
ABSTRACT
Osteoarthritis (OA) is a common difficult-to-treat condition where the goal, in the absence of disease-modifying treatments, is to alleviate symptoms such as pain and loss of function. Acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and opioids are common pharmacologic treatments for OA. Antibodies directed against nerve growth factor (NGF-Abs) are a new class of agents under clinical investigation for the treatment of OA. This narrative review describes (and uses schematics to visualize) nociceptive signaling, chronification of pain, and the mechanisms of action (MOAs) of these different analgesics in the context of OA-related pain pathophysiology. Further, the varying levels of efficacy and safety of these agents observed in patients with OA is examined, based on an overview of published clinical data and/or treatment guidelines (when available), in the context of differences in their MOAs.
1 Introduction
Osteoarthritis (OA)-related chronic pain is typically associated with deterioration of physical function and is a leading cause of global disability [Citation1]. Current OA treatment focuses on alleviating pain and improving physical function through a combination of non-pharmacological (e.g. exercise, weight management), and pharmacological approaches [Citation2–6]. Pharmacological therapy is a mainstay of OA treatment and the most commonly utilized agents include nonsteroidal anti-inflammatory drugs (NSAIDs), acetaminophen (paracetamol), and opioids [Citation2–12] (). Due to the difficulty in effectively managing OA-related pain, and since OA-related pain can manifest in multiple joints, there is a need for new treatment options and several novel systemic therapies are in various stages of development [Citation2,Citation3]. Antibodies directed against nerve growth factor (NGF-Abs), for example, have been shown to improve pain and physical function in late-stage clinical studies and are an emerging class of therapy for moderate-to-severe OA [Citation2,Citation3,Citation13,Citation14].
Table 1. Use of acetaminophen, NSAIDs, and opioids in the treatment of OA
Effective management of chronic pain is aided by an understanding of the underlying analgesic mechanism of action (MOA) of the agent(s) being utilized. This review summarizes the analgesic MOA of NSAIDs, acetaminophen, and opioids (the most common OA analgesics used currently), and of NGF-Abs (an emerging class of systemic therapy). The efficacy and safety of these agents in patients with OA (based on published treatment guidelines when available) is also examined.
2 Methods
A narrative approach was utilized due to the broad scope of the review. PubMed searches relating to pain signaling, chronification of pain, OA pain pathophysiology, NSAID MOA, acetaminophen MOA, opioid MOA, and NGF-Abs MOA were used in combination with the authors’ familiarity with relevant publications on these topics. For , phase 2/3, active-controlled clinical trials of NGF-Abs were identified via a systematic search of PubMed on 16 September 2020 for publications (in English) with the following terms in the title: ‘tanezumab,’ ‘RN624ʹ, ‘fasinumab,’ ‘REGN475ʹ, ‘fulranumab,’ or ‘JNJ-42160443ʹ. Any publication reporting a phase 2 or phase 3 OA trial with an NGF-Ab monotherapy treatment arm and an active comparator arm was included in the .
Table 2. Summary of active-controlled phase 2 and phase 3 clinical trials of NGF-Abs for the treatment of OA pain
3 Basic overview of nociceptive signaling
A basic understanding of nociceptive signaling is shown in . Cell bodies of primary nociceptive afferents (i.e. nociceptors) located in the dorsal root ganglion (DRG) have axons that bifurcate, projecting to peripheral structures and to the dorsal horn of the spinal cord. Peripheral activation of nociceptors by high-intensity stimuli (e.g. mechanical, thermal) or by mediators released in response to tissue injury/inflammation initiates a signal, propagated via fast conducting thin myelinated Aδ fibers (conveying the initial sharp pain sensation) or slow conducting unmyelinated C fibers (conveying a dull throbbing aching sensation), to the dorsal horn [Citation19,Citation20]. In the dorsal horn, nociceptors release a variety of neurotransmitters (e.g. glutamate, substance P, calcitonin gene-related peptide [CGRP], brain-derived growth factor [BDNF]) that activate second-order afferent neurons [Citation19-22]. A population of these secondary afferents projects to the thalamus, while parallel projections relay signals to medullary, pontine, and mesencephalic structures [Citation19]. In the lateral aspects of the thalamus, these ascending projections synapse with tertiary afferent neurons that project to the somatosensory cortex to provide the sensory discriminative aspects of a pain phenotype (i.e. ‘I hurt ‘here’ on a scale of 1–10ʹ) [Citation19]. Parallel pathways project into the brainstem and into more medial thalamic regions. Those pathways projecting into the medial thalamus synapse with tertiary neurons that project to areas associated with the limbic forebrain (anterior cingulate cortex, insular cortex, prefrontal cortex). These brainstem projections synapse with neurons projecting into a variety of sites including the hippocampus, amygdala, hypothalamus, basal ganglia, and cerebellum. Many of these higher order projections constitute a functional entity referred to jointly as the limbic forebrain, which are involved in the emotional, cognitive, motivational, and behavioral components of the pain experience and are considered to be responsible for the affective component of a chronic pain state [Citation19,Citation23].
Figure 1. Basic overview of nociceptive signaling. See text for details. LC = locus coeruleus; PAG = periaqueductal gray; RVM = rostral ventromedial medulla
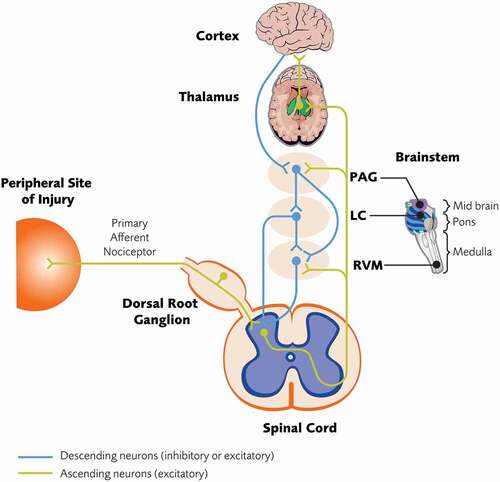
The dorsal horn is a key structure in the processing and modulation of pain signals. Second-order neurons in the dorsal horn respond to high-intensity nociceptive afferent input (marginal neurons) or to both nociceptive and non-nociceptive afferent input (wide dynamic range neurons) [Citation19]. In both cases, the encoding of stimulus intensity by these projection neurons largely depends on their discharge rate, with higher discharge rates associated with a more intense (i.e. painful) signal. However, the output function of the dorsal horn is subject to modulation via bulbospinal projections arising from brainstem structures (e.g. rostral ventromedial medulla [RVM] and locus coeruleus [LC]) that can variously serve to augment (i.e. descending facilitation) or down regulate (i.e. descending inhibition) dorsal horn excitability to modulate the firing rate of these second-order neurons [Citation20,Citation24].
4 Types of pain
Pain may be classified into different types based on its etiology. Inflammatory nociceptive pain arises from tissue injury, and/or local inflammation secondary to products release by local injury, and consequent activation of nociceptors innervating internal organs (i.e. visceral pain) or peripheral structures such as muscle, bone, and skin (i.e. somatic pain) [Citation25]. Neuropathic pain results from lesion to, or a disease affecting, the somatosensory nervous system itself and may result from direct injury (e.g. section, compression), toxic chemicals (i.e. chemotherapy), metabolic insults (i.e. diabetes), viral infections, and a myriad of autoimmune disorders mediated through innate and adaptive immune signaling [Citation26-28]. Pain may also be classified as nociplastic (i.e. pain arising from altered nociception despite no clear evidence of actual or threatened tissue damage to cause activation of peripheral nociceptors or evidence for disease or lesion of the somatosensory system) [Citation25]. Patients often present with more than one type of pain, making it challenging to differentiate underlying mechanisms [Citation29].
5 Chronification of pain
The reported magnitude of an acute pain state reflects, in part, the extent of tissue injury or inflammation. This acute pain state often resolves as the injury state resolves/heals. In some instances, however, a chronic pain state typically characterized by exaggerated responses to otherwise innocuous (i.e. allodynia) or mildly noxious (i.e. hyperalgesia) stimuli may persist after the underlying injury has apparently healed. Our understanding of the transition from an acute to a chronic pain state is limited, but is thought to involve inflammatory nociceptive cascades and the development of a neuropathic-like phenotype [Citation30].
The transition from an acute inflammatory nociceptive state to a post-inflammatory neuropathic-like phenotype likely involves several processes. One potentially important process may be increased nervous system excitability at the level of peripheral nociceptors (i.e. peripheral sensitization) and/or neurons of nociceptive pathways within the dorsal horn or neuraxis (i.e. central sensitization) [Citation31,Citation32]. Acute peripheral sensitization results from acute inflammation and subsequent release of biochemical mediators (e.g. bradykinin, H+, adenosine triphosphate [ATP], histamine, chemokines, cytokines, damage-associated molecular patterns [DAMPs], prostaglandins, growth factors) that act at the peripheral terminals of nociceptors and trigger intracellular signaling events resulting in modulation of ion channel activity, depolarization, and, thus, nociceptor sensitization [Citation19,Citation33]. Central sensitization can result from continuous high-frequency input from C fibers and presents as hyperexcitability of wide dynamic range neurons in the dorsal horn (also known as wind-up) [Citation19,Citation33]. This hyperexcitability may be due to continued neuronal depolarization through a variety of processes including 1) activation or modulation of membrane channels or receptors; 2) release of pro-nociceptive factors from microglia and astrocytes; 3) loss of local inhibitory regulation; and 4) excitatory input into the dorsal horn from bulbospinal projections [Citation19]. In animal models of chronic inflammation, afferent nociceptors, and post-ganglionic sympathetic fibers, exhibit a neuropathic-like phenotype with enhanced sprouting into affected sites in the periphery and in the DRG itself [Citation19,Citation34-36]. In addition to the processes described above, chronic nociceptive input can also lead to persistent peripheral and central sensitization by inducing changes in the engagement of innate and adaptive immune linkages within nociceptors and the DRG [Citation19]. Such adaptive immune signaling may account for a variety of anomalous pain conditions ranging from non-inflammatory joint disorders to fibromyalgia, which may be thought as having an autoimmune phenotype [Citation37].
Overall, persistent neuronal sensitization, and possibly peripheral sprouting, enhance pain responses in terms of intensity and duration, resulting in a chronically altered afferent pain signaling characterized by allodynia and/or hyperalgesia that may persist even when inflammation subsides to low-grade. Though a detailed discussion is beyond the scope of this review, it should be noted that chronic pain is often associated with changes in the sympathetic nervous system and sympathetic activity likely contributes to the overall chronic pain experience [Citation38]. The overall chronic pain experience is also affected by, and can affect, a variety of factors including genetic predisposition, patient comorbidities, previous pain experience, mood, emotional distress, coping ability, and other psychosocial or sociocultural factors [Citation39,Citation40]. Thus, the chronic pain experience may differ between patients even when the etiology is similar. Catastrophizing, for example, can lead to psychosomatic or stress-related symptoms that may exacerbate perception of chronic pain [Citation39,Citation41]. Catastrophizing encompasses helplessness (i.e. nothing will change the pain), negative amplification of pain-related thoughts through rumination (i.e. repetitive thoughts about pain), and magnification (i.e. exaggerated concern about negative consequences of pain) [Citation39]. Catastrophizing can also lead to secondary complications such as fear, irritability, anxiety, inability to cope, sleep impairment, depression, suicidal ideation, and overall diminished quality of life [Citation42-44].
6 OA-related pain pathophysiology
Most structures of the joint are densely innervated, with nociceptors present in the capsule, ligaments, menisci, periosteum, and subchondral bone [Citation45]. OA is characterized by pathological changes throughout the entire joint organ, including synovial inflammation, degeneration of ligaments and menisci, subchondral bone remodeling, and progressive loss of articular cartilage [Citation46]. Movement-induced (i.e. mechanical) pain is a hallmark symptom of OA, but pain intensity is not highly associated with the extent of anatomical damage observed upon radiological examination [Citation47,Citation48]. OA patients exhibit lower pain thresholds in response to pressure (mechanical hyperalgesia), at both affected and distal sites, than healthy controls [Citation48]. Together, these findings suggest that neuronal sensitization plays a role in chronic OA-related pain.
Pathological changes in the joint lead to release of pro-inflammatory mediators (e.g. prostaglandin E2 [PGE2], growth factors, DAMPs) that may sensitize peripheral nociceptive terminals within the joint (i.e. peripheral sensitization) and lead to increased nociceptive signaling to the dorsal horn of the spinal cord () [Citation49]. As noted earlier, increased nociceptive input can lead to sensitization of neurons within the dorsal horn (i.e. central sensitization) and may represent another mechanism contributing to OA-related pain [Citation50]. Finally, it is possible that increased nerve density (via sprouting) in OA joints contributes to pain. Sensory neurons exhibit substantial sprouting/remodeling in a murine model of inflammatory arthritis and similar remodeling causes severe chronic pain in animal models of neuronal injury [Citation34]. In addition, patients with symptomatic knee OA exhibit increased synovial nerve-fiber innervation compared with asymptomatic controls [Citation51]. Despite these observations, it is not clear whether increased nerve density within structures of the joint plays a role in chronic OA pain.
Figure 2. Overview of OA-related pain pathophysiology. Figure reproduced from: ‘Nerve growth factor antibody for the treatment of osteoarthritis pain and chronic low-back pain: mechanism of action in the context of efficacy and safety’ by Schmelz M et al [Citation22]. and is licensed under CC BY-NC-ND 4.0. DAMPs = damage-associated molecular patterns; ECM = extracellular matrix; EGF = epidermal growth factor; FGF-2 = fibroblast growth factor 2; NGF = nerve growth factor; OA = osteoarthritis; PGE2 = prostaglandin E2; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor
![Figure 2. Overview of OA-related pain pathophysiology. Figure reproduced from: ‘Nerve growth factor antibody for the treatment of osteoarthritis pain and chronic low-back pain: mechanism of action in the context of efficacy and safety’ by Schmelz M et al [Citation22]. and is licensed under CC BY-NC-ND 4.0. DAMPs = damage-associated molecular patterns; ECM = extracellular matrix; EGF = epidermal growth factor; FGF-2 = fibroblast growth factor 2; NGF = nerve growth factor; OA = osteoarthritis; PGE2 = prostaglandin E2; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor](/cms/asset/985cc54f-4393-4edf-9575-5ddebf0d00a8/ipgm_a_1949199_f0002_c.jpg)
Due to the various mediators (e.g. prostaglandins, growth factors) and processes (e.g. peripheral sensitization, central sensitization, neuronal sprouting) possibly involved in OA-related nociception, there are several pathways that can be targeted to achieve analgesia. Next, we discuss how some common treatments (NSAIDs, acetaminophen, and opioids), and a novel class of treatments in clinical development (NGF-Abs), work to alter the processing leading to an OA pain phenotype and achieve analgesia against OA-related pain.
7 NSAIDs
7.1 Analgesic MOA of NSAIDs
NSAIDs have been the cornerstone of pharmacologic OA treatment and provide analgesia via inhibition of the cyclooxygenase (COX) family of enzymes that are involved in the formation of prostaglandins in the periphery and central nervous system (CNS) () [Citation52,Citation53]. The relative contribution of peripheral and central effects to overall analgesia depends on the site of delivery and biochemical properties (e.g. ability to cross the blood–brain barrier) of specific NSAIDs [Citation52]. While some NSAIDs inhibit both COX-1 and COX-2, others selectively inhibit COX-2 [Citation53]. COX-1 is constitutively expressed in most tissues while COX-2, though constitutively expressed in some tissues, is upregulated at sites of peripheral injury and within the CNS in response to inflammation [Citation52,Citation53]. During an inflammatory response, COX enzymes drive synthesis of the prostanoid family of inflammatory mediators (e.g. prostaglandins, prostacyclins, thromboxanes) through metabolism of arachidonic acid [Citation54].
Figure 3. Analgesic mechanism of action of NSAIDs. COX = cyclooxygenase; NSAID = nonsteroidal anti-inflammatory drug; OA = osteoarthritis; PAG = periaqueductal gray; PGE2 = prostaglandin E2; PLA2 = phospholipase A2; RVM = rostral ventromedial medulla
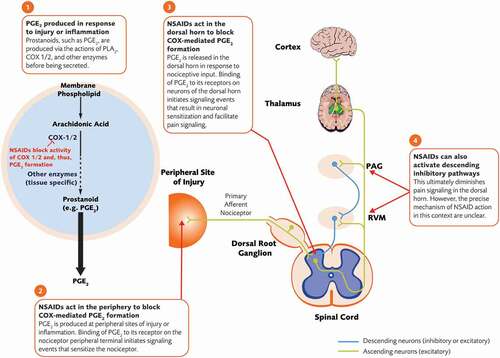
Prostaglandins, PGE2 in particular, promote the inflammatory response in the periphery and the sensitized neuronal state in the spinal cord dorsal horn [Citation54-56]. Activation of PGE2 receptors (EP2; G protein-coupled receptors that modulate cyclic adenosine monophosphate [cAMP] production and activity of protein kinases A and C) causes a change in conductance of several ion channels including sodium channels, voltage-gated potassium channels, and transient receptor potential channels [Citation55,Citation57]. At peripheral sites of inflammation, activation of PGE2 receptors on nociceptor terminals results in neuronal depolarization and sensitization, facilitating their activation by other inflammatory mediators such as bradykinin [Citation55,Citation57,Citation58].
In response to nociceptive input, COX-2 expression is upregulated in several areas of the CNS and PGE2 is released in the dorsal horn, which is considered an important site of PGE2 action [Citation52]. PGE2 is believed to have both pre- and post-synaptic effects that contribute to central sensitization of neurons in the dorsal horn [Citation52]. PGE2 acts on presynaptic nerve terminals of the nociceptor to increase the opening of voltage-gated calcium channels thereby enhancing release of pro-nociceptive neurotransmitters (e.g. glutamate, substance P, CGRP, BDNF) from nociceptors, and also acts post-synaptically to enhance depolarization of second-order neurons [Citation52]. PGE2 may also act on EP2-like receptors at the nerve terminals of inhibitory interneurons in the dorsal horn to reduce glycine release. Given the constitutive role of glycine in regulating the excitability of dorsal horn neurons, this would predictably lead to a state of enhanced excitability and facilitation of pain signaling [Citation52]. Finally, there is some evidence that NSAIDs act centrally in structures such as the periaqueductal gray (PAG) and RVM to activate descending inhibitory pain pathways [Citation59].
In the context of OA, increased levels of PGE2 have been observed in cartilage and synovial fluid of OA patients compared with that from non-OA joints [Citation60]. Notably, PGE2 has been shown to have anti-anabolic and catabolic effects on isolated human cartilage [Citation60]. In a mouse model of spontaneous OA, COX-2 activity is increased in subchondral bone compared with control animals, and pharmacologic inhibition of COX-2 activity attenuates the OA phenotype [Citation61]. Similarly, COX-2 activity is elevated in subchondral bone from human OA patients compared with control patients [Citation61]. Overall, these observations suggest a role for COX-2 and PGE2 in OA-related pain.
7.2 Efficacy and safety of NSAIDs in the context of OA
NSAIDs are commonly recommended for the management of OA (topical and oral NSAIDs for knee OA; oral NSAIDs for hip OA) due to their documented efficacy, with some caveats for oral NSAIDs due to their potential for gastrointestinal (GI), renal, and cardiovascular (CV) adverse events. American College of Rheumatology (ACR) guidelines strongly recommend the use of oral and topical NSAIDs as an initial pharmacologic therapy [Citation5]. European League Against Rheumatism (EULAR) guidelines recommend NSAIDs after a trial of acetaminophen but note that patients with GI comorbidities should be limited to COX-2-selective NSAIDs or nonselective NSAIDs in combination with a protein pump inhibitor [Citation4,Citation6]. Acetaminophen was conditionally not recommended by Osteoarthritis Research Society International (OARSI) guidelines and, similar to EULAR, conditional recommendations were made regarding use of NSAIDS in patients with GI comorbidities. OARSI guidelines also noted that NSAIDs of any class were not recommended for patients with CV comorbidities due to evidence associating NSAID use with heightened CV risk [Citation62].
The efficacy of NSAIDs for OA-related pain has been demonstrated in several meta-analyses [Citation63-66]. In one recent network meta-analysis of 76 trials of placebo, acetaminophen, and oral NSAIDs in 58,451 patients with OA of the knee or hip, for example, most NSAIDs examined improved both pain (effect sizes ranged from – 0.05 to – 0.63 across 19 arms) and function (effect sizes ranged from – 0.03 to – 0.53 across 18 arms) compared with placebo (effects sizes based on, or standardized to, a scale from 0 to 10) [Citation63]. Though response varied by NSAID and by dose, effect sizes for many arms (particularly at higher doses) exceeded the author-specified threshold for minimal clinically important difference for both pain (11 arms) and function (9 arms) [Citation63]. It should be noted many published studies of NSAIDs in OA patients are <3 months in duration and few studies are longer than 6 months, making conclusions on long-term NSAID efficacy difficult [Citation63-66]. This is reflected in treatment guidelines, which recommend oral NSAID use at the lowest doses and shortest durations possible [Citation5].
Though oral NSAIDs are typically well tolerated in most patients (common adverse events include abdominal/gastric pain, diarrhea, dyspepsia, rash, upper respiratory tract infections, nausea, and edema), serious long-term risks include GI ulceration/bleeding, CV thrombotic events, and the potential for chronic renal failure [Citation66-69]. As a result, all NSAIDs are contraindicated following coronary artery bypass graft surgery and should be used with caution in patients with, or at risk for, CV disease, GI events, or renal impairment [Citation68-70]. These serious adverse events are due to COX expression, and the role of COX-mediated signaling, in a variety of processes including gastric mucosa production/protection (predominantly COX-1), platelet aggregation (predominantly COX-1), and renal homeostasis (COX-1 and COX-2) [Citation54,Citation71]. These adverse events are applicable to all NSAID formulations (all carry the same boxed warning in the US) but topical formulations are presumed to have a more favorable safety profile than oral formulations due to localized application and the resulting low serum concentrations [Citation72]. Adverse events associated with topical NSAIDs typically involve mild and transient local skin reactions [Citation70].
8 Acetaminophen
8.1 Analgesic MOA of acetaminophen
Despite being among the most commonly used pain medications in the world, the MOA of acetaminophen is controversial and currently unknown [Citation73]. Some effects (e.g. anti-pyretic) of acetaminophen involve inhibition of centrally expressed COX-1 and COX-2 enzymes, though this is not considered the primary MOA for analgesia [Citation73].
Loss of cannabinoid receptor CB1 activity attenuates acetaminophen- or N-arachidonoylphenolamine (AM404; a acetaminophen metabolite formed in the CNS)-mediated analgesia in rodents, implicating the endocannabinoid system in acetaminophen MOA [Citation74,Citation75]. The lipid neurotransmitter anandamide is the primary endogenous agonist of the CB1 receptor, and AM404 may promote indirect activation of CB1 receptors by blocking anandamide reuptake and, possibly, metabolism [Citation76,Citation77]. Though CB1 receptors are highly expressed in many structures of the CNS (and to some extent in the peripheral nervous system), the RVM is believed to be an important site of acetaminophen action. It is possible that AM404 promotes activation of CB1 receptors (by inhibiting anandamide reuptake and/or degradation) on terminals of tonically active, gamma-aminobutyric acid (GABA)-releasing neurons projecting to the RVM from upstream structures (e.g. the PAG). The resulting decrease in GABA release from these neurons would promote activity of anti-nociceptive serotonergic fibers projecting from the RVM to the dorsal horn of the spinal cord and produce analgesia (i.e. activate descending inhibitory pathways).
Pre-clinical evidence suggests that acetaminophen may also produce analgesia through prolonged activation and desensitization of the transient receptor potential (TRP) family of nonselective cation channels, such as TRP channel subfamily vanilloid-1 (TRPV1) at peripheral and central terminals of nociceptors (and in some supraspinal structures). Alternatively, genetic and pharmacologic approaches in rodents suggest that analgesia induced by acetaminophen may represent indirect inhibition of supraspinal Cav3.2 channels (low voltage-activated T-type calcium channels known to promote inflammatory and neuropathic pain signaling) following calcium influx in response to TRPV1 activation [Citation78].
Finally, pre-clinical evidence also suggests that the analgesics effects of acetaminophen may involve inhibition of pro-nociceptive N-Methyl-d-aspartic acid/nitric oxide signaling in the spinal cord or activation of voltage-gated potassium channels in the dorsal horn [Citation73,Citation79].
8.2 Efficacy and safety of acetaminophen in the context of OA
Treatment guidelines vary in their recommendation on the use of acetaminophen for OA. EULAR guidelines recommend acetaminophen as first-line pharmacologic treatment for mild-to-moderate OA-related pain, citing demonstrated efficacy and a more favorable safety profile than other pharmacologic options [Citation4,Citation6]. ACR guidelines conditionally recommend acetaminophen only in patients with intolerance or contradiction to NSAIDs, noting very small effect sizes and hepatotoxicity concerns [Citation5]. For similar reasons, OARSI treatment guidelines do not recommend acetaminophen for OA [Citation62].
Meta-analyses demonstrate no-to-small effects for acetaminophen in patients with OA [Citation63,Citation80,Citation81]. In a recent network meta-analysis of 76 trials of placebo, acetaminophen, and oral NSAIDs in patients with OA of the knee or hip, for example, acetaminophen at doses from <2000–4000 mg had a ‘nearly null’ effect on pain (effect sizes ranged from – 0.18 to – 0.07) [Citation63].
Acetaminophen is generally considered safe for many patients, particularly in comparison with other OA treatment options such as NSAIDs and opioids [Citation4,Citation6]. Data on long-term use of acetaminophen, however, is sparse (particularly in patients >65 years of age) and mostly based on observational data [Citation82]. It is clear, though, that acetaminophen is associated with a clear dose-dependent risk of hepatotoxicity as it is the most common cause of acute liver failure in the US [Citation83]. Under therapeutic dosing conditions, acetaminophen is metabolized in the liver predominantly through glucuronidation and sulfation pathways to yield non-reactive metabolites [Citation83,Citation84]. A small amount (~10%) is oxidized by the cytochrome P450 family of enzymes to produce a hepatotoxic metabolite, NAPQI, which is rapidly detoxified via conjugation with glutathione [Citation83,Citation84]. High doses of acetaminophen saturate the glucuronidation, sulfation, and glutathione pathways, resulting in excess NAPQI that binds a variety of mitochondrial proteins and leads to inhibition of adenosine triphosphate (ATP) synthesis and cellular necrosis [Citation83,Citation84]. Though not as well established as the hepatotoxic effect, studies have suggested that acetaminophen use may also be linked to GI, CV, and renal adverse events [Citation82].
9 Opioids
9.1 Analgesic MOA of opioids
Opioids are capable of providing powerful relief of acute pain through activation of G protein-coupled opioid receptors () [Citation85,Citation86]. Three classes of classical opioid receptors lead to analgesia when activated (µ, δ, and κ), although they differ in expression (i.e. neural, non-neural, peripheral, or central), ligand selectivity, and side-effect profile [Citation85,Citation86]. Traditional opioid medications are µ-opioid agonists that alter pain processing through µ-opioid receptors expressed on peripheral nociceptors and on neurons within the dorsal horn and brain [Citation85,Citation86]. Activation of µ-opioid receptors in neurons leads (through Gi/o protein coupling) to inhibition of calcium influx, mediated by voltage-gated calcium channels and by the enhancement of inward potassium currents, and inhibition of cAMP production that results in hyperpolarization and inhibition of neurotransmitter release [Citation85,Citation86].
Figure 4. Analgesic mechanism of action of opioids. PAG = periaqueductal gray; RVM = rostral ventromedial medulla
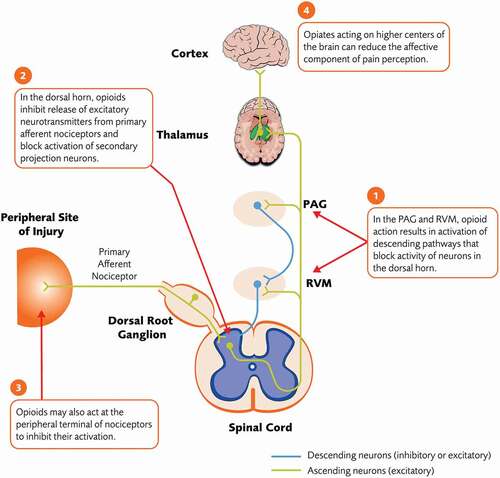
Though several structures within the CNS express µ-opioid receptors, and their activation can affect sensory discriminative or affective aspects of pain, components of the descending inhibitory pathway and the dorsal horn are thought to be important sites of opioid action in the context of analgesia [Citation86]. In the PAG, opioids act on presynaptic µ-opioid receptors to block GABA release from inhibitory interneurons, resulting in increased descending inhibitory signals from the RVM and LC to the dorsal horn [Citation86,Citation87]. Opioids also act directly in the RVM to activate descending inhibition [Citation86]. In the dorsal horn, activation of presynaptic opioid receptors at terminals of C polymodal nociceptors inhibits release of excitatory neurotransmitters, while activation of post-synaptic receptors on second-order projection neurons induces hyperpolarization and suppresses their excitability [Citation87,Citation88]. The overall effect of opioids is decreased nociceptive signaling from the dorsal horn to higher centers. Actions on opiate receptors on higher-order systems, including areas in the limbic system, may account for effects of opiates on affective components of the pain phenotype [Citation89].
Opioids may also have limited analgesic effects outside the CNS. Opioid receptors are upregulated at peripheral terminals of nociceptors in a variety of tissues outside the CNS in response to local inflammation (e.g. in OA cartilage and cultured chondrocytes) and activation of these receptors inhibits nociceptor activity, likely by promoting hyperpolarization and inhibiting release of pro-nociceptive neurotransmitters [Citation90,Citation91].
9.2 Efficacy and safety of opioids in the context of OA
Opioids are typically not recommended for the management of OA. OARSI guidelines strongly recommend against their use, while guidelines from ACR and EULAR conditionally recommend their use only when other pharmacological options have failed (with ACR guidelines favoring tramadol over non-tramadol opioids) [Citation4–6,Citation62]. These recommendations are based on both efficacy and safety concerns.
A recent systematic review of 22 randomized trials enrolling 8275 patients with OA of the knee or hip concluded that oral or transdermal opioids provide small improvements in pain (effect size = – 0.28) and function (effect size = – 0.26) compared with placebo/no treatment that are of questionable clinical relevance. Further, the review notes that most studies have treatment durations of several days to weeks and few are longer than 6 months, highlighting the sparsity of data regarding long-term use of opioids for the management of OA [Citation92]. These findings are similar to other meta-analyses of opioids for the management of OA [Citation93,Citation94].
Common opioid-related adverse events include somnolence, sedation, nausea, vomiting, dizziness, dry mouth, pruritus, and constipation [Citation95-97]. The broad association of µ-opioid receptors with systems inside and outside the CNS underlies these common events (and less common, but potentially serious, events such as respiratory depression) and limits the clinical utility of opioids, particularly over the long term. In the GI tract, for example, opioid receptor activation causes inhibition of acetylcholine release from enteric interneurons and purine/nitric oxide release from inhibitory motor neurons, leading to opioid-induced constipation [Citation98]. Activation of opioid receptors in the vestibular system and medulla leads to nausea/dizziness and vomiting, respectively, while respiratory depression is mediated through activation of µ-opioid receptors located in respiratory centers of the brainstem and/or structures that signal CO2 retention to the brainstem [Citation99,Citation100]. Traditional µ-opioid receptor agonists activate dopamine signaling in the mesolimbic system by inhibiting release of GABA from inhibitory interneurons, which can produce euphoria and lead to a powerful rewarding state in some patients [Citation101]. It is estimated that 21%–39% of chronic pain patients misuse opioids and 8%–12% experience addiction [Citation102]. Overall, approximately 10.3 million people abuse opioids in the US and over 47,000 opioid-related deaths are reported per year [Citation103]. Finally, the phenomena of opioid tolerance (the need for increased doses of opioids to achieve analgesia) and opioid-induced hyperalgesia (opioids paradoxically causing increased pain) complicates the use of opioids for chronic pain [Citation104].
Concerns regarding the long-term use of opioids for chronic OA-related pain extend to other chronic, nonmalignant, pain conditions since their primary indication is short-term use for acute or breakthrough pain.
10 NGF-Abs
10.1 Analgesic MOA of NGF-Abs
NGF-Abs provide analgesia by binding and neutralizing NGF activity [Citation21,Citation22,Citation105]. During embryogenesis, NGF is required for growth and survival of certain sensory and sympathetic neurons via its interaction with tropomyosin-related kinase A (TrkA) and the low-affinity p75 neurotrophin receptor [Citation106]. In adult organisms, NGF is known to modulate nociceptive signaling and plays a role in neuronal sprouting/remodeling in certain situations (such as after nerve injury) [Citation21,Citation105]. Levels of NGF are upregulated in several animal models of chronic pain and in patients with certain chronic pain conditions [Citation105]. In addition, subcutaneous, intradermal, or intramuscular injection of NGF causes hyperalgesia in healthy subjects [Citation107–110].
NGF’s pain-modulating effects are thought to be primarily mediated through TrkA receptors expressed on the peripheral terminal of nociceptors, though there is some evidence the p75 neurotrophin receptor may also play a role in NGF’s ability to modulate nociception [Citation21,Citation105]. NGF is released by immune and nonimmune cells at local sites of inflammation as part of the inflammatory response [Citation111]. NGF binds to TrkA on the peripheral terminal of nociceptors, which has short- and longer-term consequences on nociceptive signaling () [Citation21,Citation105]. In the short-term, NGF/TrkA signaling leads to increased local expression or activity of ion channels (e.g. TRPV1; sodium, calcium, and potassium channels) at the peripheral terminal that leads to rapid nociceptor depolarization and sensitization (i.e. peripheral sensitization) [Citation21,Citation105]. In the longer-term, the NGF/TrkA signaling complex is internalized and retrogradely transported from the nociceptor peripheral terminal to the cell body in the DRG [Citation112]. In the DRG, NGF/TrkA signaling leads to increased synthesis of pro-nociceptive peptides (e.g. substance P, CGRP, BDNF), ion channels (acid-sensing ion channels, sodium, and calcium channels), and receptors (e.g. bradykinin receptor 2, NMDA receptor) [Citation21,Citation105]. These components are transported to the peripheral or central (dorsal horn) nociceptor terminals where they contribute to peripheral and central sensitization, respectively. In addition to actions directly at the peripheral nociceptor terminal, NGF binds TrkA on inflammatory cells to stimulate the release of additional mediators known to promote peripheral nociceptor sensitization including histamine, 5-HT, PGE2, and NGF itself [Citation22,Citation105]. Finally, NGF/TrkA signaling may contribute to pain hypersensitivity by promoting neuronal sprouting and, thus, increasing TrkA+ nerve-fiber density (both sensory and sympathetic fibers) at peripheral (e.g. sites of injury) and/or central (e.g. dorsal horn of the spinal cord) sites [Citation105].
Figure 5. Pronociceptive effects of NGF. For simplicity, and since NGF’s pain modulating effects are thought to be primarily mediated through TrkA receptors, the low affinity neurotrophin p75 receptor has been omitted in this diagram. 5-HT = serotonin; ASIC = acid-sensing ion channels; BDNF = brain-derived neurotrophic factor; Ca = calcium; CGRP = calcitonin gene-related peptide; DRG = dorsal root ganglion; K = potassium; Na = sodium; NGF = nerve growth factor; NMDA = N-methyl-D-aspartate; PGE2 = prostaglandin E2; TrkA = tropomyosin receptor kinase A; TRPV1 = transient receptor potential cation channel subfamily V member 1
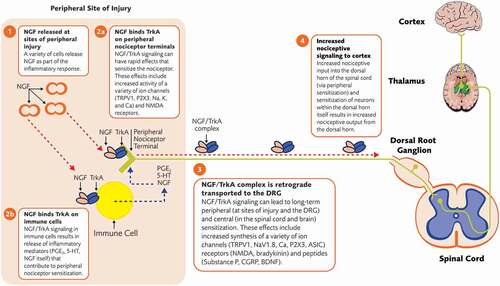
In the context of OA, synovial specimens from patients with advanced OA show increased levels of NGF compared with non-OA specimens [Citation113]. A similar increase in NGF is seen in synovial specimens with painful chondropathy compared with specimens from patients with non-painful chondropathy [Citation113]. These findings suggest that NGF and the presence of pain are correlated in OA joints. A variety of cells within the OA joint are possible sources of NGF, including synovial fibroblasts and chondrocytes [Citation114,Citation115]. Murine cartilage explants, for example, synthesize and release NGF in response to mechanical stress in vitro [Citation114]. NGF, TrkA, and overall density of nerve fibers are increased in the synovium of the knee in patients with painful OA compared with asymptomatic controls [Citation51]. Overall, these observations suggest a role, to some extent, for NGF in OA-related pain.
10.2 Efficacy and safety of NGF-Abs in the context of OA
Since, at the time of this writing, NGF-Abs are in clinical development and not covered in current OA treatment guidelines, a more detailed overview (compared to other treatments) of efficacy and safety is provided. The most comprehensive systematic review of NGF-Ab efficacy for the treatment of moderate-to-severe OA (pain score of ≥4 or 5 on a 10-point scale) of the knee or hip was published in 2015 and included 13 phase 2 or 3 randomized, placebo-controlled trials of tanezumab (n = 10), fasinumab (n = 1), and fulranumab (n = 2) [Citation13]. The review concluded that intravenous administration of NGF-Abs (every 4 or 8 weeks) provides superior efficacy to placebo, based on improvements in measures of pain and physical function [Citation13].
More recent phase 3 randomized, placebo-controlled trials of NGF-Abs have also been published confirming efficacy of NGF-Abs in patients with OA. Subcutaneous (SC) administration of tanezumab (2.5 mg at baseline and Week 8, or 2.5 mg at baseline and 5 mg at Week 8) significantly improved co-primary endpoints of pain, function, and overall patient global assessment of OA (PGA-OA) compared with placebo after 16 weeks of treatment [Citation116]. In a separate study of SC tanezumab (2.5 mg or 5 mg every 8 weeks for 24 weeks), the 5 mg dose significantly improved the three co-primary endpoints compared with placebo, while the 2.5 mg dose improved pain and function (but not PGA-OA) [Citation117]. In one study of SC fasinumab (1 mg, 3 mg, 6 mg, or 9 mg every 4 weeks for 16 weeks), all doses significantly improved pain (primary endpoint) and function (secondary endpoint) versus placebo at Week 16 [Citation118]. It should be noted that these recent (post-2015) studies of tanezumab and fasinumab demonstrated efficacy of NGF-Abs despite enrollment criteria requiring patients to have moderate-to-severe OA and a history of inadequate response to standard analgesics for OA-related pain (e.g. acetaminophen, opioids, NSAIDs). Finally, four studies of SC fulranumab were terminated early due to a partial clinical hold (see section on NGF-Ab safety below) [Citation119]. Though improvements in pain and function were observed at Week 16 in a pooled analysis of these trials, only 34% (n = 84) of patients completed the treatment phase due to study termination, and firm conclusions on efficacy could not be made (fulranumab development was discontinued in 2016).
NGF-Abs, mostly tanezumab, have demonstrated greater improvements in pain compared with NSAIDs and opioids in several head-to-head clinical trials in patients with moderate-to-severe OA of the knee or hip (). Only one of these studies, however, was conducted post-2015 and included design elements to mitigate joint safety risks (e.g. lower doses of tanezumab in patients with a history of inadequate response to standard OA analgesics and on a stable dose of oral NSAIDs prior to randomization) [Citation120]. In this study, all treatment groups (SC tanezumab 2.5 mg, SC tanezumab 5 mg, oral NSAID) provided improvements in pain and function that were clinically meaningful in a large proportion of patients and sustained over 56 weeks of treatment [Citation120]. Magnitude of improvement with tanezumab was at least the same as NSAIDs at all time points and periodically greater than NSAIDs at time points on or before Week 16 [Citation120]. Conclusions on comparative efficacy after Week 16 were limited by a trial designed primarily to assess safety (e.g. discontinuation of non-responders, use of rescue medication).
NGF-Ab programs were subject to partial clinical holds in 2010 and 2012 due to reports of joint safety events in clinical trials and possible changes in sympathetic neuronal morphology in pre-clinical studies, respectively [Citation121,Citation122]. Subsequent studies have not found evidence for an adverse sympathetic effect of NGF-Abs in humans or non-human primates, and NGF-Ab clinical testing resumed in 2015 with trials designed to mitigate joint safety risks (e.g. lower doses, comprehensive joint screening and monitoring procedures, enrolling patients with inadequate response to other treatments, restrictions on concomitant NSAID use) [Citation122,Citation123]. A recent pooled analysis of all placebo-controlled trials of SC tanezumab (2.5–10 mg every 8 weeks) in patients with moderate-to-severe OA conducted to date (n = 3) demonstrated that tanezumab was well tolerated in most patients [Citation124]. While adverse events were mostly mild in severity across all dose groups, tanezumab 2.5 mg demonstrated a more favorable safety profile than higher doses [submitted manuscript]. Adverse events of abnormal peripheral sensation (predominantly paresthesia and hypoesthesia) were more frequently reported in tanezumab treatment groups (ranging from 5.1% in the 2.5 mg group to 12.8% in the 10 mg group) than in the placebo group (2.2%), and have consistently been observed across all trials of NGF-Abs [Citation125] [submitted manuscript]. These events, however, are mostly mild and typically resolve over time, particularly at the tanezumab 2.5 mg dose [submitted manuscript]. Joint safety events, predominantly rapidly progressive OA (RPOA), have also been consistently reported across trials of NGF-Abs [Citation121]. RPOA is characterized by pain and joint space narrowing, due to chondrolysis, observed upon radiographic examination [Citation126]. RPOA type 1 is defined in the tanezumab program as a significant loss of joint space width ≥2 mm (predicated on optimal joint positioning) within approximately 1 year, without gross structural failure; RPOA type 2 is defined in the tanezumab program as abnormal bone loss or destruction, including limited or total collapse of at least one subchondral surface, that is not normally present in conventional end-stage OA [Citation126]. The overall occurrence of RPOA (predominantly RPOA type 1) in recent SC trials of the tanezumab (at doses from 2.5 to 5 mg) is low and dose-dependent (2.5 mg = 1.9%; 5 mg = 2.8%) [submitted manuscript]. In these recent trials, 6%–7% of tanezumab-treated patients underwent a total joint replacement, but these rates were not significantly different from placebo (4.5%) [submitted manuscript].
The mechanisms underlying RPOA are not clear. Pre-clinical studies have not demonstrated a risk for accelerated OA and repeated administration of high-dose NGF-Ab does not impact healthy joint or bone tissue from rodents or monkeys [Citation127,Citation128]. It has been proposed that RPOA observed following NGF-Ab treatment could be due to analgesic arthropathy (overuse of the joint due to effective analgesia). This is unlikely, however, as pain severity in the index joint was similar among patients with and without RPOA in an analysis of the pre-2015 tanezumab program [Citation129]. Another proposed mechanism is neuropathic arthropathy (overuse of the joint due to nerve damage and loss of pain sensation). This explanation is also unlikely, since loss of protective pain sensitivity is not evident in tanezumab-treated patients [Citation129]. Other possible, but unconfirmed, explanations for RPOA in NGF-Ab programs include preexisting bone integrity issues in patients or a role for NGF in bone repair and cartilage formation [Citation129].
In March 2021, the U.S. Food and Drug Administration Joint Arthritis Advisory Committee and Drug Safety and Risk Management Advisory Committee voted on a single question focused on whether a proposed risk evaluation and mitigation strategy (REMS) for tanezumab (including physician and patient education, radiographic monitoring for joint-safety events, and avoiding the concomitant use of NSAIDs) will ensure its benefits outweigh its risks. The Committee voted 1 in favor and 19 against. At the time of this writing, however, a final decision from the U.S. Food and Drug Administration on the Biologics License Application for tanezumab is pending.
11 Other treatments for OA-Related pain
Though NSAIDs, acetaminophen, and opioids are among the most commonly used analgesics for OA-related pain, we would be remiss not briefly discuss a few additional non-surgical treatments that are also used including intra-articular corticosteroids (IACS), intra-articular hyaluronic acid (IAHA), and duloxetine.
IACS relieve OA-related pain through anti-inflammatory and immunosuppressive actions [Citation130]. The action of corticosteroids on nuclear steroid receptors results in changes in gene transcription that has a variety of effects including (but not limited to) a decrease vascular permeability, inhibition of the migration and accumulation of inflammatory cells in the joint, and a decrease in the production of inflammatory mediators [Citation130]. OARSI treatment guidelines conditionally recommend IACS to all types of patients with knee (but not hip) OA, noting that they may provide short-term pain relief [Citation62]. In contrast, ACR guidelines strongly recommend IACS injections in patients with knee or hip OA (with ultrasound guidance for the hip), noting short-term efficacy in knee OA [Citation5]. ACR further recommends IACS over IAHA due to higher quality evidence relative to IAHA (despite a lack of head-to-head studies) [Citation5]. A recent case series, however, suggests that structural joint events such as rapid progression of OA, subchondral insufficiency fracture, and rapid joint destruction is observed in patients who have received IACS and further research is needed to determine the role of IACS in these events [Citation131].
Hyaluronic acid is naturally found in joints and provides viscoelasticity to synovial fluid [Citation130,Citation132]. Analgesic effects of IAHA have been reported at and beyond 12 weeks of treatment, despite a relatively short presence in the joint following administration [Citation62]. The analgesic mechanisms of IAHA are unclear, but pre-clinical studies have offered several possibilities. The most studied mechanism involves chondroprotection, but other possibilities include promotion of proteoglycan and glycosaminoglycan synthesis, anti-inflammatory actions, lubrication and shock absorption, protection of subchondral bone, and effects on nociceptors within the joint [Citation132]. OARSI treatment guidelines conditionally recommend IAHA for all types of patients with knee (but not hip) OA, citing beneficial analgesic effects and a more favorable safety profile than repeated intra-articular injections of corticosteroids [Citation62]. In contrast, however, ACR guidelines recommend against the use of IAHA in patients with OA of the knee (conditionally) or hip (strongly) [Citation5]. They note that the best quality evidence (based on meta-analysis of low bias studies) available for IAHA demonstrates effects sizes that approach zero compared to saline [Citation5]. As a result, ACR only advocates for the use of IAHA in patients with knee OA who have exhausted other treatment options such as NSAIDs and IACS [Citation5].
Duloxetine is a selective serotonin norepinephrine reuptake inhibitor (SNRI) that exhibits analgesic properties, most likely via enhancement of serotonergic and noradrenergic signaling within the CNS [Citation133]. Duloxetine is approved for chronic musculoskeletal pain and has been shown to have modest to moderate effects on OA-related pain [Citation134]. OARSI treatment guidelines conditionally recommend duloxetine for OA patients with widespread pain or comorbid depression due to its beneficial effects on mood and widespread pain conditions such as fibromyalgia [Citation62]. Likewise, ACR treatment guidelines conditionally (due to tolerability concerns) recommend duloxetine for patients with knee or hip OA [Citation5].
12 Conclusion and future directions
Among the variety of pharmacologic agents available to treat OA-related pain, NSAIDs, acetaminophen, and opioids are among the most commonly utilized. Acetaminophen and opioids, however, offer little efficacy for chronic OA-related pain. Further, potentially serious adverse events are associated with NSAIDs (e.g. gastrointestinal and cardiovascular events), acetaminophen (e.g. hepatotoxicity), and opioids (e.g. respiratory depression, abuse, misuse). As a result, and despite their widespread use, treatment guidelines typically do not recommend opioids and only conditionally recommend acetaminophen for chronic OA-related pain. Thus, there is an unmet need for novel, safe, and effective treatments. NGF-Abs are an emerging treatment for OA-related pain with a novel MOA (i.e. inhibition of NGF activity) (). Clinical trials have demonstrated efficacy of NGF-Abs for moderate-to-severe OA pain in patients with a history of inadequate response to other analgesics, but concerns persist over the risk of joint-related events (particularly RPOA) associated with NGF-Abs. Additional research is needed to understand the mechanism(s) of RPOA in general and in the context of NGF-Ab treatment.
Table 3. Summary of the analgesic mechanisms of action of NSAID, acetaminophen, opioids, and NGF antibodies
In addition to NGF-Abs, other agents that target nociceptive pathways are being investigated for the management of OA-related pain including TrkA inhibitors, voltage-gated sodium channel inhibitors, TRPV1 antagonists, peripheral opioid receptor agonists with improved safety profiles, agents that target bradykinin B2 receptors, and agents that target cannabinoid receptors [Citation135]. Several disease-modifying agents that target articular cartilage or subchondral bone, and regenerative stem cell or gene-therapy approaches are also being investigated [Citation136].
Declaration of funding
The study was sponsored by Pfizer and Eli Lilly and Company.
Declaration of financial/ other disclosures
PM has served as a consultant to Pfizer and has received anti-NGF antibody from Rinat (Pfizer) for research use. JH and LV are full-time employees of, and own stock in, Eli Lilly & Company. SD and MS are full-time employees of, and own stock/options in, Pfizer Inc. The remaining authors report no potential conflicts. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Geolocation information
United States
Acknowledgments
Medical writing support was provided by Matt Soulsby, PhD, CMPP, of Engage Scientific Solutions and was funded by Pfizer and Eli Lilly & Company.
References
- Cross M, Smith E, Hoy D, et al. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1323–1330.
- Cao P, Li Y, Tang Y, et al. Pharmacotherapy for knee osteoarthritis: current and emerging therapies. Expert Opin Pharmacother. 2020;21(7):797–809.
- Zhang W, Robertson WB, Zhao J, et al. Emerging trend in the pharmacotherapy of osteoarthritis. Front Endocrinol (Lausanne). 2019;10:431.
- Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a task force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62(12):1145–1155.
- Kolasinski SL, Neogi T, Hochberg MC, et al. 2019 American College of Rheumatology/Arthritis Foundation guideline for the management of osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2020;72(2):149–162.
- Zhang W, Doherty M, Arden N, et al. EULAR evidence based recommendations for the management of hip osteoarthritis: report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2005;64(5):669–681.
- Spitaels D, Mamouris P, Vaes B, et al. Epidemiology of knee osteoarthritis in general practice: a registry-based study. BMJ Open. 2020;10(1):e031734.
- Birtwhistle R, Morkem R, Peat G, et al. Prevalence and management of osteoarthritis in primary care: an epidemiologic cohort study from the Canadian Primary Care Sentinel Surveillance Network. CMAJ Open. 2015;3(3):E270–275.
- Zeng C, Zhang W, Doherty M, et al. Initial analgesic prescriptions for osteoarthritis in the United Kingdom, 2000-2016. Rheumatology (Oxford). 2021;60(1):147–159.
- Trentalange M, Runels T, Bean A, et al. Analgesic prescribing trends in a national sample of older veterans with osteoarthritis: 2012-2017. Pain. 2019;160(6):1319–1326.
- Akazawa M, Mimura W, Togo K, et al. Patterns of drug treatment in patients with osteoarthritis and chronic low back pain in Japan: a retrospective database study. J Pain Res. 2019;12:1631–1648.
- Nalamachu SR, Robinson RL, Viktrup L, et al. Multimodal treatment patterns for osteoarthritis and their relationship to patient-reported pain severity: a cross-sectional survey in the United States. J Pain Res. 2020;13:3415–3425.
- Schnitzer TJ, Marks JA. A systematic review of the efficacy and general safety of antibodies to NGF in the treatment of OA of the hip or knee. Osteoarthritis Cartilage. 2015;23(Suppl 1):S8–17.
- Yang S, Huang Y, Ye Z, et al. The efficacy of nerve growth factor antibody for the treatment of osteoarthritis pain and chronic low-back pain: a meta-analysis. Front Pharmacol. 2020;11:817.
- Mayorga AJ, Wang S, Kelly KM, et al. Efficacy and safety of fulranumab as monotherapy in patients with moderate to severe, chronic knee pain of primary osteoarthritis: a randomised, placebo- and active-controlled trial. Int J Clin Pract. 2016;70(6):493–505.
- Ekman EF, Gimbel JS, Bello AE, et al. Efficacy and safety of intravenous tanezumab for the symptomatic treatment of osteoarthritis: 2 randomized controlled trials versus naproxen. J Rheumatol. 2014;41(11):2249–2259.
- Spierings EL, Fidelholtz J, Wolfram G, et al. A phase III placebo- and oxycodone-controlled study of tanezumab in adults with osteoarthritis pain of the hip or knee. Pain. 2013;154(9):1603–1612.
- Schnitzer TJ, Ekman EF, Spierings EL, et al. Efficacy and safety of tanezumab monotherapy or combined with non-steroidal anti-inflammatory drugs in the treatment of knee or hip osteoarthritis pain. Ann Rheum Dis. 2015;74(6):1202–1211.
- Woller SA, Eddinger KA, Corr M, et al. An overview of pathways encoding nociception. Clin Exp Rheumatol. 2017;35 Suppl 107(Suppl 5):40–46.
- Garland EL. Pain processing in the human nervous system: a selective review of nociceptive and biobehavioral pathways. Prim Care. 2012;39(3):561–571.
- Mantyh PW, Koltzenburg M, Mendell LM, et al. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology. 2011;115(1):189–204.
- Schmelz M, Mantyh P, Malfait AM, et al. Nerve growth factor antibody for the treatment of osteoarthritis pain and chronic low-back pain: mechanism of action in the context of efficacy and safety. Pain. 2019;160(10):2210–2220.
- Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14(7):502–511.
- Todd AJ. Identifying functional populations among the interneurons in laminae I-III of the spinal dorsal horn. Mol Pain. 2017;13:1744806917693003.
- International Association for the Study of Pain. IASP terminology [Internet]. [updated 2017 Dec 14. cited 2020 Oct 20]. Available from: https://www.iasp-pain.org/terminology
- Colloca L, Ludman T, Bouhassira D, et al. Neuropathic pain. Nat Rev Dis Primers. 2017;3(1):17002.
- Nlm Q, Santin JR, Stoeberl LC, et al. Pharmacological treatment of chemotherapy-induced neuropathic pain: pPARγ agonists as a promising tool. Front Neurosci. 2019;13:907.
- Schomberg D, Ahmed M, Miranpuri G, et al. Neuropathic pain: role of inflammation, immune response, and ion channel activity in central injury mechanisms. Ann Neurosci. 2012;19(3):125–132.
- Stanos S, Brodsky M, Argoff C, et al. Rethinking chronic pain in a primary care setting. Postgrad Med. 2016;128(5):502–515.
- Xu Q, Yaksh TL. A brief comparison of the pathophysiology of inflammatory versus neuropathic pain. Curr Opin Anaesthesiol. 2011;24(4):400–407.
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895–926.
- Graven-Nielsen T, Arendt-Nielsen L. Peripheral and central sensitization in musculoskeletal pain disorders: an experimental approach. Curr Rheumatol Rep. 2002;4(4):313–321.
- Kidd BL, Urban LA. Mechanisms of inflammatory pain. Br J Anaesth. 2001;87(1):3–11.
- Jimenez-Andrade JM, Mantyh PW. Sensory and sympathetic nerve fibers undergo sprouting and neuroma formation in the painful arthritic joint of geriatric mice. Arthritis Res Ther. 2012;14(3):R101.
- Chung K, Chung JM. Sympathetic sprouting in the dorsal root ganglion after spinal nerve ligation: evidence of regenerative collateral sprouting. Brain Res. 2001;895(1–2):204–212.
- Ghilardi JR, Freeman KT, Jimenez-Andrade JM, et al. Neuroplasticity of sensory and sympathetic nerve fibers in a mouse model of a painful arthritic joint. Arthritis Rheum. 2012;64(7):2223–2232.
- Totsch SK, Sorge RE. Immune system involvement in specific pain conditions. Mol Pain. 2017;13:1744806917724559.
- Schlereth T, Birklein F. The sympathetic nervous system and pain. Neuromolecular Med. 2008;10(3):141–147.
- Pulvers K, Hood A. The role of positive traits and pain catastrophizing in pain perception. Curr Pain Headache Rep. 2013;17(5):330.
- Neogi T. The epidemiology and impact of pain in osteoarthritis. Osteoarthritis Cartilage. 2013;21(9):1145–1153.
- Quartana PJ, Campbell CM, Edwards RR. Pain catastrophizing: a critical review. Expert Rev Neurother. 2009;9(5):745–758.
- Edwards RR, Smith MT, Kudel I, et al. Pain-related catastrophizing as a risk factor for suicidal ideation in chronic pain. Pain. 2006;126(1):272–279. .
- Semeru GM, Halim MS. Acceptance versus catastrophizing in predicting quality of life in patients with chronic low back pain. Korean J Pain. 2019;32(1):22–29.
- Campbell CM, Buenaver LF, Finan P, et al. Sleep, pain catastrophizing, and central sensitization in knee osteoarthritis patients with and without insomnia. Arthritis Care Res (Hoboken). 2015;67(10):1387–1396.
- McDougall JJ. Arthritis and pain Neurogenic origin of joint pain. Arthritis Res Ther. 2006;8(6):220.
- Loeser RF, Goldring SR, Scanzello CR, et al. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–1707.
- Bedson J, Croft PR. The discordance between clinical and radiographic knee osteoarthritis: a systematic search and summary of the literature. BMC Musculoskelet Disord. 2008;9(1):116.
- Suokas AK, Walsh DA, McWilliams DF, et al. Quantitative sensory testing in painful osteoarthritis: a systematic review and meta-analysis. Osteoarthritis Cartilage. 2012;20(10):1075–1085.
- Miller RE, Miller RJ, Malfait A-M. Osteoarthritis joint pain: the cytokine connection. Cytokine. 2014;70(2):185–193.
- Lluch E, Torres R, Nijs J, et al. Evidence for central sensitization in patients with osteoarthritis pain: a systematic literature review. Eur J Pain. 2014;18(10):1367–1375.
- Kc R, Li X, Kroin JS, et al. PKCδ null mutations in a mouse model of osteoarthritis alter osteoarthritic pain independently of joint pathology by augmenting NGF/TrkA-induced axonal outgrowth. Ann Rheum Dis. 2016;75(12):2133–2141.
- Burian M, Geisslinger G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol Ther. 2005;107(2):139–154.
- Kirkby NS, Chan MV, Zaiss AK, et al. Systematic study of constitutive cyclooxygenase-2 expression: role of NF-κB and NFAT transcriptional pathways. Proc Natl Acad Sci U S A. 2016;113(2):434–439.
- Smyth EM, Grosser T, Wang M, et al. Prostanoids in health and disease. J Lipid Res. 2009;50(Suppl):S423–428.
- Meves H. The action of prostaglandins on ion channels. Curr Neuropharmacol. 2006;4(1):41–57.
- Svensson CI, Yaksh TL. THESPINAL P HOSPHOLIPASE-C YCLOOXYGENASE-PROSTANOID CASCADE IN NOCICEPTIVE PROCESSING. Annu Rev Pharmacol Toxicol. 2002;42(1):553–583.
- Prostaglandin KA. E2 and pain--an update. Biol Pharm Bull. 2011;34(8):1170–1173.
- Petho G, Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev. 2012;92(4):1699–1775.
- Vanegas H, Vazquez E, Tortorici V. NSAIDs, opioids, cannabinoids and the control of pain by the central nervous system. Pharmaceuticals (Basel). 2010;3(5):1335–1347.
- Li X, Ellman M, Muddasani P, et al. Prostaglandin E2 and its cognate EP receptors control human adult articular cartilage homeostasis and are linked to the pathophysiology of osteoarthritis. Arthritis Rheum. 2009;60(2):513–523.
- Tu M, Yang M, Yu N, et al. Inhibition of cyclooxygenase-2 activity in subchondral bone modifies a subtype of osteoarthritis. Bone Res. 2019;7(1):29.
- Bannuru RR, Osani MC, Vaysbrot EE, et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthritis Cartilage. 2019;27(11):1578–1589.
- Da Costa BR, Reichenbach S, Keller N, et al. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: a network meta-analysis. Lancet. 2017;390(10090):e21–33.
- Bannuru RR, Schmid CH, Kent DM, et al. Comparative effectiveness of pharmacologic interventions for knee osteoarthritis: a systematic review and network meta-analysis. Ann Intern Med. 2015;162(1):46–54.
- Osani MC, Vaysbrot EE, Zhou M, et al. Duration of symptom relief and early trajectory of adverse events for oral nonsteroidal antiinflammatory drugs in knee osteoarthritis: a systematic review and meta-analysis. Arthritis Care Res (Hoboken). 2020;72(5):641–651.
- Ton J, Perry D, Thomas B, et al. PEER umbrella systematic review of systematic reviews: management of osteoarthritis in primary care. Can Fam Physician. 2020;66(3):e89–98.
- Wongrakpanich S, Wongrakpanich A, Melhado K, et al. A comprehensive review of non-steroidal anti-inflammatory drug use in the elderly. Aging Dis. 2018;9(1):143–150.
- CELEBREX® (celecoxib) US prescribing information [Internet]. New York, NY: Pfizer Inc. [updated 2018 June; cited 2020 Oct 21]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/020998s050lbl.pdf
- Voltaren® (diclofenac sodium enteric-coated tablets) US prescribing information [Internet]. Parsippany, NJ: novartis Consumer Health, Inc. [updated 2011 Feb; cited 2020 Oct 21]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/019201s039s040lbl.pdf
- Voltaren® gel (diclofenac sodium topical gel) US prescribing information [Internet] . Parsippany, NJ: novartis Consumer Health, Inc. [updated 2009 July. cited 2020 Oct 21]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022122s006lbl.pdf
- Sostres C, Gargallo CJ, Lanas A. Nonsteroidal anti-inflammatory drugs and upper and lower gastrointestinal mucosal damage. Arthritis Res Ther. 2013;15(Suppl 3):S3.
- Honvo G, Leclercq V, Geerinck A, et al. Safety of topical non-steroidal anti-inflammatory drugs in osteoarthritis: outcomes of a systematic review and meta-analysis. Drugs Aging. 2019;36(Suppl S1):45–64.
- Przybyła GW, Szychowski KA, Paracetamol GJ. An old drug with new mechanisms of action. Clin Exp Pharmacol Physiol. 2021;48(1):3–19.
- Mallet C, Daulhac L, Bonnefont J, et al. Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain. 2008;139(1):190–200.
- Klinger-Gratz PP, Ralvenius WT, Neumann E, et al. Acetaminophen relieves inflammatory pain through CB(1) cannabinoid receptors in the rostral ventromedial medulla. J Neurosci. 2018;38(2):322–334.
- Giuffrida A, Beltramo M, Piomelli D. Mechanisms of endocannabinoid inactivation: biochemistry and pharmacology. J Pharmacol Exp Ther. 2001;298(1):7–14.
- Glaser ST, Kaczocha M. Cyclooxygenase-2 mediates anandamide metabolism in the mouse brain. J Pharmacol Exp Ther. 2010;335(2):380–388.
- Kerckhove N, Mallet C, François A, et al. Ca(v)3.2 calcium channels: the key protagonist in the supraspinal effect of paracetamol. Pain. 2014;155(4):764–772.
- Ray S, Salzer I, Kronschläger MT, et al. The paracetamol metabolite N-acetylp-benzoquinone imine reduces excitability in first- and second-order neurons of the pain pathway through actions on KV7 channels. Pain. 2019;160(4):954–964.
- Machado GC, Maher CG, Ferreira PH, et al. Efficacy and safety of paracetamol for spinal pain and osteoarthritis: systematic review and meta-analysis of randomised placebo controlled trials. BMJ. 2015;350(mar31 2):h1225.
- Towheed TE, Maxwell L, Judd MG, et al. Acetaminophen for osteoarthritis. Cochrane Database Syst Rev. 2006; (1):CD004257. DOI:https://doi.org/10.1002/14651858.CD004257.pub2.
- Conaghan PG, Arden N, Avouac B, et al. Safety of paracetamol in osteoarthritis: what does the literature say? Drugs Aging. 2019;36(Suppl S1):7–14.
- Yoon E, Babar A, Choudhary M, et al. Acetaminophen-induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol. 2016;4(2):131–142.
- Ramachandran A, Jaeschke H. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J Clin Trans Res. 2017;3(Suppl 1):157–169.
- Al-Hasani R, Bruchas Michael R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115(6):1363–1381.
- McDonald J, Lambert D. Opioid receptors. BJA Educ. 2014;15(5):219–224.
- Yaksh TL. Pharmacology and mechanisms of opioid analgesic activity. Acta Anaesthesiol Scand. 1997;41(1):94–111.
- Yaksh TL, Fisher CJ, Hockman TM, et al. Current and future issues in the development of spinal agents for the management of pain. Curr Neuropharmacol. 2017;15(2):232–259.
- Zubieta J-K, Ketter TA, Bueller JA, et al. Regulation of human affective responses by anterior cingulate and limbic µ-opioid neurotransmission. Arch Gen Psychiatry. 2003;60(11):1145–1153.
- Iwaszkiewicz KS, Schneider JJ, Hua S. Targeting peripheral opioid receptors to promote analgesic and anti-inflammatory actions. Front Pharmacol. 2013;4:132.
- Stein C, Lang LJ. Peripheral mechanisms of opioid analgesia. Curr Opin Pharmacol. 2009;9(1):3–8.
- Da Costa BR, Nüesch E, Kasteler R, et al. Oral or transdermal opioids for osteoarthritis of the knee or hip. Cochrane Database Syst Rev. 2014(9):CD003115
- Osani MC, Lohmander LS, Bannuru RR. Is there any role for opioids in the management of knee and hip osteoarthritis? A systematic review and meta-analysis. In Arthritis Care Res (Hoboken). 2020.
- Welsch P, Petzke F, Klose P, et al. Opioids for chronic osteoarthritis pain: an updated systematic review and meta-analysis of efficacy, tolerability and safety in randomized placebo-controlled studies of at least 4 weeks double-blind duration. Eur J Pain. 2020;24(4):685–703.
- Tramadol hydrochloride US prescribing information [Internet]. [updated 2010 May; cited 2020 Oct 21]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022370s000lbl.pdf
- DILAUDID® (hydromorphone hydrochloride) US prescribing information [Internet]. Stamford, CT: purdue Pharma L.P. [updated 2016 Dec. cited 2020 Oct 21]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/019891s024%2C019892s029lbl.pdf
- OXYCONTIN® (oxycodone hydrochloride) US prescribing information [Internet]. Stamford, CT: purdue Pharma L.P. [updated 2015 Aug cited 2020 Oct 21]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022272s027lbl.pdf
- Galligan JJ, Akbarali HI. Molecular physiology of enteric opioid receptors. Am J Gastroenterol Suppl. 2014;2(1):17–21.
- Smith HS, Laufer A. Opioid induced nausea and vomiting. Eur J Pharmacol. 2014;722:67–78.
- Pattinson KT. Opioids and the control of respiration. Br J Anaesth. 2008;100(6):747–758.
- Kosten TR, George TP. The neurobiology of opioid dependence: implications for treatment. Sci Pract Perspect. 2002;1(1):13–20.
- Vowles KE, McEntee ML, Julnes PS, et al. Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain. 2015;156(4):569–576.
- US Department of Health and Human Services. What is the U.S. opioid epidemic? [Internet]. 2019 [updated 2020 Sept 2019; cited 2020 Oct 20]. Available from: https://www.hhs.gov/opioids/about-the-epidemic/
- Colvin LA, Bull F, Hales TG. Perioperative opioid analgesia-when is enough too much? A review of opioid-induced tolerance and hyperalgesia. Lancet. 2019;393(10180):1558–1568.
- Barker PA, Mantyh P, Arendt-Nielsen L, et al. Nerve Growth Factor Signaling and Its Contribution to Pain. J Pain Res. 2020;13:1223–1241.
- Aloe L, Rocco ML, Balzamino BO, et al. Nerve growth factor: a focus on neuroscience and therapy. Curr Neuropharmacol. 2015;13(3):294–303.
- Petty BG, Cornblath DR, Adornato BT, et al. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann Neurol. 1994;36(2):244–246.
- Rukwied R, Mayer A, Kluschina O, et al. NGF induces non-inflammatory localized and lasting mechanical and thermal hypersensitivity in human skin. Pain. 2010;148(3):407–413.
- Andresen T, Nilsson M, Nielsen AK, et al. Intradermal injection with nerve growth factor: a reproducible model to induce experimental allodynia and hyperalgesia. Pain Pract. 2016;16(1):12–23.
- Hayashi K, Shiozawa S, Ozaki N, et al. Repeated intramuscular injections of nerve growth factor induced progressive muscle hyperalgesia, facilitated temporal summation, and expanded pain areas. Pain. 2013;154(11):2344–2352.
- Minnone G, De Benedetti F. Bracci-Laudiero L. NGF and its receptors in the regulation of inflammatory response. Int J Mol Sci. 2017;18(5):1028.
- Marlin MC, Li G. Biogenesis and function of the NGF/TrkA signaling endosome. Int Rev Cell Mol Biol. 2015;314:239–257.
- Stoppiello LA, Mapp PI, Wilson D, et al. Structural associations of symptomatic knee osteoarthritis. Arthritis Rheumatol. 2014;66(11):3018–3027.
- Pecchi E, Priam S, Gosset M, et al. Induction of nerve growth factor expression and release by mechanical and inflammatory stimuli in chondrocytes: possible involvement in osteoarthritis pain. Arthritis Res Ther. 2014;16(1):R16.
- Manni L, Lundeberg T, Fiorito S, et al. Nerve growth factor release by human synovial fibroblasts prior to and following exposure to tumor necrosis factor-alpha, interleukin-1 beta and cholecystokinin-8: the possible role of NGF in the inflammatory response. Clin Exp Rheumatol. 2003;21(5):617–624.
- Schnitzer TJ, Easton R, Pang S, et al. Effect of tanezumab on joint pain, physical function, and patient global assessment of osteoarthritis among patients with osteoarthritis of the hip or knee: a randomized clinical trial. JAMA. 2019;322(1):37–48.
- Berenbaum F, Blanco FJ, Guermazi A, et al. Subcutaneous tanezumab for osteoarthritis of the hip or knee: efficacy and safety results from a 24-week randomised phase III study with a 24-week follow-up period. Ann Rheum Dis. 2020;79(6):800–810.
- Dakin P, DiMartino SJ, Gao H, et al. The efficacy, tolerability, and joint safety of fasinumab in osteoarthritis pain: a phase IIb/III double-blind, placebo-controlled, randomized clinical trial. Arthritis Rheumatol. 2019;71(11):1824–1834.
- Kelly KM, Sanga P, Zaki N, et al. Safety and efficacy of fulranumab in osteoarthritis of the hip and knee: results from four early terminated phase III randomized studies. Curr Med Res Opin. 2019;35(12):2117–2127.
- Hochberg MC, CJ A, TJ S, et al. Long-term safety and efficacy of subcutaneous tanezumab versus nonsteroidal antiinflammatory drugs for hip or knee osteoarthritis: a randomized trial. Arthritis Rheumatol. 2021 [In Press].
- Hochberg MC. Serious joint-related adverse events in randomized controlled trials of anti-nerve growth factor monoclonal antibodies. Osteoarthritis Cartilage. 2015;23(Suppl 1):S18–21.
- Brown M, Koltzenburg M, Nguyen H, et al. Tanezumab does not cause sympathetic nervous system dysfunction in clinical osteoarthritis studies (P3.303). Neurology. 2015;84(Suppl14):3.303.
- Belanger P, Butler P, Butt M, et al. From the cover: evaluation of the effects of tanezumab, a monoclonal antibody against nerve growth factor, on the sympathetic nervous system in adult cynomolgus monkeys (Macaca fascicularis): a stereologic, histomorphologic, and cardio functional assessment. Toxicol Sci. 2017;158(2):319–333.
- Berenbaum F, Schnitzer TJ, Kivitz AJ, et al. General Safety and Tolerability of Subcutaneous Tanezumab for Osteoarthritis: A Pooled Analysis of 3 Randomized, Placebo-Controlled Trials. 2021 May 10. doi:https://doi.org/10.1002/acr.24637
- Bannwarth B, Kostine M. Nerve growth factor antagonists: is the future of monoclonal antibodies becoming clearer? Drugs. 2017;77(13):1377–1387.
- Miller CG, Guermazi A, Roemer F. The current status of imaging in anti-NGF clinical trials. Osteoarthritis Cartilage. 2015;23(Suppl 1):S3–7.
- Gropp KE, Carlson CS, Evans MG, et al. Effects of monoclonal antibodies against nerve growth factor on healthy bone and joint tissues in mice, rats, and monkeys: histopathologic, biomarker, and microcomputed tomographic assessments. Toxicol Pathol. 2018;46(4):408–420.
- LaBranche TP, Bendele AM, Omura BC, et al. Nerve growth factor inhibition with tanezumab influences weight-bearing and subsequent cartilage damage in the rat medial meniscal tear model. Ann Rheum Dis. 2017;76(1):295–302 .
- Hochberg MC, Tive LA, Abramson SB, et al. When is osteonecrosis not osteonecrosis? Adjudication of reported serious adverse joint events in the tanezumab clinical development program. Arthritis Rheumatol. 2016;68(2):382–391.
- Ayhan E, Kesmezacar H, Akgun I. Intraarticular injections (corticosteroid, hyaluronic acid, platelet rich plasma) for the knee osteoarthritis. World J Orthop. 2014;5(3):351–361.
- Kompel AJ, Roemer FW, Murakami AM, et al. Intra-articular Corticosteroid Injections in the Hip and Knee: perhaps Not as Safe as We Thought? Radiology. 2019;293(3):656–663.
- Altman RD, Manjoo A, Fierlinger A, et al. The mechanism of action for hyaluronic acid treatment in the osteoarthritic knee: a systematic review. BMC Musculoskelet Disord. 2015;16(1):321.
- CYMBALTA® (duloxetine) US Prescribing Information [Internet] . [ cited 2021 June 7]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021427s052lbl.pdf
- Weng C, Xu J, Wang Q, et al. Efficacy and safety of duloxetine in osteoarthritis or chronic low back pain: a Systematic review and meta-analysis. Osteoarthritis Cartilage. 2020;28(6):721–734.
- Grässel S, Muschter D Recent advances in the treatment of osteoarthritis. F1000Res. 2020;9:F1000 Faculty Rev-1325.
- Latourte A, Kloppenburg M, Richette P. Emerging pharmaceutical therapies for osteoarthritis. Nat Rev Rheumatol. 2020;16(12):673–688.