meal, within 2 h of waking. The CST study protocol was approved by site-specific or central institutional review boards and all subjects provided written informed consent to participate in the study before enrollment.
The CST protocol required that subjects maintained a stable diabetes treatment regimen for ≥30 days prior to randomization and subjects had to remain on their established baseline antihyperglycemic treatments without any changes to the regimen (including no changes to the insulin regimen type) except adjustments in the dosages of their baseline diabetes medications if needed during the first 12 weeks of the study. However, the CST protocol allowed for changes to the baseline diabetes medication regimen (including changes to the composition/type of insulin regimen and/or adding or stopping medication(s) including insulin) after week 12 of the study though without employing a specific algorithm for such adjustments. Therefore, due to the inability to accurately account for all such insulin changes occurring after the initial 12-week study period, the primary analysis for the present study evaluated the treatment effects of B-QR versus placebo from baseline to week 12 of therapy. As a secondary analysis, the treatment effects of B-QR versus placebo were analyzed at study weeks 24 and 52 (end of study), during which time changes in both insulin dose and insulin regimen were allowed. All analyses in the current study are original and different from any previously reported results from the CST.
HbA1c levels, FPG levels, and TDID were assessed at baseline and week 12 for the primary analysis and at weeks 24 and 52 for the secondary analyses. FBG and HbA1c assays were performed at a central lab. HbA1c was measured using high-performance liquid chromatography. Insulin dose information was as obtained and recorded in the case report forms collected at the baseline and weeks-12, 24 and 52 study visits.
2.2. Statistical analysis
The primary statistical analyses were performed to assess (a) between study group (B-QR vs. placebo) differences in the changes from baseline to week 12 in HbA1c, FPG, and TDID; (b) within-group changes in HbA1c, FPG, and TDID from baseline to week 12; and (c) between group comparison of percent of subjects achieving goal HbA1c of ≤7% after 12 weeks of treatment. These analyses were performed in the entire cohort of 60 subjects (44 B-QR, 16 placebo), which included all subjects on basal–bolus insulin plus metformin regardless of their baseline TDID. In addition, to more closely mimic the study subjects in the previous pilot study[Citation38] discussed above, the between-group and within-group changes in HbA1c, FPG, and TDID were analyzed in a subset of the cohort limited to subjects who were on high-dose insulin defined as TDID ≥ 70 units at baseline (N = 36:27 B-QR, 9 placebo). Further, a sensitivity analysis of the treatment effect was conducted in both the total and high-dose insulin study populations wherein the analysis was restricted to subjects with TDID change within ≤5 units/day from baseline. Secondary statistical analyses included analysis of the within and between group changes in HbA1c, FPG, and TDID at study week 24 and study week 52.
Between-group differences were analyzed using 2-tailed Student’s t-tests and within-group comparisons were analyzed using paired samples t-tests. Fisher’s exact tests were performed to evaluate the between-group differences in the percent of subjects reaching HbA1c goal of ≤7.0. Statistical analyses were performed using SPSS version 19.0 (Armonk, NY: IBM Corp). The significance level was set at p < 0.05. Data are presented as mean ± standard error of the mean (SEM) except categorical variables shown as numbers and percent.
Safety analyses were conducted as previously described for the CST [Citation17]. As per the CST study protocol, hypoglycemia was defined as characteristic symptoms of hypoglycemia with blood glucose 59 mg/dL or less or if blood glucose was not checked, prompt resolution of symptoms with treatment (food intake, subcutaneous glucagon, or intravenous glucose) or any glucose measurement of 49 mg/dL or less, with or without symptoms. The intensity of hypoglycemia was defined as severe if all three of the following criteria were met including patient being unable to treat himself/herself, exhibiting neuroglycopenic symptoms, and either a blood glucose of 49 mg/dL or less or if the blood glucose was not measured, reversal of clinical manifestations with oral carbohydrates, subcutaneous glucagon, or intravenous glucose. Events that did not meet all three criteria for severe hypoglycemia were characterized as mild–moderate.
3. Results
3.1. Baseline characteristics
The baseline characteristics of the study population are shown in . The study subjects in the B-QR and placebo treatment arms were well matched at baseline with no significant differences in average age, BMI, duration of diabetes, blood pressure, lipids, renal function, and gender or race distributions (p > 0.05 for all). There were also no significant differences between the two groups in baseline HbA1c (p = 0.40), FPG (p = 0.31), or TDID (p = 0.48). The characteristics of the subset of the study cohort limited to those on high-dose insulin defined as TDID ≥70 units at baseline were similar to the whole cohort except for higher mean TDID doses with no significant between treatment group differences within this subset (see for details).
Table 1. Baseline characteristics.
3.2. Effect of B-QR versus placebo on glycemic control in the total study population
After 12 weeks of study drug treatment, mean HbA1c decreased significantly in the B-QR treated group from 8.31 ± 0.13 at baseline to 7.58 ± 0.19 at week 12 (p < 0.001) and trended up in the placebo-treated group from 8.10 ± 0.19 at baseline to 8.5 ± 0.30 at week 12 (p = 0.14) yielding a significant between group difference in mean change in % HbA1c of −1.13 (p < 0.001) (). When stratified by gender, the between group difference in the change in HbA1c from baseline held in favor of B-QR for both males (N = 50, −1.03, p = 0.007) and females (N = 10, −1.4, p = 0.008). Similarly, the results remained significant in favor of B-QR when stratified by age for both age <60 (N = 33, −1.4, p = 0.01) and age ≥60 (N = 27, −0.85, p = 0.006). Within each treatment arm, differences in the change in % HbA1c were not statistically significantly between men versus women or age <60 versus ≥60.
Table 2. Effect of bromocriptine-QR as add-on therapy in subjects with suboptimal glycemic control (HbA1c >7%) on treatment with metformin plus basal–bolus insulin.
The within-group change in FPG from baseline to week 12 was −12.2 ± 9.1 mg/dL (from 161.8 ± 8.6 at baseline to 149.7 ± 7.8 at week 12, p = 0.19) in the B-QR group and 27.1 ± 16.1 mg/dL (from 144.8 ± 13.6 at baseline to 171.9 mg/dL ± 20.6 at week 12, p = 0.11) in the placebo group, with neither of these within-group changes being statistically significant but yielding a statistically significant between-group difference of −39.2 ± 17.9 mg/dL (p = 0.03) in favor of B-QR ().
The sensitivity analysis restricted to subjects with ≤5 units TDID change from baseline (N = 39:25 B-QR; 14 placebo) yielded similar findings as the primary analysis (). In this subset, mean % HbA1c decreased significantly in the B-QR-treated group by −0.85 from 8.07 ± 0.19 at baseline to 7.22 ± 0.18 at week 12 (p < 0.001) and trended up in the placebo-treated group by 0.43 from 8.13 ± 0.22 at baseline to 8.56 ± 0.34 at week 12 (p = 0.16), yielding a significant between-group difference in mean change in % HbA1c of −1.28 (p = 0.001) associated with B-QR therapy relative to placebo (). The within-group change in FPG from baseline to week 12 was −23.6 ± 12.2 mg/dL (from 167.5 ± 11.4 at baseline to 143.9 ± 8.0 at week 12, p = 0.06) in the B-QR group and 27.9 ± 17.98 mg/dL (from 144.5 ± 15.1 at baseline to 172.4 ± 23.6 mg/dL at week 12, p = 0.14) in the placebo group, with neither of these within-group changes being statistically significant but yielding a statistically significant between-group difference of −51.6 ± 21.1 mg/dL (p = 0.02) in favor of B-QR. There were no significant within-group changes or between-group difference in the change in TDID (see for details).
Table 3. Effect of bromocriptine-QR as add-on therapy in subjects with suboptimal glycemic control (HbA1c > 7%) on treatment with metformin plus high-dose basal–bolus insulin (total daily insulin dose ≥ 70 units/day) at baseline.
3.3. Effect of B-QR versus placebo on glycemic control in the subset of subjects on high-dose insulin (TDID ≥70 units at baseline)
When the above analyses were limited to only subjects within the study cohort who were on ≥70 units TDID at baseline (N = 36:27 B-QR; 9 placebo), the mean HbA1c decreased significantly from 8.42 ± 0.17% at baseline to 7.47 ± 0.23 (p < 0.001) in the B-QR group and trended up from 8.36 ± 0.24 at baseline to 8.90 ± 0.49 at week 12 (p = 0.2) in the placebo group, yielding a significant between-group difference in the change in HbA1c of −1.49 (p = 0.001) ().
The mean FPG change from baseline was −11 ± 10 mg/dL (from 157.9 ± 11.2 at baseline to 146.9 ± 8.7 at week 12, p = 0.30) in the B-QR group and 43 ± 27 mg/dL (from 169.2 ± 20.2 at baseline to 212.6 ± 27.9 at week 12, p = 0.1) in the placebo group with neither of these within-group changes being statistically significant but yielding a significant between-group difference of −54 mg/dL (p = 0.028) in favor of B-QR ().
The mean TDID change from baseline was not statistically significant within or between the two treatment groups (see for details).
Sensitivity analysis restricted to a subset of subjects within this high-dose insulin cohort that had ≤5 units TDID change from baseline (N = 20:13 B-QR; 7 placebo) revealed similar findings () with the mean % HbA1c decreasing significantly by −1.03 in the B-QR-treated group from 8.18 ± 0.28 at baseline to 7.15 ± 0.28 at week 12 (p < 0.001) and trending up by 0.64 in the placebo-treated group from 8.49 ± 0.29 at baseline to 9.13 ± 0.6 at week 12 (p = 0.24), yielding a significant between-group difference in the change in %HbA1c of −1.67 (p = 0.001). In this subgroup, the within-group change in FPG from baseline to week 12 was −11.3 ± 13.97 mg/dL (from 158.9 ± 17.1 at baseline to 147.6 ± 9.5 at week 12, p = 0.43) in the B-QR group and 49.7 ± 33.3 mg/dL (from 175.6 ± 24.7 at baseline to 225.3 ± 34.8 mg/dL at week 12, p = 0.19) in the placebo group, with neither of these within-group changes being statistically significant and yielding a relative between-group difference of −61.0 ± 30.7 mg/dL which was also not statistically significant (p= 0.06).
3.4. Effect of B-QR versus placebo in percent of subjects achieving goal HbA1c ≤7%
Among subjects treated with B-QR, 16 out of the 44 (36.4%) subjects achieved HbA1c ≤7% at week 12 compared to 0 of the 16 subjects in the placebo group (Fisher’s exact 2-sided p = 0.003). In the high-dose insulin subgroup, 10 out of the 27 B-QR-treated (37%) versus 0 of the 9 placebo-treated subjects achieved HbA1c ≤7% (Fisher’s exact 2-sided p = 0.039).
3.5. Secondary analyses of B-QR versus placebo treatment effect on glycemic control at study weeks 24 and 52
After 24 weeks of study drug treatment (N = 59:43 B-QR, 16 placebo), mean HbA1c decreased significantly in the B-QR-treated group from 8.27 ± 0.13 at baseline to 7.76 ± 0.25 at week 24 (p = 0.03) and trended up in the placebo-treated group from 8.10 ± 0.19 at baseline to 8.64 ± 0.29 at week 24 (p = 0.03), yielding a significant between-group difference in mean change in % HbA1c of −1.05 (p = 0.01) (, Panel A). Similar to the primary analysis at week 12, there was a non-statistically significant decrease in mean FPG from baseline in the B-QR group (−13.6 ± 9.96 mg/dL, p = 0.18) and an increase in FPG from baseline in the placebo group (23.6 ± 9.9 mg/dL, p = 0.03), yielding a statistically significant between-group difference in the mean change in FPG from baseline of −37.18 (p = 0.04). The mean TDID change was −4.5 ± 3.4 units in the B-QR group and 4.2 ± 6.9 units in the placebo group, yielding a between-group difference of −8.7 units (95% CI −22.5, 5.1) in favor of lower TDID in the B-QR group but neither of the within group changes nor the between-group difference in TDID were statistically significant.
Figure 1. Effect of B-QR vs Placebo on HbA1c in Subjects on Basal-Bolus (BB) Insulin Plus Metformin (Met).
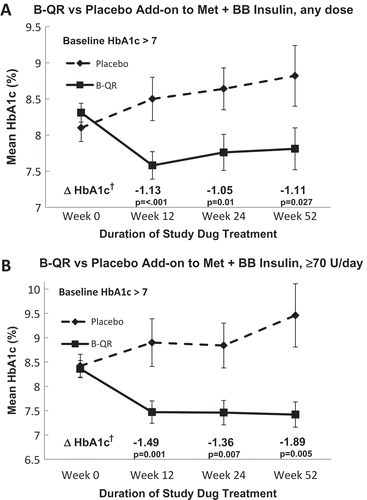
When the above analyses were limited to subjects within the study cohort who were on ≥70 units TDID at baseline and completed 24 weeks of study drug treatment (N = 36:27 B-QR; 9 placebo), the mean HbA1c decreased significantly from 8.33 ± 0.17% at baseline to 7.46 ± 0.25 (p = 0.001) in the B-QR group and trended up from 8.36 ± 0.24 at baseline to 8.84 ± 0.46 at week 24 (p = 0.19) in the placebo group, yielding a significant between-group difference in the change in % HbA1c of −1.36 (p = 0.007) (, Panel B). The change in FPG was −14.9 ± 13.2 (p = 0.27) in the B-QR group and 7.8 ± 14.1 (p = 0.6) in the placebo group, yielding a between-group difference of −22.7 ± 19.3 mg/dL, which was not statistically significant (p = 0.36). The TDID change was −3.4 ± 5 units in the B-QR group and 4.4 ± 12.3 units in the placebo, yielding a between-group difference of −7.8 ± 11.1 units (95% CI −30.4, 14.8) in favor of lower dose in the B-QR group but neither of the within group changes nor the between-group difference were statistically significant.
Among subjects completing 52 weeks of study drug treatment (N = 45: 32 B-QR, 13 placebo), mean HbA1c trended down in the B-QR treated group from 8.25 ± 0.16 at baseline to 7.81 ± 0.29 at week 52 (p = 0.13) and trended up in the placebo-treated group from 8.15 ± 0.21 at baseline to 8.82 ± 0.42 at week 52 (p = 0.045), yielding a significant between-group difference in the mean change in HbA1c from baseline of −1.11 (p = 0.027) (, Panel A). The changes in FPG from baseline to week 52 were not statistically significant within either treatment groups (−7.8 ± 11.3 mg/dL, p = 0.5 in the B-QR group and 25 ± 13.9 mg/dL, p = 0.096 in the placebo group) and the between-group difference in the mean change in FPG (−32.8 ± 19.8 mg/dL, p = 0.1) was also not statistically significant. The within-group changes and between-group difference in the change in TDID from baseline were not statistically significant (mean change in TDID 0.3 ± 5.5 units in the B-QR group; 4.2 ± 7 units in the placebo group; between-group difference −3.8 ± 9.7 units).
Among subjects in the high-dose insulin subset (≥70 units TDID at baseline) completing 52 weeks of study drug (N = 28: 21 B-QR, 7 placebo), HbA1c decreased significantly from 8.31 ± 0.2 to 7.42 ± 0.26 (p = 0.01) in the B-QR treated group and trended up in the placebo group from 8.46 ± 0.25 to 9.46 ± 0.65 (p = 0.07), yielding a significant between-group difference in the mean change in % HbA1c of −1.89 (95% CI −3.2, −0.61; p = 0.005) (, Panel B). The within-group changes in mean FPG from baseline to week 52 and the mean between-group difference in the change in FPG from baseline were not statistically significant (−17.6 ± 14.7 mg/dL, p = 0.2 in the B-QR group, 35.9 ± 15.9 mg/dL, p = 0.07 in the placebo group, between-group difference −53.4 ± 27.2 mg/dL, p = 0.06). The within-group changes and between-group difference in the change in mean TDID were not statistically significant (mean change in TDID 1.1 ± 7.6 units in the B-QR group; 0.29 ± 12.5 units in the placebo group, between-group difference 0.86 ± 15 units).
3.6. Safety
There was no significant difference in adverse events associated with B-QR therapy compared to placebo. Episodes of hypoglycemia were infrequent and mild–moderate in severity. Between baseline and study week 12, 4 out of the 44 subjects (9%) treated with B-QR had one episode of mild–moderate hypoglycemia each compared to 2 out of the 16 subjects (12.5%) treated with placebo with one episode each of mild–moderate hypoglycemia during the 12-week study period. Between study weeks 12 and 24, there were 2 episodes of mild hypoglycemia in the B-QR group and one episode of hypoglycemia, considered moderate, in the placebo group. From 24 to 52 weeks, there were 3 episodes of mild–moderate hypoglycemia in the B-QR group, but 2 of these were associated with other reasons for hypoglycemia, including one subject who developed mild hypoglycemia after having taken AM insulin dose while fasting and another patient with an episode of moderate hypoglycemia in the setting of having not eaten the night before. Overall, between baseline to week 52, there were a total of 9 episodes of hypoglycemia occurring in 8 subjects in the B-QR group, but with 2 of the episodes likely due to other reasons as described above and a total of 3 episodes in 2 subjects in the placebo group. There were no severe/serious episodes of hypoglycemia in either treatment group.
4. Discussion
The results of this study demonstrate that in T2DM subjects whose glycemia was poorly controlled on metformin plus basal–bolus insulin therapy, the addition of B-QR resulted in significant improvement of glycemic control (−1.13 reduction in HbA1c with B-QR relative to placebo [p < 0.001]; –0.73 reduction in HbA1c from baseline of 8.3% within the B-QR group [p < 0.001]). This B-QR effect was even more pronounced in subjects whose glycemia was poorly controlled on high dose (≥70 U/day) basal–bolus insulin therapy (−1.49 reduction in HbA1c with B-QR relative to placebo [p = 0.001]; −0.95 reduction in HbA1c from baseline of 8.4% within the B-QR group [p < 0.001]). Moreover, 36% of B-QR treated versus 0% of placebo-treated subjects achieved an HbA1c ≤7.0. No significant within or between-group differences were observed in the change from baseline in TDID. Importantly, a sensitivity analysis restricted to individuals with minimal insulin dose change during the study reaffirmed the study findings in the total study population (both in the any baseline TDID and baseline TDID ≥70 units/day groups). The findings of this randomized, double-blind, placebo-controlled study corroborate and are in agreement with those of a previous open-label, pilot study of B-QR therapy in T2DM subjects on metformin plus high-dose basal–bolus insulin [Citation38]. Furthermore, consistent with previous studies of B-QR therapy impact on glycemic control [Citation23,Citation38,Citation39], the improvement in HbA1c observed within B-QR-treated subjects in this study cannot be fully attributed to a reduction in fasting glucose levels (−0.73 and −0.95 percentage points reductions from baseline in HbA1c, p < 0.001 for both with −12 and −11 mg/dL reduction from baseline in mean FPG, p = NS for the any baseline TDID and ≥70 units/day baseline TDID groups, respectively) (see for details). In the previous pilot study mentioned above, the HbA1c reduction with B-QR was coupled with no significant change in FPG levels but a 32% reduction in postprandial blood glucose response to a mixed-meal tolerance test. While postprandial blood glucose levels were not available for analysis in the present study, the similar pattern of significant HbA1c lowering without significant change in fasting glucose levels within the B-QR-treated group suggests that the HbA1c reduction may be mostly driven by lowering of postprandial blood glucose levels.
The preponderance of available evidence from preclinical and clinical mechanistic studies indicates that a majority (but not the total) of the improvement in glycemic control observed with B-QR therapy in T2DM subjects results from a drug-induced improvement in postprandial glucose metabolism and hyperglycemia [Citation21–Citation23,Citation35–Citation39]. Interestingly, this B-QR effect on postprandial hyperglycemia is expressed at meal times across the day [Citation22,Citation23] and well after (e.g. 24 h later) the short pulsed duration of bromocriptine into the circulation following its circadian-timed morning administration [Citation37,Citation38], which supports the preclinical data suggesting a ‘resetting’ of aberrant neuroendocrine responsiveness to meal-time feeding to improve postprandial insulin sensitivity with this therapy [Citation27–Citation36]. Mechanistic insights into this metabolic phenomenon can be gained from a series of neurophysiological studies that established a facilitatory role for the circadian peak in dopaminergic input activity to the body pacemaker clock system (suprachiasmatic nuclei [SCN]) in the maintenance of normal insulin sensitivity and glucose tolerance in animal models of insulin resistance [Citation26–Citation34] described as follows.
The normal circadian peak in neuronal dopaminergic input activity to the SCN is diminished in insulin-resistant states and selectively reducing such peak activity in normal animals by targeted biochemical disruption of dopaminergic function at the SCN induces the insulin-resistant and glucose-intolerant condition [Citation26,Citation32]. Moreover, high fat feeding that induces insulin resistance/glucose intolerance results in a marked reduction in the circadian peak of dopaminergic activity at the SCN [Citation40]. Importantly, in animals made insulin resistant by such high fat feeding, a 1-min administration of dopamine to the SCN made once daily for a 2-week period at the time of day that dopamine activity peaks at the SCN of normal insulin-sensitive animals reverses the insulin resistance/glucose intolerance while animals are maintained on the high fat diet [Citation27]. Such circadian-timed dopamine treatment at the SCN reduces abnormally elevated noradrenergic (NA) input activity to the ventromedial hypothalamus (VMH) and the paraventricular nuclei (PVN) and is coupled to decreases in elevated neuropeptide Y (NPY) and corticotropin releasing hormone (CRH) at the PVN [Citation25–Citation28,Citation31], two neurophysiological conditions precipitating the insulin resistance syndrome [Citation25,Citation26,Citation28,Citation30,Citation33,Citation41–Citation43] in part by inducing overactivation of sympathetic tone and hypothalamic–pituitary axis (HPA) drive to the viscera and vasculature that potentiate the syndrome [Citation43–Citation47]. That is, a diminution of the circadian peak dopaminergic activity at the SCN is a neuromodulatory signal that initiates output signals from the SCN to other hypothalamic nuclei (e.g. VMH and PVN) programing them to increase sympathetic drive to the liver and adipose, increase HPA activity and alter neuroendocrine functions (increase glucagon secretion, increase plasma leptin levels and resistance) that favor increased hepatic glucose production and lipid synthesis, adipose lipolysis, hyperinsulinemia and decreased peripheral uptake of glucose (in part from increased triglyceride storage in muscle), ultimately leading to insulin resistance without a requirement for any change in food consumption (reviewed in Refs. [25, 26]).
Likewise, circadian-timed administration of the dopamine agonist, bromocriptine (either systemically or intracerebroventricularly) to insulin resistant rodents at a time of day precedent to the circadian peak in dopaminergic activity at the hypothalamic clock (SCN) center observed in normal insulin-sensitive animals (and which is diminished in insulin resistant states as mentioned above) improves insulin resistance/glucose intolerance particularly insulin-mediated glucose disposal assessed during a hyperglycemic–euinsulinemic clamp (postprandial glucose environment) [Citation28–Citation31,Citation35]. Such circadian-timed bromocriptine treatment also reduces abnormally elevated NA and serotonergic (S) input activity to the VMH [Citation28,Citation31] and CRH and NPY activities at the PVN [Citation30], which are all neuromodulatory activities that, as described above, act to potentiate insulin resistance and glucose intolerance [Citation28–Citation31,Citation41–Citation43]. In particular, elevated VMH NA input activity functions to attenuate appropriate glucose and free fatty acid nutrient sensing at meal time [Citation34] that in turn potentiates postprandial insulin resistance and reduced glucose disposal [Citation48–Citation50]. Furthermore, such metabolic effects of increased NA input activity to the VMH can be exacerbated by concurrent increased serotonergic input activity to the VMH [Citation42].
The simple rationale of combining the administration of an insulin sensitizer with insulin is an effort to simultaneously treat two main pathologies of T2DM, namely insulin resistance and deficient plasma insulin levels, as has been previously reviewed [Citation51]. While the rationale of such a combination is straightforward respecting optimizing glucose control in T2DM patients with inadequate glycemic control on basal–bolus insulin plus metformin, the options currently available for an effective and safe insulin sensitizer are very limited. The use of TZDs, the only other currently available option for an insulin sensitizer, is limited by safety concerns particularly edema and heart failure leading to the Food and Drug Administration including a warning in the prescription information for rosiglitazone and pioglitazone [Citation14–Citation16]. Inasmuch as improving insulin resistance with TZD therapy is accompanied with increased risk of bone fracture, edema, weight gain, and CHF [Citation8–Citation16] and its effects on edema and CHF may be further exacerbated among insulin-treated T2DM subjects [Citation8,Citation13–Citation16], safe and effective therapies are needed in this T2DM patient population, particularly those patients on high-dose insulin with few treatment options to improve insulin action and hyperglycemia whose diabetes is difficult to manage. The combination of B-QR with basal–bolus insulin represents a rather unique aspect of this therapeutic complementary strategy in that B-QR therapy appears to induce improvement primarily in postprandial responsiveness to insulin as discussed above and this effect is combined with a meal-time insulin treatment to improve postprandial dysglycemia. It should be appreciated, however, that other meal-time, insulin-independent, effects of circadian-timed B-QR therapy may also be operative in its effects to reduce postprandial hyperglycemia, such as affecting glucose-mediated glucose disposal, gastric absorption, reduction of elevated diurnal plasma prolactin level, and/or leptin action [Citation25,Citation26,Citation28]. The results of this small study suggest that a larger, corroborative trial of B-QR impact on glycemic control in T2DM subjects on metformin plus basal/bolus insulin is warranted.
Insulin resistance, postprandial dysglycemia, postprandial hyperlipidemia, and elevated sympathetic nervous system activity are each risk factors for cardiovascular disease (CVD) [Citation44–Citation46,Citation52–Citation58]. As discussed above, circadian-timed B-QR therapy improves insulin sensitivity and postprandial dysglycemia. Circadian-timed bromocriptine therapy also reduces elevated postprandial lipids, sympathetic tone, inflammation, and endothelial nitric oxide synthase uncoupling [Citation22,Citation26,Citation28,Citation59], all major risk factors for CVD [Citation44–Citation46,Citation52–Citation54,Citation58]. B-QR therapy has been associated with a 40–55% CVD hazard ratio rate reduction in T2DM subjects in the CST [Citation17,Citation18] potentially via its influences to reduce insulin resistance, postprandial dysglycemia hyperlipidemia, and elevated sympathetic tone. Though no specific data are presently available regarding B-QR therapy’s impact on CVD outcomes in insulin-requiring T2DM subjects, available evidence suggests that this is an investigation worthy of attention as CVD is a major pathology in such patients.
The limitations of this study include the relatively short 12-week duration, lack of measures of beta-cell function and insulin action to assess mechanisms involved in the B-QR effect and the lack of a prespecified treatment algorithm for adjusting the insulin doses during the study to assess B-QR impact on daily insulin requirement concurrent with impact on dysglycemia. However, a sensitivity analysis limiting the study population to those subjects that had minimal (<5 units) change in their TDID reaffirmed the study findings within the total study population. These study findings corroborate with a high degree of similarity, the findings of a pilot investigation of B-QR’s effect on glycemic control in a similar study population. These findings collectively strongly suggest that further investigation of B-QR therapy in a larger study population of T2DM subjects whose dysglycemia is poorly controlled on metformin plus (high-dose) basal–bolus insulin therapy to evaluate its impact on glycemic control, insulin sparing effect, beta-cell function, and insulin action is warranted. Such B-QR therapy may represent a safe and effective means of improving glycemic control in this T2DM patient population whose dysglycemia is difficult to manage but the evidence however is limited and further large trials are required to evaluate/confirm this effect and such trials are warranted.
5. Conclusion
B-QR therapy for a 12–52-week period in T2DM subjects whose glycemia was poorly controlled on metformin plus basal–bolus insulin, including those subjects on daily high-dose insulin (≥70 units/day), improved glycemic control relative to placebo.
Declaration of interest
B Chamarthi is a consultant and part-time employee of VeroScience LLC. AH Cincotta serves as the President and Chief Scientific Officer and is a shareholder of VeroScience LLC. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- Ferrannini E, Gastaldelli A, Miyazaki Y, et al. Beta-cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab. 2005;90:493–500.
- Khunti K, Davies M, Majeed A, et al. Hypoglycemia and risk of cardiovascular disease and all-cause mortality in insulin-treated people with Type 1 and Type 2 diabetes: a cohort study. Diabetes Care. 2015;38:316–322.
- UK Hypoglycaemia Study Group. Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia. 2007;50:1140–1147.
- McNay EC, Teske JA, Kotz CM, et al. Long-term, intermittent, insulin-induced hypoglycemia produces marked obesity without hyperphagia or insulin resistance: a model for weight gain with intensive insulin therapy. Am J Physiol Endocrinol Metab. 2013;304:E131–8.
- Pontiroli AE, Miele L, Morabito A. Increase of body weight during the first year of intensive insulin treatment in type 2 diabetes: systematic review and meta-analysis. Diabetes Obes Metab. 2011;13:1008–1019.
- Russell-Jones D, Khan R. Insulin-associated weight gain in diabetes–causes, effects and coping strategies. Diabetes Obes Metab. 2007;9:799–812.
- Ross SA, Tildesley HD, Ashkenas J. Barriers to effective insulin treatment: the persistence of poor glycemic control in type 2 diabetes. Curr Med Res Opin. 2011;27(Suppl3):13–20.
- Yki-Järvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118.
- Chaggar PS, Shaw SM, Williams SG. Review article: thiazolidinediones and heart failure. Diab Vasc Dis Res. 2009;6:146–152.
- Horita S, Nakamura M, Satoh N, et al. Thiazolidinediones and edema: recent advances in the pathogenesis of thiazolidinediones-induced renal sodium retention. PPAR Res. 2015;2015:1–7.
- Aubert RE, Herrera V, Chen W, et al. Rosiglitazone and pioglitazone increase fracture risk in women and men with type 2 diabetes. Diabetes Obes Metab. 2010;12:716–721.
- Billington EO, Grey A, Bolland MJ. The effect of thiazolidinediones on bone mineral density and bone turnover: systematic review and meta-analysis. Diabetologia. 2015;58:2238–2246.
- Clar C, Royle P, Waugh N. Adding pioglitazone to insulin containing regimens in type 2 diabetes: systematic review and meta-analysis. PLoS One. 2009;4:e6112.
- Scheen AJ. Combined thiazolidinedione-insulin therapy: should we be concerned about safety? Drug Saf. 2004;27:841–856.
- Actos (pioglitazone hydrochloride) prescribing information [online]. cited 2017 Jan. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021073s043s044lbl.pdf; http://www.actos.com/pi.pdf
- Avandia (rosiglitazone maleate) prescribing information [online] . cited 2017 Jan. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021071s034lbl.pdf; www.avandia.com
- Gaziano JM, Cincotta AH, O’Connor CM, et al. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care. 2010;33:1503–1508.
- Gaziano JM, Cincotta AH, Vinik A, et al. Effect of bromocriptine-QR (a quick-release formulation of bromocriptine mesylate) on major adverse cardiovascular events in type 2 diabetes subjects. J Am Heart Assoc. 2012;1:e002279.
- Vinik AI, Cincotta AH, Scranton RE, et al. Effect of bromocriptine-QR on glycemic control in subjects with uncontrolled hyperglycemia on one or two oral anti-diabetes agents. Endocr Pract. 2012;18:931–943.
- Defronzo RA. Bromocriptine: a sympatholytic, d2-dopamine agonist for the treatment of type 2 diabetes. Diabetes Care. 2011;34:789–794.
- Pijl H, Ohashi S, Matsuda M, et al. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care. 2000;23:1154–1161.
- Cincotta AH, Meier AH, Cincotta JM. Bromocriptine improves glycaemic control and serum lipid profile in obese Type 2 diabetic subjects: a new approach in the treatment of diabetes. Expert Opin Investig Drugs. 1999;8:1683–1707.
- Scranton R, Cincotta A. Bromocriptine–unique formulation of a dopamine agonist for the treatment of type 2 diabetes. Expert Opin Pharmacother. 2010;11:269–279.
- Monti JM, Monti D. The involvement of dopamine in the modulation of sleep and waking. Sleep Med Rev. 2007;11:113–133.
- Cincotta A. Hypothalamic role in the insulin resistance syndrome. In: Hansen B, Shaffrir E, eds. Resistance and Insulin Resistance Syndrome. London: Taylor and Francis; 2002. p. 271–312.
- Raskin P, Cincotta AH. Bromocriptine-QR therapy for the management of type 2 diabetes mellitus: developmental basis and therapeutic profile summary. Expert Rev Endocrinol Metab. 2016;11:113–148.
- Luo S, Ezrokhi M, Yang L, et al. Circdian dopamine activity at the biological clock regulates insulin sensitivity via the hypothalamus [abstract]. Diabetes. 2016;65(Suppl1):A489.
- Ezrokhi M, Luo S, Trubitsyna Y, et al. Neuroendocrine and metabolic components of dopamine agonist amelioration of metabolic syndrome in SHR rats. Diabetol Metab Syndr. 2014;6:104.
- Cincotta AH, MacEachern TA, Meier AH. Bromocriptine redirects metabolism and prevents seasonal onset of obese hyperinsulinemic state in Syrian hamsters. Am J Physiol. 1993;264:E285–93.
- Bina KG, Cincotta AH. Dopaminergic agonists normalize elevated hypothalamic neuropeptide Y and corticotropin-releasing hormone, body weight gain, and hyperglycemia in ob/ob mice. Neuroendocrinology. 2000;71:68–78.
- Luo S, Meier AH, Cincotta AH. Bromocriptine reduces obesity, glucose intolerance and extracellular monoamine metabolite levels in the ventromedial hypothalamus of Syrian hamsters. Neuroendocrinology. 1998;68:1–10.
- Luo S, Luo J, Meier A. Dopaminergic neurotoxin administration to the area of the suprachiasmatic nuclei induces insulin resistance. Neuroreport. 1997;8:3495–3499.
- Luo S, Luo J, Cincotta AH. Suprachiasmatic nuclei monoamine metabolism of glucose tolerant versus intolerant hamsters. NeuroReport. 1999;10:2073–2077.
- Luo M, Ezrokhi M, Trubitsyna Y, et al. Intrahypothalamic circuitry regulating hypothalamic fuel sensing to induce insulin sensitivity or insulin resistance [abstract]. Diabetologia. 2008;51(Suppl1):S59.
- Ezrokhi M, Luo S, Trubitsyna Y, et al. Weighted effects of bromocriptine treatment on glucose homeostasis during hyperglycemic versus euglycemic clamp conditions in insulin resistant hamsters: bromocriptine as a unique postprandial insulin sensitizer. J Diab Metab. 2012;S2:007.
- Moore MC, Smith M, Farmer B, et al. Timed daily bromocriptine mesylate (BC) administration improves glucose disposal in a canine diet-induced model of impaired glucose tolerance [abstract]. Diabetologia. 2014;57(Suppl1):S350.
- Cincotta AH, Meier AH. Bromocriptine (Ergoset) reduces body weight and improves glucose tolerance in obese subjects. Diabetes Care. 1996;19:667–670.
- Roe ED, Chamarthi B, Raskin P. Impact of bromocriptine-QR therapy on glycemic control and daily insulin requirement in type 2 diabetes mellitus subjects whose dysglycemia is poorly controlled on high-dose insulin: a pilot study. J Diabetes Res. 2015;2015:834903.
- Florez H, Scranton R, Farwell WR, et al. Randomized clinical trial assessing the efficacy and safety of bromocriptine-qr when added to ongoing thiazolidinedione therapy in patients with type 2 diabetes mellitus. J Diabetes Metab. 2011;2:142.
- Luo S, Zhang Y, Ezrokhi M, et al. High-Fat feeding abolishes the insulin-sensitizing peak in circadian dopamine activity at the biological clock [abstract]. Diabetes. 2014;63(Suppl1):A470.
- Cincotta AH, Luo S, Zhang Y, et al. Chronic infusion of norepinephrine into the VMH of normal rats induces the obese glucose-intolerant state. Am J Physiol Regulatory Integrative Comp. Physiol. 2000;278:R435–44.
- Luo S, Luo J, Cincotta AH. Chronic ventromedial hypothalamic infusion of norepinephrine and serotonin promotes insulin resistance and glucose intolerance. Neuroendocrinology. 1999;70:460–465.
- Zheng H, Liu X, Li Y, et al. Attenuated dopaminergic tone in the paraventricular nucleus contributing to sympathoexcitation in rats with Type 2 diabetes. Am J Physiol Regul Integr Comp Physiol. 2014;306:R138–48.
- Tentolouris N, Liatis S, Katsilambros N. Sympathetic system activity in obesity and metabolic syndrome. Ann N Y Acad Sci. 2006;1083:129–152.
- Thorp AA, Schlaich MP. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res. 2015;2015:341583.
- Lambert GW, Straznicky NE, Lambert EA, et al. Sympathetic nervous activation in obesity and the metabolic syndrome–causes, consequences and therapeutic implications. Pharmacol Ther. 2010;126:159–172.
- Chiodini I, Adda G, Scillitani A. Cortisol secretion in patients with type 2 diabetes: relationship with chronic complications. Diabetes Care. 2007;30:83–88.
- Lam TK, Pocai A, Gutierrez-Juarez R, et al. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med. 2005;11:320–327.
- Pocai A, Lam TK, Obici S, et al. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest. 2006;116:1081–1091.
- Jordan SD, Könner AC, Brüning JC. Sensing the fuels: glucose and lipid signaling in the CNS controlling energy homeostasis. Cell Mol Life Sci. 2010;67:3255–3273.
- Raskin P. Why insulin sensitizers but not secretagogues should be retained when initiating insulin in type 2 diabetes. Diabetes Metab Res Rev. 2008;24:3–13.
- Bell DSH. Focusing on cardiovascular disease in type 2 diabetes mellitus: an introduction to bromocriptine-QR. Postgradmed. 2012;124:121–135.
- Cersosimo E, DeFronzo RA. Insulin resistance and endothelial dysfunction: the road map to cardiovascular diseases. Diabetes Metab Res Rev. 2006;22:423–436.
- Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12:144–153.
- O’Keefe JH, Bell DS. Postprandial hyperglycemia/hyperlipidemia (postprandial dysmetabolism) is a cardiovascular risk factor. Am J Cardiol. 2007;100:899–904.
- Garber AJ. Postprandial dysmetabolism and the heart. Heart Fail Clin. 2012;8:563–573.
- Node K, Inoue T. Postprandial hyperglycemia as an etiological factor in vascular failure. Cardiovasc Diabetol. 2009;8:23.
- Grassi G. Sympathetic overdrive and cardiovascular risk in the metabolic syndrome. Hypertens Res. 2006;29:839–847.
- Ezrokhi M, Trubitsyna Y, Luo S, et al. Timed dopamine agonist treatment ameliorates both vascular nitrosative/oxidative stress pathology and aortic stiffness in arteriosclerotic, hypertensive SHR rats [abstract]. Diabetes. 2010;59(Suppl1):A67.