
Abstract
For the first time, we report of a Swedish family of five individuals with a TTR Glu54Leu (p. Glu74Leu) mutation in the transthyretin gene. This mutation has been previously described a few times in the literature, but no phenotypic or clinical description has been done before. The most common mutation in the Swedish population is TTRVal30Met and is mostly found in the Northern part of Sweden. Interestingly, the TTRGlu54Leu mutation was found in the same endemic area. The main phenotype of the TTR Glu54Leu patients is severe cardiomyopathy, which resulted in heart transplantation for the index person. As previously seen for ATTR amyloidosis patients with mainly cardiomyopathy, the amyloid fibrils consisted of a mixture of full-length and fragmented TTR species. However, western blot analyses detected a previously unrecognized band, indicating that these patients may have a third, so far unrecognized, fibril composition type that is distinct from the usual type A band pattern.
Introduction
Hereditary transthyretin (ATTRv) amyloidosis is an autosomal dominantly inherited disease. More than 120 mutations in the transthyretin (TTR) gene have been reported, and the majority is associated with development of systemic amyloidosis.
The phenotype of ATTRv amyloidosis varies between different mutations, and also within mutations. The most common manifestations of the disease are a peripheral axonal neuropathy and/or cardiomyopathy. However, mutations characterized by central nervous manifestations of the disease, often in combination with ocular symptoms (oculo-meningeal) are also recognized.
TTR circulates as a homo-tetramer and functions as a carrier for thyroxine and retinol-binding protein in serum and cerebrospinal fluid. Dissociation of the tetramer to monomers is believed to lead to misfolding and reassembly of the misfolded monomers into insoluble TTR amyloid (ATTR) fibrils that are deposited in different tissues [Citation1,Citation2].
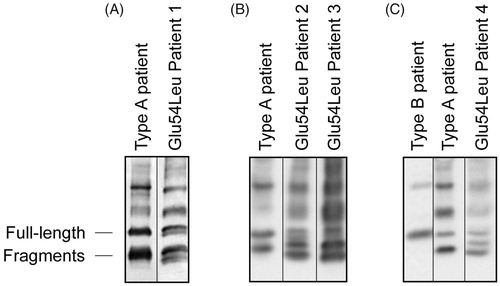
ATTR fibrils either consist of a mixture of full-length TTR and large amounts of C-terminal TTR fragments (fibril type A) or contain mainly full-length TTR molecules (fibril type B). Western blot analysis using an antibody directed against the C-terminal part of TTR (amino acid 50-127) is able to separate between these two fibril composition types. With this antibody, the typical western blot band pattern for type B fibrils is characterised by absence of clear bands below the band corresponding to full-length TTR, while for type A fibrils, a clear and sometimes relatively wide band corresponding to C-terminal fragments is seen below the full-length TTR band (). This particular type A pattern has been reported in all ATTRwt amyloidosis patients [Citation3], in the vast majority of non-Val30Met ATTR amyloidosis cases investigated [Citation4], as well as in a subset of ATTRVal30Met cases who show progressive cardiomyopathy with a general later age of onset [Citation5]. Type A and B ATTR fibrils do not only differ in regards to composition but also structure, which can be seen when studying the amyloid deposits in polarized light after Congo red staining as well as in electron microscope [Citation3,Citation5].
Figure 1. Western blot analysis of adipose tissue. All four of the ATTR Glu54Leu amyloidosis patients we have investigated showed a unique western blot pattern of TTR.
The most common of the Swedish TTR variants causing ATTRv amyloidosis is the Val30Met variant. It is clustered to an area in northern Sweden. Additional cases of TTR mutations have been found in the Swedish population e.g. Val30Leu, Phe33Leu, Ala45Ser, Gly53Glu, Leu55Gln, Gly57Arg, Tyr69His, and His88Arg.
Most TTR mutations have so far only been described in a few cases, which make it difficult to ascertain a common phenotype. Now, a family with the ATTR Glu54Leu (p. Glu74Leu) variant has been discovered, living in the Val30Met endemic area in northern Sweden. The TTR Glu54Leu variant has been mentioned a few times before in the literature. This variant has been listed in reviews [Citation6–8] and patients have participated in research [Citation9,Citation10] but their disease phenotype has never been described in detail.
We now present five cases of ATTR Glu54Leu amyloidosis including their pedigree and clinical presentation. Interestingly, we also found that these patients present a new amyloid fibril type.
Methods
The diagnoses were determined by histopathological examination of tissue samples stained by Congo red and examined in polarized light. Identification of the amyloid precursor protein and amyloid fibril composition was done by western blot on abdominal subcutaneous adipose tissue biopsies, as described in [Citation5], with the exception of using NuPAGE 4-12% Bis-Tris gels with MES SDS running buffer (Fisher Thermo Scientific) according to the manufacturers’ description, for the blots shown in . An in-house produced polyclonal antisera against an in vitro expressed sequence of TTR50-127 was used in the western blot analyses [Citation5,Citation11]. The TTR Glu54Leu mutation was identified in all cases by sequencing of all four exons of the TTR-gene, using traditional Sanger sequencing.
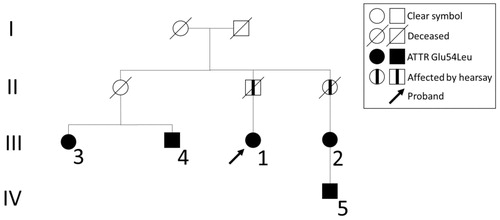
Figure 2. Family pedigree. Relationship of the affected individuals in the Swedish Glu54Leu ATTR family. Arabic numerals refer to the case numbers in the text while the roman numerals refer to the level of generation.
The patients’ medical records were reviewed for information concerning disease onset and symptoms, as well as the outcome of routine clinical investigations of neurological and heart manifestations of the disease. Technetium-99m 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD-) scintigraphy was used to detect TTR amyloid cardiomyopathy [Citation12].
Clinical report
Patient 1
The proband, a female born 1956, sought medical attention at the age of 57 after experiencing increased coughing and shortness of breath the last three months. She had undergone a surgery for a carpal tunnel syndrome 8 years earlier. She smoked until onset of coughing, and had experienced pain in her feet the last year, and used acetylsalicylic acid at bedtime as painkiller. Her weight had been stable, though she had experienced increased bowel discomfort with a tendency of alternating between loose and hard stools.
Her symptoms became acute as she developed atrial fibrillation. She was investigated for suspected heart failure by echocardiographic examination at Piteå Hospital, which disclosed a marked left ventricular hypertrophy with an inter-ventricular septal thickness of 21–22 mm and posterior wall thickness of 18 mm. The ejection fraction was preserved at 55%, but a restrictive velocity pattern across the mitral valve was noted. In addition, findings of low voltage of precordial ECG recording and an abnormal R-wave progression in V1–V3 raised suspicion of an amyloid cardiomyopathy, and the patient was referred to the amyloid team at Piteå Hospital with suspected ATTR amyloidosis.
In the evaluation by the amyloid team at Piteå Hospital, no family history of diagnosed ATTRv amyloidosis was noted. However, her father underwent surgery for carpal tunnel syndrome and had been diagnosed with heart enlargement and died of heart failure at the age of 63. Several of the father´s siblings had similar symptoms, but none had been diagnosed with ATTR cardiomyopathy. Her mother had a myocardial infarction but is alive without any symptoms of heart failure.
The proband has a dizygotic twin sister, who suffers from Parkinson’s disease, and a healthy brother. Another sister had died of malignancy.
At the physical examination, she exhibited no edema, and heart and lung auscultation were normal. However, decreased temperature sensation distally in both legs and in the fingers was noted. The plantar reflexes were absent, but her muscle strength was preserved and no impairment of walking capability was present.
An abdominal fat pad biopsy showed large amounts of amyloid deposits, which by western blot were identified to be composed of transthyretin. Genetic testing with sequencing of all of the four exons unexpectedly showed a mutation formerly unknown in Sweden: Glu54Leu (p.Glu74Leu). The patient was submitted to the Amyloidosis Centre at Umeå University Hospital for further evaluation. The findings are summarized in and .
Table 1. Summary of symptoms and findings for each patient.
Table 2. Cardiac biomarkers and echocardiographic findings for each patient.
The patient underwent sequential heart and liver transplantations in 2014 and 2015, respectively, without complications and is currently alive 5 years after onset of disease. Unfortunately, the patient continued deteriorating regarding the symptoms from the gastrointestinal tract as well as polyneuropathy, both subjectively and objectively.
Patient 2
Woman born 1950, first cousin to the proband. She had temporalis arteritis, amaurosis fugax, hypothyreodism, atrial fibrillation, anaemia, spinal stenosis, bilateral subdural hematoma (both old and new bleedings), hypertension, polyneuropathy and heart failure. The patient was submitted to Amyloidosis Centre at Umeå University Hospital for further evaluation. The outcome is summarized in . She underwent cardiac catheterization which showed elevated filling pressure biventricularly, and echocardiography () showed a hypertrophic left ventricle with pronounced reduced contractions and pronounced impaired systolic function. Comorbidities made her unsuitable for heart transplantation. An abdominal fat pad biopsy showed the presence of transthyretin amyloid, and genetic testing confirmed the TTR Glu54Leu mutation. She was diagnosed by family screening.
Patient 3
Woman born 1942, first cousin to the proband. She suffered from obstructive lung disease and symptoms of heart failure; summary of symptoms in . The abdominal fat pad biopsy showed transthyretin amyloid deposits. DNA testing demonstrated the TTR Glu54Leu mutation. She declined further evaluations. She was diagnosed by family screening.
Patient 4
Man born 1945, first cousin to the proband and brother to patient 3. He suffered from rather advanced rheumatoid arthritis and symptoms of heart failure and was wheelchair bound. An abdominal fat pad biopsy was positive for transthyretin amyloidosis, and the genetic testing confirmed the TTR Glu54Leu mutation. He was submitted to the Amyloidosis Centre at Umeå University Hospital for further evaluation and the outcome is summarized in . He is obese and the neurophysiological examination was rather difficult to evaluate due to advanced rheumatoid arthritis. Echocardiography () showed hypertrophy of the heart, dilated atrium on both the right and left side, however, preserved systolic function. DPD-scintigraphy showed uptake in the septum and left ventricle. The patient showed pronounced edema in both lower extremities. Troponin I was 51 ng/L (reference <15) and proBNP 2000 ng/L (<150). Exercise ECG was difficult to interpret (due to advanced rheumatoid arthritis) but did not show any signs of ischemic heart disease. Resting ECG showed sinus rhythm with first degree AV block, and incomplete left bundle branch block.
The neurography and EMG examination revealed a classic axonal neuropathy with distal dominance.
The patient did not show any clinical symptoms of gastrointestinal dysfunction or any bladder dysfunction. He was diagnosed because he showed symptoms of the TTR disease and initially was suspected as ATTRV30M.
Patient 5
Man born 1973, son to patient 2. He suffered from obstructive sleep apnea and hypopnea syndrome. He had no symptoms from the heart or neuropathic pain or gastrointestinal symptoms. He had undergone surgery for a carpal tunnel syndrome. Because his mother carried the Glu54Leu mutation, he was also tested for the mutation, which was confirmed. An abdominal fat pad biopsy was negative, but the DPD-scintigraphy was positive. Echocardiography () showed a septum thickness of 16 mm, and a normal global systolic left ventricular function. There was no sign of elevated filling pressure. Heart rate variability (HRV) was normal at rest and in standing, and all other HRV parameters were normal. Spirometry and ergospirometry showed ordinary workload expressed in watts, ordinary heart rate response, and systolic blood pressure was at the upper normal limit during heavy work. Long-term ECG showed no bradyarrhythmia. A summary from the evaluation is shown in . He was diagnosed by family screening.
Genealogy
All five cases of ATTR Glu54Leu amyloidosis are relatives and a family pedigree that connects all cases could be constructed ().
Western blot pattern and amyloid fibril composition
Abdominal adipose tissue biopsies showed amyloid in four of the five individuals included in the study. Western blot analysis on the amyloid positive biopsies showed the amyloid to be of TTR nature.
Interestingly, all four of the ATTR Glu54Leu amyloidosis patients we have investigated showed a unique western blot pattern with the anti-TTR50-127 antibody. Instead of one band corresponding to fragmented TTR, two bands were clearly visible, and in some, a third weaker band near the band corresponding to full-length TTR was visible as well ().
Discussion
We have identified five cases of the TTR mutation Glu54Leu (p.Glu74Leu) characterised by amyloid cardiomyopathy and in some cases neuropathy. To the best of our knowledge, this is the first report describing the phenotype of this new mutation. Glu54 is located in the short D-strand (amino acids 53-55) of TTR. Changes in the amino acid composition of the D-strand have been predicted to be highly amyloidogenic [Citation13].
Several reported mutations in the D-strand and also in the position 54 cause aggressive forms of ATTRm amyloidosis [Citation14–17].
The binding of thyroxine (T4) in the two T4 binding pockets of TTR has been suggested to be mediated by polar interactions with Glu54 and Lys15 of TTR and thus stabilizing the TTR tetramer [Citation18,Citation19]. The structure of the entrance to the T4 binding pocket has been shown to be altered in TTR Glu54Gly and Glu54Lys [Citation20]. These mutations caused TTR to be less stabilized by T4 compared to wild-type TTR, which indicates an important role of Glu54 in TTR tetramer stability. Hypothetically this could mean that patients with Glu54 mutations could be less likely to benefit from TTR stabilization by small molecules such as Diflunisal and Tafamidis.
Moreover, Glu54 forms a hydrogen bond with His56 [Citation21]. A substitute of Glu54 could result in a shift in the positions of Glu54/Leu55/His56 with a loss of the D-strand structure which could affect the hydrogen bonding between the D-strand and the A-strand [Citation22]. Hence, the change from the acidic hydrophilic glutamic acid to the non-polar hydrophobic leucine in position 54 could potentially affect the structure of TTR in several ways and subsequently cause dissociation of the tetramer into monomers.
Western blot analysis for determining amyloid fibril composition type revealed a previously unseen band pattern. Instead of detecting either no fragmented bands (as in type B fibrils) or one fragmented band (as in type A fibrils) below the band corresponding to full-length TTR, all four patients with the Glu54Leu mutation showed two or three fragmented bands.
It is believed that the C-terminal fragments seen in western blots for typical type A ATTR amyloid fibrils start around amino acid 50, as fragments beginning at amino acid positions 46, 49, and 52 have been reported to be present in high amounts and even being the major species in such ATTR fibrils [Citation23–25]. Marcoux et al. have suggested that the cleavage and release of a 49–127 fragment is a result of proteolysis/fibrogensis pathway [Citation26]. In this work, it is not known which stretches of the TTR sequence the western blot bands seen in the Glu54Leu patients correspond to, more than that they must belong to TTR50-127 since it is detected by the antibody which recognizes this part of TTR. It is possible that this new type of western blot band pattern is correlated to a third type of fibril composition, with possible differences in fibril structure as well as mechanism of fibrillogenesis. Further studies are needed to answer such questions.
In summary, we report five persons from the same family with the rare ATTRm Glu54Leu amyloidosis in the Swedish population. The phenotype is characterised by cardiomyopathy with a rather early onset, and the amyloid deposits seem to have a unique fibril composition type not encountered before. To date, the best treatment available is a combined heart and liver transplantation; however, medical treatment with patisiran or inotersen could be an option also for these patients.
Ethics approval and consent for publication
Ethical approval was obtained by the Ethics Committee of Umeå University (Sweden), Dnr 2018-329-32M. The patient provided written informed consent for publication.
Acknowledgments
Special thanks to Professor Ole B. Suhr, who contributed with his extensive knowledge in the field.
Disclosure statement
The authors have no financial or other conflicts of interest to disclose.
Data availability statement
Public data sharing is not applicable to this article but portions of the data reported may be available through the corresponding author upon request.
Additional information
Funding
References
- Lai Z, Colon W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 1996;35:6470–6482.
- Quintas A, Vaz DC, Cardoso I, et al. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J Biol Chem. 2001;276:27207–27213.
- Bergstrom J, Gustavsson A, Hellman U, et al. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J Pathol. 2005;206:224–232.
- Ihse E, Rapezzi C, Merlini G, et al. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid. 2013;20:142–150.
- Ihse E, Ybo A, Suhr O, et al. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J Pathol. 2008;216:253–261.
- Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36:411–423.
- Nuvolone M, Obici L, Merlini G. Transthyretin-associated familial amyloid polyneuropathy-current and emerging therapies. Eur Neurol Rev. 2012;7:14–21.
- Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35:E2403–E2412.
- Pilebro B, Suhr OB, Naslund U, et al. (99m)Tc-DPD uptake reflects amyloid fibril composition in hereditary transthyretin amyloidosis. Ups J Med Sci. 2016;121:17–24.
- Quarta CC, Gonzalez-Lopez E, Gilbertson JA, et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J. 2017;38:1905–1908.
- Bergstrom J, Murphy CL, Weiss DT, et al. Two different types of amyloid deposits-apolipoprotein A-IV and transthyretin-in a patient with systemic amyloidosis. Lab Invest. 2004;84:981–988.
- Rapezzi C, Quarta CC, Guidalotti PL, et al. Role of (99m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011;4:659–670.
- Goldsteins G, Andersson K, Olofsson A, et al. Characterization of two highly amyloidogenic mutants of transthyretin. Biochemistry. 1997;36:5346–5352.
- Yamamoto K, Hsu SP, Yoshida K, et al. Familial amyloid polyneuropathy in Taiwan: identification of transthyretin variant (Leu55->Pro). Muscle Nerve. 1994;17:637–641.
- Togashi S, Watanabe H, Nagasaka T, et al. An aggressive familial amyloidotic polyneuropathy caused by a new variant transthyretin Lys 54. Neurology. 1999;53:637–639.
- Busse A, Sanchez MA, Monterroso V, et al. A severe form of amyloidotic polyneuropathy in a Costa Rican family with a rare transthyretin mutation (Glu54Lys). Am J Med Genet A. 2004;128A:190–194.
- Kim HS, Kim SM, Kang SW, et al. An aggressive form of familial amyloid polyneuropathy caused by a Glu54Gly mutation in the transthyretin gene. Eur J Neurol. 2005;12:657–659.
- Wojtczak A, Cody V, Luft JR, et al. Structures of human transthyretin complexed with thyroxine at 2.0 A resolution and 3',5'-dinitro-N-acetyl-L-thyronine at 2.2 A resolution. Acta Crystallogr D Biol Crystallogr. 1996;52:758–765.
- Klabunde T, Petrassi HM, Oza VB, et al. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–321.
- Miyata M, Sato T, Mizuguchi M, et al. Role of the glutamic acid 54 residue in transthyretin stability and thyroxine binding. Biochemistry. 2010;49:114–123.
- Yokoyama T, Mizuguchi M, Nabeshima Y, et al. Hydrogen-bond network and pH sensitivity in transthyretin: neutron crystal structure of human transthyretin. J Struct Biol. 2012;177:283–290.
- Sebastiao MP, Saraiva MJ, Damas AM. The crystal structure of amyloidogenic Leu55 –> Pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J Biol Chem. 1998;273:24715–24722.
- Liepnieks JJ, Wilson DL, Benson MD. Biochemical characterization of vitreous and cardiac amyloid in Ile84Ser transthyretin amyloidosis. Amyloid. 2006;13:170–177.
- Gustavsson A, Jahr H, Tobiassen R, et al. Amyloid fibril composition and transthyretin gene structure in senile systemic amyloidosis. Lab Invest. 1995;73:703–708.
- Westermark P, Sletten K, Johansson B, et al. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA. 1990;87:2843–2845.
- Marcoux J, Mangione PP, Porcari R, et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015;7:1337–1349.