
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The selected acyclic halogenated monoterpenes derived from red macroalgae are investigated in terms of their isolation, their reactions with oxidants (ozone, OH radical) and the impacts of the first-generation oxidation products on the atmosphere. The yields of these monoterpenes from marine algae are small and highly variable (0.01–6.7%), the fractions can then enter the atmosphere through different meteorologically driven processes. The loss of the monoterpenes is mostly driven by OH radical, however, 1,6-dibromo-2,7-dichloro-3,7-dimethyl-oct-3-ene and 4-bromo-8-chloro-3,7-dimethyl-octa-2,6-dienal undergo significant loss via reaction with ozone (>50%). The atmospheric lifetimes of the compounds investigated range from ca. 1.5–10 h. Given their short lifetimes, these compounds can be rapidly oxidised to form a range of stable compounds. The investigation of first-generation products from the OH-initiated oxidation of 5,6-dichloro-2-dichloromethyl-6-methyl-octa-1,3,7-triene, 1,6-dibromo-2,7-dichloro-3,7-dimethyl-oct-3-ene and 4-bromo-8-chloro-3,7-dimethyl-octa-2,6-dienal showed the formation of stable organic hydroperoxides, alcohols and carbonyl compounds which can have significant impact on cloud condensation nuclei as contributors of secondary organic aerosols. Release of free halogen can occur from subsequent product oxidation, notably photolysis of carbonyl type species and these are briefly considered.
INTRODUCTION
The chemical structures and bioactivities of over 80 acyclic halogenated monoterpenes, isolated from marine red macroalgae (Rhodophyta), have been reported in a series of comprehensive reviews of the marine natural product chemical literature over the past four decades (Faulkner, Citation1984; Faulkner, Citation2001; Kladi et al., Citation2004; Lever et al., Citation2020; Carrol et al., Citation2020). The wide range of bioactivities associated with these compounds have been regularly reviewed (Cabrita et al., Citation2010; Paul and Pohnert, Citation2011; Cikoš et al., Citation2019) and include antibacterial, antifungal, antiviral, anti-inflammatory, antiproliferative, antitumour, anthelmintic, antioxidant, antifouling, antifeedant, cytotoxic, ichthyotoxic and insecticidal bioactivities. These volatile, marine secondary metabolites are produced in significant quantities by red macroalgae (Maliakal et al., Citation2001; Barahona and Rorrer, Citation2003; Mann et al., Citation2007). However, the variable factors that influence the production of marine monoterpenes in macroalgae are poorly understood (Hay, Citation1996; Wise, Citation2003; Kladi et al., Citation2004; Cikoš et al., Citation2019) and include inter alia: changes in season, reproductive phase of the macroalgae, levels of herbivory and/or microbial infection and availability of nutrients.
Of interest to us, is the possible fate of these volatile halogenated monoterpenes when they enter the atmosphere, either directly when intertidal algae are exposed to the atmosphere during low tide, or indirectly when these compounds diffuse into the water column and then into the atmosphere via wind driven processes (Liss and Slater, Citation1974; Hackenberg et al., Citation2017). In this paper, we postulate what can be happened to these secondary metabolites if they reach the atmosphere and consequently what impact they can have on local and regional air composition.
Terpenes are known to have short atmospheric lifetimes as they react rapidly with atmospheric oxidants (e.g. OH radical, O3, Cl radical and NO3 radical). The measurements (Hallquist et al., Citation2009; Jimenez et al., Citation2009; Ehn et al., Citation2014; Jokinen et al., Citation2015; Berndt et al., Citation2016) and modelling studies (Utembe et al., Citation2011; Khan et al., Citation2017) suggested that the oxidised species formed from the reactions of terpenes with oxidants present in the atmosphere can yield secondary organic aerosol (SOA) in the atmosphere. Thus, the marine terpenes have the potential to significantly influence local climate via formation of SOA which can act as nuclei for water droplet formation and ultimately generate clouds (Kim et al., Citation2017), which, in turn, will reflect solar radiation and contribute to climate cooling in the lower troposphere. Although halogenation of terpenes will tend to have a deactivating effect on the reactivity with tropospheric oxidants to some extent (Atkinson, Citation1987), these atmospheric oxidative reactions of the marine secondary metabolites considered here are still likely to be sufficiently rapid to provide possible routes to SOA formation. In addition, the oxidation mechanisms also result in the release of Cl and Br atoms into the atmosphere. Both halogens participate in cyclical reactive processes that destroy ground level ozone, which is beneficial, as at this altitude ozone is a potent greenhouse gas and a powerful oxidant causing damage to plants (Vainonen and Kangasjärvi, Citation2015) and animals (Zhang et al., Citation2019) and for Cl in particular, also contribute to the initiation of VOC oxidation.
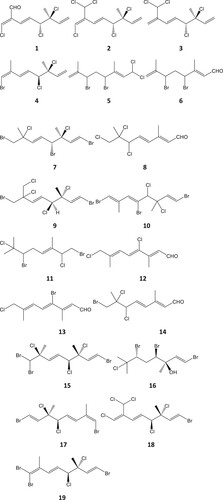
Over 40 halogenated monoterpenes have been reported from the common intertidal red algal genus Plocamium and in this paper, we have predicted the likely major atmospheric degradation routes of a selection of 19 acyclic halogenated monoterpenes (1–19, see ) isolated from four Plocamium species (P. cornutum, P. brasiliense, P. cartilagineum and P. corallorhiza) collected from different regions of the world (Mynderse and Faulkner, Citation1975; Abreu and Galindro, Citation1996; Mann et al., Citation2007; Afolayan et al., Citation2009; Vasconcelos et al., Citation2010); these compounds possess a range of halogenation extent and hence reactivity. We have used established methods discussed in detail in a later section to estimate their rate coefficients for reaction with the OH radical and ozone, which can consequently afford an approximation of the lifetime of the parent compound. The first-generation oxidised products and the very low volatility highly oxidised multifunctional (HOM) products formed from RO2 isomerisation/auto-oxidation including H-shift reactions of RO2 have been investigated concerning their contributions to SOA formation. We have estimated the fraction of the stable possible first-generation oxidation and HOM products from selected oxidation pathways and their vapour pressure. From these hypothetical data we assess the role which these compounds and their products play on aerosol formation and halogen release. The assessment methods used to estimate kinetic and mechanistic information do not distinguish between diastereoisomers and the compounds selected here are a single example of, in some cases, multiple diastereoisomers present in Plocamium and other red macroalgal species.
Figure 1. Halogenated acyclic monoterpenes isolated from four Plocamium species: 1–4 from P. cornutum (South Africa; Afolayan et al., Citation2009); 5–8 from P. corallorhiza (South Africa, Mann et al., Citation2007; Afolayan et al., Citation2009,) 9, 10 from P. brasiliense (Brazil; Vasconcelos et al., Citation2010); 11 from P. cartilagineum (Portugal, Abreu and Galindro, Citation1996); 12–14 from P. corallorhiza (South Africa, Mann et al., Citation2007) and 15–19 from P. cartilagineum (California; Mynderse and Faulkner, Citation1975).
METHODS
Gas-phase rate coefficient estimation methods
OH radical reactions
The rate coefficients for reactions with OH radicals were estimated using the empirically-based structure–activity relationship (SAR) method of Kwok and Atkinson (Citation1995). The SAR method is based upon a number of experimentally determined rate coefficients for volatile organic compounds. The SAR consists of a set of generic rate coefficients for H-atom abstraction and OH addition at a given carbon atom, which are defined in relation to the extent of substitution by alkyl groups. The rate coefficients are further modified to take account of the influence of neighbouring oxygenated and halogenated substituents. In the present work, the following additional assumptions were made:
The generic rate coefficient for OH addition to CH(-R) = CH(-R) structures was taken to be the average of that given for cis- and trans- isomers in Kwok and Atkinson (Citation1995).
The substituent factor given for -CH2Cl groups, in relation to OH addition, was taken to apply to all -CH2X, -CHX2, -CH(X)- and -C(X)< groups, where X = Cl and Br.
Owing to a lack of kinetic data, the Kwok and Atkinson (Citation1995) method does not cover reactions with conjugated trienes, for which there is one example in the current set of compounds (12). In the present work, the following additional assumptions were made:
On the basis of the limited reported data for 1,3,5-hexatriene and 1,3,5-cycloheptatriene (Atkinson and Arey, Citation2003), a rate coefficient of 1.0 × 10−10 cm3 molecule−1 s−1 was assumed representative of OH addition to trienes with no more than one alkyl (R) substitution at each terminal carbon, i.e. CH(-R) = CHCH = CHCH = CH2 and CH(-R) = CHCH = CHCH = CH(-R).
Based on the SARs defined for conjugated dienes (Kwok and Atkinson, Citation1995), additional substitution of each R group was assumed to increase the rate coefficient by a factor of 1.35, up to a maximum rate coefficient of 3 × 10−10 cm3 molecule−1 s−1. For the reference structure CH(-R) = C(-R)C(-R) = CHCH = C(-R)2, containing three additional R substituents (being relevant to Compound N), a rate coefficient of 2.46 × 10−10 cm3 molecule−1 s−1 was therefore used.
O3 reactions
The estimation method used for the reactions with O3 was also derived from an empirically-based SAR method, developed by Atkinson, as reported in Calvert et al. (Citation2000). This is more limited than the OH method, considering only addition of O3 to monoalkenes and unconjugated polyalkenes. The related generic rate coefficients are very similar to those applied with the Master Chemical Mechanism, MCM (Jenkin et al., Citation1997). The method was supplemented here to allow rough estimation of addition rate coefficients to conjugated dienes and trienes, and the impact of substituent groups, as summarised in and . These supplementary methods are subject to some uncertainty, owing to the highly limited dataset of relevant compounds.
Table 1. Group rate constants used for addition of O3 to alkenes and conjugated dienes and trienes.
Table 2. Substituent factors used for addition of O3 to alkenes and conjugated dienes and trienes.
Vapour pressure estimation methods
The vapour pressure of the halogenated monoterpenes and their oxidation products were estimated using the method of Nannoolal et al. (Citation2008) in conjunction with species boiling points estimated by the method of Nannoolal et al. (Citation2004). This had been previously identified (Booth et al., Citation2010) as one of the best estimation methods for highly functionalised, low volatility compounds. A brief summary is repeated here: The method is based on group contributions and includes 207 primary and secondary groups and group interactions. First, this is used to calculate a normal boiling point, Tb for the compound of interest:
(1)
(1) where Ni is the number of groups of type i, Ci the group contribution of group i, a, b, c are adjustable parameters from a linear regression of the equation to experimental boiling points, and n is the total number of atoms in the molecule (except hydrogen). The boiling point estimate is discussed in further detail in Nannoolal et al. (Citation2004). Secondly, using the boiling point as an input, the vapour pressure is calculated by extrapolating this down to the temperature of interest. The vapour pressure is given by:
(2)
(2) where dB adjusts the slope of the vapour pressure curve and is calculated by a group contribution method:
(3)
(3) where the first term in the brackets is the sum of the primary and secondary group contributions, and the second term is the group interaction:
(4)
(4) where Ci–j = Cj − I and m, n are the total number of inter-acting groups and the number of (non-hydrogen) atoms in the molecule, respectively. The vapour pressure estimate is discussed in further detail in Nannoolal et al. (Citation2008). The vapour pressures were calculated from SMILE strings generated for each compound and processed on the University of Manchester multiphase System online Property prediction (UManSysProp) facility (Barley and McFiggans, Citation2010; O’Meara et al., Citation2014).
Method for gas-aerosol partitioning of the oxidation products
The representation of SOA formation is based on partitioning between the vapour and condensed phases of the species formed from the considered halogenated monoterpenes over the 1st generations of oxidation. The adsorption coefficient value, 6.2 × 10−3 m3 μg−1 s−1, estimated for collision of the gas molecules with a monodispersed aerosol of 50 nm diameter, is multiplied with condensed organic material concentrations to calculate adsorption rate coefficient of gas molecules. The desorption rate coefficient of aerosol is calculated using the adsorption rate and partitioning coefficients which is calculated using Pankow’s expression (Pankow, Citation1994) and subcooled vapour pressures of the gas molecules. The equilibrium partitioning coefficient is defined as
(5)
(5) where γ is the activity coefficient,
is the saturated vapour pressure, MW is the molecular weight of the species, R is the molar gas constant and T is the temperature.
Assuming γ = 1 as shown in Jenkin (Citation2004) and using the calculated vapour pressure of the 1st generation stable oxidation products as discussed in section 2.3, we calculated Kp which will determine the extent of gas-aerosol partitioning of these stable species as shown in Odum et al. (Citation1996).
(6)
(6) where Ca, Cg and Co are the concentrations of the aerosol phase oxidation product, gas phase oxidation product and the total concentration of condensed organic materials (in µg m−3), respectively.
RESULTS AND DISCUSSION
Summary of Kinetic results
The estimated gas-phase rate coefficients for the reactions of the selected halogenated terpenes with OH radical and ozone (O3) is summarised in , together with the estimated vapour pressure and atmospheric lifetime. The rate coefficients of Cl atoms with terpenes are about one order of magnitude larger than for OH (Timerghazin and Ariya, Citation2001), but the Cl atom concentration originating from heterogeneous reaction cycles involving sea-salt is believed to generate concentrations ∼1% of that of OH in the marine boundary layer. Thus, the contribution of Cl atoms in the oxidation process of halogenated terpenes is assumed to be minor compared with that for OH. However, a few indirect experiments suggest that Cl atom concentrations could be higher in marine locations and so Cl atom removal may be significant in certain locations (e.g. Spicer et al., Citation1998). Some of the halogenated terpenes in (1,6,8,12,13,14) contain an aldehydic group which can undergo photolysis in the presence of solar radiation, but previous studies (O’Connor et al., Citation2006; Jiménez, et al., Citation2007; Mellouki et al., Citation2015) showed that photolysis loss for unsaturated aldehydes (even for conjugated aldehydes) is minor compared with the loss process by OH radical. Thus, the reaction of halogenated monoterpenes with Cl and the photolysis loss has not been considered in our kinetic analysis. Rate coefficients for reaction with OH radical vary between ∼ 2 × 10−11 cm3 molecule−1 s−1 (around 1 in 5 successful collisions) to ∼ 1 × 10 −11 cm3 molecule−1 s−1 (around 1 in 10 successful collisions). Most compounds (90%) in the selected cohort of 19 halogenated marine monoterpenes presented here have predicted atmospheric lifetimes > 2 and < 10 h (). The vapour pressures of the compounds are found to be in the range of 0.08–15.6 Pa. The fractional contribution from reaction with OH radical and O3 () suggests that O3-initiated oxidation can be considered a minor atmospheric oxidation process for this majority of halogenated marine monoterpenes and therefore be omitted as a simplification in atmospheric models. The short lifetimes of the compounds suggest that they can affect local and regional atmospheric composition depending on their emission rates and the products formed, the extent of which will have a dependence on meteorology (e.g. wind speed and wind direction). Although OH radical dominates loss of the halogenated monoterpenes, compound 11(1,6-dibromo-2,7-dichloro-3,7-dimethyl-oct-3-ene) and compound 13(4-bromo-8-chloro-3,7-dimethyl-octa-2,6-dienal) behave differently, where their losses via reaction with O3 are around 60 and 50%, respectively.
Table 3. Summary of compounds assessed and their estimated rate coefficients and lifetimes based on an OH radical concentration of 1 × 106 molecule cm−3 and an ozone concentration of 30 ppbv (∼ 7.5 × 1011 molecule cm−3 at the Earth’s surface).
Table 4. The table shows the contributions of OH- and O3-initiated oxidation, assuming [O3]/[.OH] = 7.5 × 105.
Possible influence of halogenated, marine monoterpenes on aerosol production
The understanding of marine monoterpene emissions and the characterisation of their degradation products are important to constrain global predictions of cloud condensation nuclei (CCN) sources, properties and abundances in the marine environment. Myriokefalitakis et al. (Citation2010) showed that the annual global marine SOA from monoterpene oxidation is minor compared with that from the oxidation of DMS and marine amines. However, Kim et al. (Citation2017) found a mean monoterpene sea-to-air emission fluxes of 2.6 × 107 molecules cm−2 s−1, translated to ∼5 ppt monoterpenes which could be responsible for the nucleation rates of particles 1.7 nm in diameter (J1.7) of <10−3 cm−3 s−1. Hu et al. (Citation2013) also showed that the monoterpene-derived SOA concentrations were as large as 11 ng m−3 during phytoplankton blooms. We investigated the possible influence of halogenated marine monoterpenes on aerosol formation to determine their importance in marine atmospheric chemistry.
In order to estimate the impact of the atmospheric oxygenation products of halogenated monoterpenes on aerosol production in the intertidal zone during, for example, low tide, a number of factors need to be considered.
First, an estimate of the biomass of monoterpene producing species of alga occurring in the intertidal zone. In an extensive comparative and quantitative study of the trophic structure of rocky intertidal communities of the Cape Peninsular, South Africa, McQuaid and Branch (Citation1985) postulated a mean weighted biomass of primary producers (macroalgae) with a dried mass range of 197–710 gm−2 on exposed beaches around the Cape Peninsular and a diversity of 9–31 species of macroalgae in each m2 quadrat. P. cornutum, the source of 1–4, was collected from the exposed beaches near Kalk Bay on the Cape Peninsular (Afolayan et al., Citation2009) in a study area, for which McQuaid and Branch had earlier established an average macroalgal biomass of 304 gm−2 and recorded 31 different macroalgal species. The contribution of P. cornutum to the macroalgal diversity in this study area was not quantified by McQuaid and Branch (Citation1985).
Second, estimating the amount of monoterpene present in a marine alga. The volatility of marine monoterpenes hampers an accurate measurement of the yield of these compounds per dry mass of their algal source (macroalgae can contain as much as 85% seawater, John Bolton, Personal Communication) as significant amounts of these compounds can be lost during the drying and isolation process. Therefore, isolated yields of marine algal monoterpenes following the contemporary lyophilisation, extraction and chromatography protocols reported in the marine natural products literature, is considered an underestimate of the in vivo concentrations of these compounds.
The isolated yields of compounds 1–19 per dry mass of alga () showed a range of yields (0.01–6.73%) of monoterpenes is produced from the sample collected from different marine environments. Interestingly, the specimens of P. cornutum collected at Noordhoek, Port Elizabeth (750 kms due east of Kalk Bay), afforded predominantly 2 in a significantly higher yield of 6.7% per dry mass of macroalgae (1.14 g of compound 2 from 16.93 g dry mass) than that collected from Kalk Bay on the west coast of South Africa (yield of 0.71% from 26.02 g dry mass) due to the geographical variation of halogenated metabolites (Afolayan et al., Citation2009).
Table 5. Isolated yields of halogenated monoterpenes per dry mass of alga.
Possible atmospheric oxidation products of compounds 3, 11 and 13
Of the 19 acyclic, halogenated, macroalgal monoterpenes presented in , we have focussed our attention on three short-lived compounds (3, 11 and 13). We postulate that these compounds can influence aerosol production on either a localised or even a macro scale when the three algal species that produce these compounds are exposed during low tide.
In order to understand the atmospheric implications of three halogenated monoterpenes, one must consider the chemistry and the impacts associated with their degradation products. Major mechanistic features of the OH- and O3-initiated chemistry of compounds 3, 11 and 13 with the shortest predicted atmospheric lifetimes of 2.1, 2 and 1.5 h, respectively are summarised schematically in the section that follows. In all cases, the OH initiated chemistry is generally dominated by addition to one or more alkene, diene or triene groups, although a significant H abstraction route also contributes for those species possessing a -CHO group (compound 13). For each parent compound (or group of parent compounds), the structures of likely first-generation oxidation products are illustrated, following classic atmospheric chemistry reaction sequences after the initiation reactions. For simplicity, in the current assessment, the formation of only one peroxy radical (RO2) is presented following OH addition to alkene or conjugated diene or triene structures. In each case, the presented RO2 is judged to be the major contributor to the isomeric set of RO2 radicals that can potentially be formed (consisting of 2, 6 and 12 isomers in the cases un-symmetric alkenes, conjugated dienes and conjugated trienes, respectively).
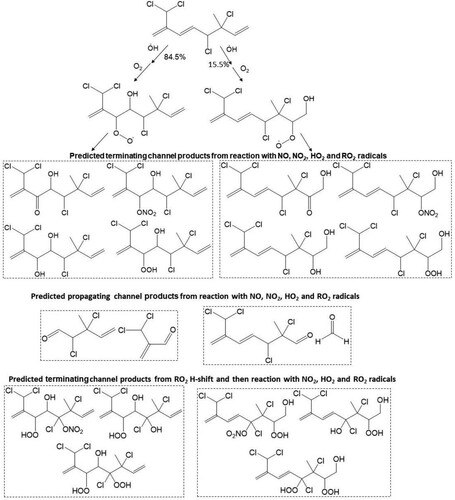
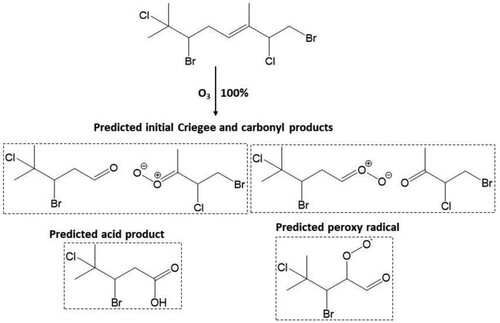
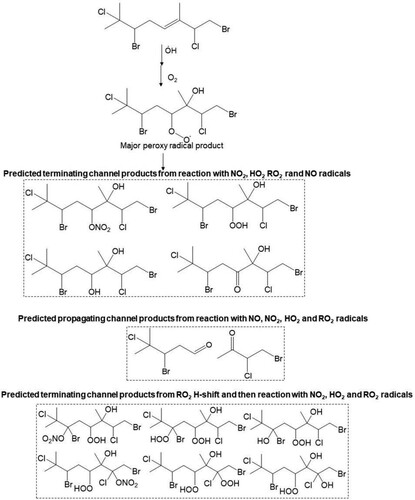
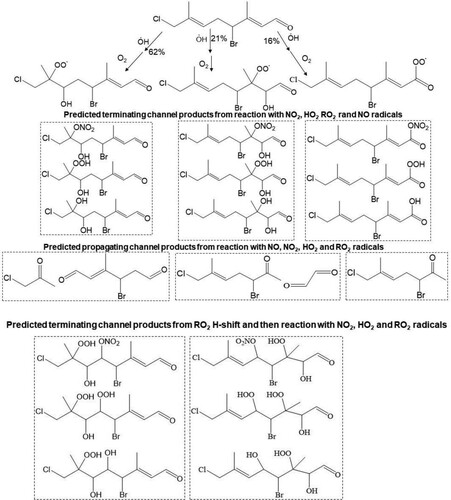
For compound 3, OH radical oxidation almost exclusively dominates with a postulated gas phase lifetime of 2.1 h. The predicted product analysis from the OH radical oxidation of this compound suggests the formation of all first-generation terminating products, e.g. organic hydroperoxides, alcohols, carbonyl compounds, organic nitrates which retain all four chlorine atoms (). Similar types of products are formed from the OH initiated oxidation of compounds 11 and 13 with predicted atmospheric lifetimes of ≤2 h which contain all chlorine/bromine atoms ( and ). However, the rapid unimolecular RO2 H-shift (autooxidation) through 6-member ring transition states can form very low volatility highly oxidised multifunctional (HOM) products shown in , and . Conversely, O3 initiated oxidation can play a significant role in the oxidation of compounds 11 and 13. Both carbonyl and acid products are postulated to form from bimolecular reactions of the stabilised Criegee intermediate (SCI) formed from the ozonolysis of compounds 11 and 13. In addition, the peroxy radical is hypothesised to form via the hydroperoxide Criegee decomposition mechanism which is available for one Criegee intermediate. The predicted products from O3 initiated oxidation of 11 and 13 are presented in and .
Scheme 1. Predicted major peroxy radicals, and terminating and propagating channel products from the reaction of atmospheric OH radical with the Δ3 and Δ7 olefins in 3. Both cohorts of channel products are proposed from reaction of RO2, with NO, NO2, HO2 and RO2 radicals.
Scheme 2. Predicted major Criegee and carbonyl products, acid product and peroxy radical formed from reaction of atmospheric O3 at the Δ5 olefin in 11.
Scheme 3. Proposed major peroxy radical, and terminating and propagating channel products from the reaction of atmospheric OH radical with the Δ5 olefin in 11. Both cohorts of channel products are proposed from reaction of RO2, with NO, NO2, HO2 and RO2 radicals.
Scheme 4. Predicted major Criegee and carbonyl products, acid product and peroxy radicals formed from reaction of atmospheric O3 at the Δ2 and Δ6 olefins in 13.
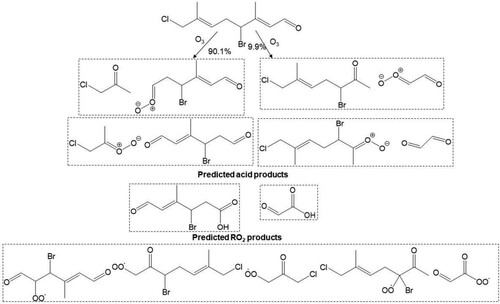
Scheme 5. Predicted major peroxy radicals (RO2), and terminating and propagating channel products from the reaction of atmospheric OH radical with the Δ2, Δ6 and Δ8 olefins in 13. Both cohorts of channel products are proposed from reaction of RO2, with NO, O2, O2 and R’O2 radicals.
The NOx level is low (typically less than <10 ppt) in the marine boundary layer, thus peroxy-peroxy radical self- or/and cross-reactions will dominate leading to the formation of either carbonyl and alcohol or organic hydroperoxide (ROOH). The peroxy radical self-reactions act to slow the production of ozone (Lightfoot et al., Citation1992), thus impacting the oxidation cycle in the marine environment. Depending on the volatility, organic hydroperoxides, alcohols and carbonyl compounds can be partitioned with the particle phase to form SOA (Paulot et al., Citation2009; Utembe et al., Citation2011) which can play an important role by affecting the number and composition of CCN in the marine boundary layer.
Using the fraction yields of monoterpenes from dry algae (), OH radical oxidation mechanisms of monoterpenes (, and ), their first generation OH-oxidation products yields ( and , and ), the OH radical oxidation rate coefficients (), steady-state concentrations of OH, HO2 and RO2 radicals taken from STOCHEM model run (Khan et al., Citation2021) and the rate coefficients for their respective reactions taken from MCM (http://mcm.leeds.ac.uk), we calculated the amount of 1st generation oxidation products of monoterpenes 3, 11 and 13 produced up to 3 h after release from 1 g dry algae (). The analysis shows that 1 g dry algae can produce up to 2 × 1020 molecules organic hydroperoxides formed from compound (11). We found 1.6352 g wet mass of P. cornutum with a surface area of ∼0.0057 m2 which was used to calculate the wet mass of 688 kg per 2400 m2 in Kalk Bay. Using this, Kalk Bay can produce 1.2 × 1026 molecules organic hydroperoxides formed from compound (3). We also calculated the equilibrium partition coefficients, Kp from the vapour pressure of these oxidation products which determine the percentage of gas and aerosol phases of the compounds in the partitioning if the total mass of the condensed organic materials is 1.0 μg/m3 (see ). The data suggests that the OH-initiated 1st generation oxidation leads to produce low volatility, highly oxygenated products (e.g. organic hydroperoxides, alcohols, carbonyl compounds) which can condense thereby enhancing SOA formation. With the inherent heterogeneity of ocean fluxes, the halogenated monoterpenes emissions are high enough along with their high reactivity and SOA yield to contribute an important source of submicron marine SOA (Shaw et al., Citation2010; Kim et al., Citation2017; Yu and Li, Citation2021). A back-of-envelope calculation shows that to generate an increase in SOA concentration of 1 µg/m3 from the 1st generation OH-initiated oxidation product, organic hydroperoxides, the halogenated monoterpene (3), (11) and (13) concentrations need to be at least 14, 191 and 4100 ppt, respectively. Whilst thousands of ppt may be unlikely, 10–100s of ppt can be possible.
Table 6. The vapour pressure of the 1st generation OH-initiated oxidation products produced from monoterpenes 3, 11 and 13 and the percentage of gas and particle phase contribution in the formation of the SOA.
The ozonolysis reactions of the halogenated monoterpenes degrade the compounds into smaller molecules, e.g. carbonyl compounds and peroxy radicals (see and ). These peroxy radicals can also form stable organic hydroperoxides, alcohols and carbonyl compounds, but their vapour pressures are not low enough for being a contributor of SOA. However, organic hydroperoxides are a temporary reservoir of HOx, thus allowing the air to travel away from the marine boundary layer, thus contributing HOx upon photolysis or undergoes a reaction with OH radical. Further oxidation of the products (e.g. photolysis of carbonyl compounds containingBr or Cl atom) can release Cl and Br radicals into the atmosphere. We showed a simplified example of the photolysis of halogenated carbonyl compound produced from the ozone-initiated oxidation of halogenated monoterpenes, where halogen can be freed by the following reaction sequence (Equtions 7 to 10):
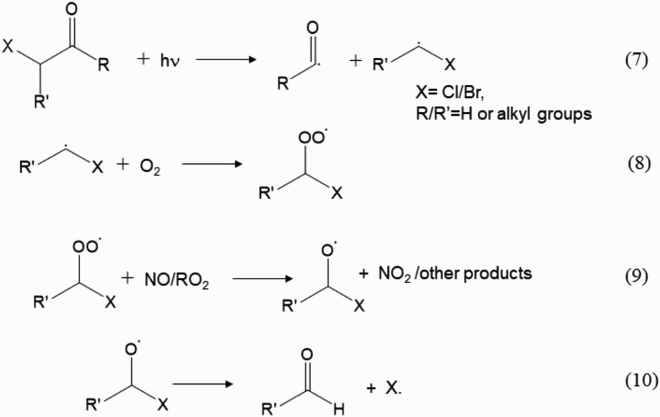
Both halogens can participate in cycles that destroy tropospheric level ozone in the marine boundary layer (Simpson et al., Citation2015). In addition, Cl radical can also react with CH4, another potent greenhouse gas in the atmosphere, reducing the lifetime of this gas. Therefore, when acyclic halogenated marine monoterpenes enter the atmosphere a cursory analysis suggests that they will be potential climate coolers on local and regional scales.
CONCLUSION
A suite of acyclic halogenated monoterpenes produced from marine macroalgae is investigated in terms of their atmospheric lifetimes (due to reactions with OH and ozone), and the atmospheric impacts of the stable products formed from their 1st generation oxidation. The lifetimes of the selected monoterpenes are found to be in the range of 1.5–10 h, thus they have an environmental impact on the marine boundary layer through the formation of stable products such as organic hydroperoxides, alcohols, carbonyl compounds and organic acids. OH-initiated oxidation compounds (3), (11) and (13) can produce organic hydroperoxides up to 7.35 × 1019, 1.99 × 1020 and 3.65 × 1018 molecules from 1 g dry algae which can partition with the aerosol phase due to their low vapour pressures resulting in the contribution of SOA formation. The oxidation of compounds (11) and (13) by ozone is significantly high, contributing up to 60 and 50%, respectively to the total loss process, the oxidation products produced from these channels can free chlorine and bromine radicals by photolysis and further oxidations. The measurements of the emission flux of these halogenated monoterpenes from marine red macroalgae, kinetics and products for the oxidation reactions of these monoterpenes by OH radical and ozone will refine our estimation and constrain the role of oxidation products of halogenated monoterpenes in SOA formation and tropospheric ozone depletion.
DATA AVAILABILITY STATEMENT
The methodological data will be available from the corresponding author on request.
Additional information
Funding
References
- Abreu, P.M. & Galindro, J.M. 1996. Polyhalogenated monoterpenes from Plocamium cartilagineum from the Portuguese coast. Journal of Natural Products 59: 1159–1162.
- Afolayan, A.F., Mann, M.G.A., Lategan, C.A., Smith, P.J., Bolton, J.J. & Beukes, D.R. 2009. Antiplasmodial halogenated monoterpenes from the marine red alga Plocamium cornutum. Phytochemistry 70: 597–600.
- Atkinson, R. 1987. A structure-activity relationship for the estimation of rate constants for the gas-phase reactions of OH radicals with organic compounds. International Journal of Chemical Kinetics 19: 799–828.
- Atkinson, R. & Arey, J. 2003. Atmospheric degradation of volatile organic compounds. Chemical Reviews 103: 4605–4638.
- Barahona, L.F. & Rorrer, G.L. 2003. Isolation of halogenated monoterpenes from bioreactor-cultured microplantlets of the macrophytic red algae Ochtodes secundiramea and Portieria hornemannii. Journal of Natural Products 66: 743–751.
- Barley, M.H. & McFiggans, G. 2010. The critical assessment of vapour pressure estimation methods for use in modelling the formation of atmospheric organic aerosol. Atmospheric Chemistry and Physics 10: 749–767.
- Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjaer, R.V., Kjaergaard, H.G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M. & Ehn, M. 2016. Hydroxyl radical-induced formation of highly oxidized organic compounds. Nature Communications 7: 13677.
- Booth, A.M., Barley, M.H., Topping, D.O., McFiggans, G. & Garforth, A. 2010. Solid state and sub-cooled liquid vapour pressures of substituted dicarboxylic acids using Knudsen effusion mass spectrometry (KEMS) and differential scanning calorimetry. Atmospheric Chemistry and Physics 10: 4879–4892.
- Cabrita, M.T., Vale, C. & Rauter, A.P. 2010. Halogenated compounds from Marine Algae. Marine Drugs 8: 2301–2317.
- Calvert, J.G., Atkinson, R., Kerr, J.A., Madronich, S., Moortgat, G.K., Wallington, T.J. & Yarwood, G. 2000. The mechanisms of atmospheric oxidation of alkenes. New York, Oxford University Press. ISBN 0-19-513177-0.
- Carrol, A.R., Copp, B.R., Davis, R.A., Keyzers, R.A. & Prinsep, M.R. 2020. Marine natural products. Natural Product Reports 37: 175–223.
- Cikoš, A.-M., Jurin, M., Což-Rakovac, R., Jokiċ, S. & Jerkoviċ, I. 2019. Update on monoterpenes from red macroalgae: isolation, analysis and bioactivity. Marine Drugs 17: 537–557.
- Ehn, M., Thornton, J.A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L.B., Jørgensen, S., Kjaergaard, H.G., Canagaratna, M., Maso, M.D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D.R., Wildt, J. & Mentel, T.F. 2014. A large source of low-volatility secondary organic aerosol. Nature 506: 476–479.
- Fantechi, G., Jensen, N.R., Hjorth, J. & Peeters, J. 1998. Mechanistic studies of the atmospheric oxidation of methyl butenol by OH radicals, ozone and NO3 radicals. Atmospheric Environment 32: 3547–3556.
- Faulkner, D.J. 1984. Marine natural products: metabolites of marine algae and herbivorous Marine Molluscs. Natural Product Reports 1: 251–280.
- Faulkner, D.J. 2001. Marine natural products (1999). Natural Product Reports 18: 1–49.
- Hackenberg, S.C., Andrews, S.J., Airs, R.L., Arnold, S.R., Bouman, H.A., Cummings, D., Lewis, A.C., Minaeian, J.K., Reifel, K.M., Small, A., Tarran, G.A., Tilstone, G.H. & Carpenter, L.J. 2017. Basin-scale observations of monoterpenes in the Arctic and Atlantic Oceans. Environmental Science and Technology 51: 10449–10458.
- Hallquist, M., Wenger, J.C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N.M., George, C., Goldstein, A.H., Hamilton, J.F., Herrmann, H., Hoffmann, T., Linuma, Y., Jang, M., Jenkin, M.E., Jimenez, J.L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, T.F., Monod, A., Prevot, A.S.H., Seinfeld, J.H., Surratt, J.D., Szmigielski, R. & Wildt, J. 2009. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmospheric Chemistry and Physics 9: 5155–5236.
- Hay, M.E. 1996. Marine chemical ecology: what’s known and what’s next? Journal of Experimental Marine Biology and Ecology 200: 103–134.
- Hu, Q.-H., Xie, Z.-Q., Wang, X.-M., Kang, H., He, Q.-F. & Zhang, P. 2013. Secondary organic aerosols over oceans via oxidation of isoprene and monoterpenes from Arctic to Antarctic. Scientific Reports 3: 2280.
- Jenkin, M.E. 2004. Modelling the formation and composition of secondary organic aerosol from α- and β-pinene ozonolysis using MCM v3. Atmospheric Chemistry and Physics 4: 1741–1757.
- Jenkin, M.E., Saunders, S.M. & Pilling, M.J. 1997. The tropospheric degradation of volatile organic compounds: a protocol for mechanism development. Atmospheric Environment 31: 81–104.
- Jimenez, J.L., Canagaratna, M.R., Donahue, N.M., Prevot, A.S.H., Zhang, Q., Kroll, J.H., DeCarlo, P.F., Allan, J.D., Coe, H., Ng, N.L., Aiken, A.C., Docherty, K.S., Ulbrich, I.M., Grieshop, A.P., Robinson, A.L., Duplissy, J., Smith, J.D., Wilson, K.R., Lanz, V.A., Hueglin, C., Sun, Y.L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J.M., Collins, D.R., Cubison, M.J., Dunlea, E.J., Huffman, J.A., Onasch, T.B., Alfarra, M.R., Williams, P.I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J.Y., Zhang, Y.M., Dzepina, K., Kimmel, J.R., Sueper, D., Jayne, J.T., Herndon, S.C., Trimborn, A.M., Williams, L.R., Wood, E.C., Middlebrook, A.M., Kolb, C.E., Baltensperger, U. & Worsnop, D.R. 2009. Evolution of organic aerosols in the atmosphere. Science 326: 1525–1529.
- Jiménez, E., Lanza, B., Martinez, E. & Albaladejo, J. 2007. Daytime tropospheric loss of hexanal and trans-2-hexenal: OH kinetics and UV photolysis. Atmospheric Chemistry and Physics 7: 1565–1574.
- Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A.B., Worsnop, D.R., Kulmala, M., Ehn, M. & Sipilä, M. 2015. Production of extremely low volatile organic compounds from biogenic emissions: measured yields and atmospheric implications. Proceedings of the National Academy of Sciences 112: 7123–7128.
- Khan, M.A.H., Jenkin, M.E., Foulds, A., Derwent, R.G., Percival, C.J. & Shallcross, D.E. 2017. A modeling study of secondary organic aerosol formation from sesquiterpenes using the STOCHEM global chemistry and transport model. Journal of Geophysical Research Atmospheres 122: 4426–4439.
- Khan, M.A.H., Schlich, B.-L., Jenkin, M.E., Cooke, M.C., Derwent, R.G., Neu, J.L., Percival, C.J. & Shallcross, D.E. 2021. Changes to simulated global atmospheric composition resulting from recent revisions to isoprene oxidation chemistry. Atmospheric Environment 244: 117914.
- Kim, M.J., Novak, G.A., Zoerb, M.C., Yang, M., Blomquist, B.W., Huebert, B.J., Cappa, C.D. & Bertram, T.H. 2017. Air-Sea exchange of biogenic volatile organic compounds and the impact on aerosol particle size distributions: air-sea exchange of biogenic VOCs. Geophysical Research Letters 44: 3887–3896.
- Kladi, M., Vagias, C. & Roussis, V. 2004. Volatile halogenated metabolites from marine red algae. Phytochemistry Reviews 3: 337–366.
- Knott, M.G., Mkwananzi, H., Arendse, C.E., Hendrickes, D.T., Bolton, J.J. & Beukes, D.R. 2005. Plocoralides A-C, polyhalogenated monoterpenes from the marine alga Plocamium corallorhiza. Phytochemistry 66: 1108–1112.
- Kwok, E.S.C. & Atkinson, R. 1995. Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: an update. Atmospheric Environment 29: 1685–1695.
- Leather, K.E., McGillen, M.R., Ghalaieny, M., Shallcross, D.E. & Percival, C.J. 2011. Temperature-dependent kinetics for the ozonolysis of selected chlorinated alkenes in the gas phase. International Journal of Chemical Kinetics 43: 120–129.
- Lever, J., Brkljača, R., Kraft, G. & Urban, S. 2020. Natural products of marine macroalgae from South Eastern Australia, with emphasis on the Port Phillip bay and heads regions of Victoria. Marine Drugs 18: 142.
- Lightfoot, P.D., Cox, R.A., Crowley, J.N., Destriau, M., Hayman, G.D., Jenkin, M.E., Moortgat, G.K. & Zabel, F. 1992. Organic peroxy-radicals-Kinetics, spectroscopy and tropospheric chemistry. Atmospheric Environment. Part A. General Topics 26: 1805–1961.
- Liss, P.S. & Slater, P.G. 1974. Flux of gases across the air-sea interface. Nature 247: 181–184.
- Maliakal, S., Cheney, D.P. & Rorrer, G.L. 2001. Halogenated monoterpene production in regenerated plantlet cultures of Ochtodes Secundiramea (Rhodophyta, Cryptonemiales). Journal of Phycology 37: 1010–1019.
- Mann, M.G.A., Mkwananzi, H.B., Antunes, E.M., Whibley, C.E., Hendricks, D.T., Bolton, J.J. & Beukes, D.R. 2007. Halogenated monoterpene aldehydes from the South African Marine Alga Plocamium corallorhiza. Journal of Natural Products 70: 596–599.
- Mcquaid, C.D. & Branch, G.M. 1985. Trophic structure of rocky intertidal communities: response to wave action and implications for energy flow. Marine Ecology Progress Series 22: 153–161.
- Mellouki, A., Wallington, T.J. & Chen, J. 2015. Atmospheric chemistry of oxygenated volatile organic compounds: Impacts on air quality and climate. Chemical Reviews 115: 3984–4014.
- Mynderse, J.S. & Faulkner, D.J. 1975. Polyhalogenated monoterpenes from the Red Alga Plocamium cartilagineum. Tetrahedron 31: 1963–1967.
- Myriokefalitakis, S., Vignati, E., Tsigaridis, K., Papadimas, C., Sciare, J., Mihalopoulos, N., Facchini, M.C., Rinaldi, M., Dentener, F.J., Ceburnis, D., Hatzianastasiou, N., O’Dowd, C.D., van Weele, M. & Kanakidou, M. 2010. Global modeling of the oceanic source of organic aerosols. Advance Meteorology 2010: 939171.
- Nannoolal, Y., Rarey, J. & Ramjugernath, D. 2008. Estimation of pure component properties: Part 3. Estimation of the vapour pressure of non-electrolyte organic compounds via group contributions and group interactions. Fluid Phase Equilibria 269: 117–133.
- Nannoolal, Y., Rarey, J., Ramjugernath, D. & Cordes, W. 2004. Estimation of pure component properties: Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions. Fluid Phase Equilibria 226: 45–63.
- O’Connor, M.P., Wenger, J.C., Mellouki, A., Wirtz, K. & Muñoz, A. 2006. The atmospheric photolysis of E-2-hexenal, Z-3-hexenal and E,E-2,4-hexadienal. Physical Chemistry Chemical Physics 8: 5236–5246.
- Odum, J.R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R.C. & Seinfeld, J.H. 1996. Gas/particle partitioning and secondary organic aerosol yields. Environmental Science and Technology 30: 2580–2585.
- O’Meara, S., Booth, A.M., Barley, M.H., Topping, D. & McFiggans, G. 2014. An assessment of vapour pressure estimation methods. Physical Chemistry and Chemical Physics 16: 19453–19469.
- Pankow, J.F. 1994. An absorption model of gas-particle partitioning involved in the formation of secondary organic aerosol. Atmospheric Environment 28: 189–193.
- Paul, C. & Pohnert, G. 2011. Production and role of volatile halogenated compounds from marine algae. Natural Product Reports 28: 186–195.
- Paulot, F., Crounse, J.D., Kjaergaard, H.G., Kürten, A., St. Clair, J.M., Seinfeld, J.H. & Wennberg, P.O. 2009. Unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science 325: 730–733.
- Shaw, S.L., Gantt, B. & Meskhidze, N. 2010. Production and emissions of marine isoprene and monoterpenes: a review. Advances in Meteorology 2010: 408696.
- Simpson, W.R., Brown, S.S., Saiz-Lopez, A., Thornton, J.A. & von Glasow, R. 2015. Tropospheric halogen chemistry: sources, cycling and impacts. Chemical Reviews 115: 4035–4062.
- Spicer, C.W., Chapman, E.G., Finlayson-Pitts, B.J., Plastridge, R.A., Hubbe, J.M., Fast, J.D. & Berkowitz, C.M. 1998. Unexpectedly high concentrations of molecular chlorine in coastal air. Nature 394: 353–356.
- Timerghazin, Q.K. & Ariya, P.A. 2001. Kinetics of the gas-phase reaction of atomic chlorine with selected monoterpenes. Physical Chemistry Chemical Physics 3: 3981–3986.
- Utembe, S.R., Cooke, M.C., Archibald, A.T., Shallcross, D.E., Derwent, R.G. & Jenkin, M.E. 2011. Simulating secondary organic aerosol in a 3-D Lagrangian chemistry transport model using the reduced Common Representative Intermediates mechanism (CRI v2-R5). Atmospheric Environment 45: 1604–1614.
- Vainonen, J.P. & Kangasjärvi, J. 2015. Plant signalling in acute ozone exposure. Plant Cell & Environments 38: 240–252.
- Vasconcelos, M.A., Ferreira, W.J., Pereira, R.C., Cavalcanti, D.N. & Teixera, V.L. 2010. Chemical constituents from the red alga Plocamium brasiliense (Greville) M. Howe and W.R. Taylor. Biochemical Systematics and Ecology 38: 119–121.
- Wise, M.L. 2003. Monoterpene biosynthesis in marine algae. Phycologia 42: 370–377.
- Yu, Z. & Li, Y. 2021. Marine volatile organic compounds and their impacts on marine aerosol—a review. Science of The Total Environment 768: 145054.
- Zhang, J., Wei, Y. & Fang, Z. 2019. Ozone pollution: a major health hazard worldwide. Frontiers in Immunology 10: 2518.