
Abstract
Objective: Infliximab is important in the therapeutic arsenal of Crohn’s disease (CD). However, its effect on mucosal barrier function is not fully understood. Adherent-invasive Escherichia coli (AIEC) are important in CD pathophysiology, but the transmucosal uptake routes are partly unknown. We investigated effects of infliximab on uptake of colon-specific AIEC HM427 across CD colonic mucosa.
Materials and methods: Endoscopic biopsies from non-inflamed colon of seven patients with CD, before and after two infliximab infusions, and eight non-inflammation controls, were mounted in Ussing chambers. Paracellular permeability (51Cr-EDTA) and transmucosal passage of GFP-expressing HM427 were studied. Mechanisms of HM427 transepithelial transport were investigated in Caco-2 monolayers treated with TNF, in the presence of infliximab and/or endocytosis inhibitors.
Results: Before infliximab treatment, colonic passage of HM427 [CD: 2475 CFU (450–3000); controls 1163(225–1950)] and 51Cr-EDTA permeability were increased in CD (p < .05), but were restored to control levels by infliximab (CD: 150 (18.8–1069)). In TNF-exposed Caco-2 monolayers HM427 transport and lipid rafts/HM427 co-localization was decreased by infliximab. The lipid raft inhibitor methyl-β-cyclodextrin decreased HM427 transport.
Conclusion: Infliximab restored the colonic barrier to AIEC in CD; an effect partially mediated by blocking lipid rafts in epithelial cells. This ability likely contributes to infliximab’s clinical efficacy in colonic CD.
Introduction
Crohn’s disease (CD) is a chronic inflammatory bowel disease of unknown etiology that is characterized by a disruption of the intestinal barrier and increased permeability. The increased intestinal permeability reflects a combined defect of transcellular and paracellular barrier function, which partly is mediated by TNF [Citation1]. In vitro studies in epithelial cell monolayers have demonstrated that TNF mediates increased paracellular permeability, which can be characterized by decreased transepithelial electrical resistance, as well as disruption of tight junction proteins [Citation2]. Moreover, it has been shown that TNF signaling may be involved in the increased transcytosis of protein antigen in the ileum of CD [Citation3].
Infliximab is a monoclonal IgG1 antibody, which can bind and neutralize TNF. Apart from neutralizing soluble and membrane bound TNF, the mechanisms of action of anti-TNF antibodies are not completely understood. Moreover, inflammatory bowel disease (IBD) patients receiving infliximab therapy demonstrate a normalized rate of epithelial cell apoptosis [Citation4], improved permeability to small molecules [Citation5], and restored expression of genes related to permeability [Citation6]. These studies suggest an effect of infliximab on epithelial function, but the interaction between infliximab and TNF at the epithelial cell level has been very little studied. Fischer et al showed effects of adalimumab on TNF-induced changes of paracellular permeability and tight junction function in cell monolayers [Citation5], but the effects of anti-TNF treatment on transcellular permeability pathways have not previously been reported.
Numerous studies have proven the involvement of luminal factors and bacteria in the development of CD [Citation2,Citation7]. Altered intestinal microbiota composition and increased amounts of pathogenic bacteria have been found in or at the mucosa of CD patients [Citation8] In particular, adherent-invasive Escherichia coli (AIEC) that have been isolated from intestinal mucosa of CD patients have been in focus [Citation9,Citation10]. The genomic evolution during the last decade has revealed the importance of the innate barrier to microorganisms in the genetic susceptibility to IBD [Citation11]. Despite the current paradigm of a disturbed innate barrier to microbes causing CD, there are to date no reports on the mucosal barrier to bacteria following anti-TNF treatment.
Here, we investigate passage of the CD-associated AIEC strain HM427 (known to be associated with the colonic mucosa of CD patients [Citation10] across non-inflamed colonic mucosa before and after treatment with infliximab in CD patients with colonic disease involvement.
Materials and methods
Patients and ethics
Seven CD patients admitted to the University Hospital of Linköping, Sweden, and eight non-IBD controls, respectively, was included following informed consent and with approval from the Regional Ethics Board in Linköping. The seven patients (1 woman, 6 men; 30 (24–37) years; ) had CD with colonic location (Montreal classification L2 or L3) and a clinical indication for treatment with anti-TNF. Colonic biopsies (n = 8–10 per patient) for ex vivo studies were obtained from macroscopically non-inflamed sigmoid colon during ileocolonoscopy immediately before initiating treatment with infliximab [Remicade® 5 mg per kg body weight (BW) at 0 and 2 weeks], as well as at a second ileocolonoscopy 4–6 weeks after the first infusion. Biopsies from the sigmoid colon of eight healthy individuals [four healthy volunteers and four otherwise healthy individuals undergoing control colonoscopy for colonic adenomas; age 36 (range 25–81) years] were used as controls.
Table 1. Patient characteristics.
Ussing chamber experiments
Biopsies were transported to the laboratory. Six biopsies per individual were mounted in modified Ussing chambers (Harvard apparatus Inc.) as previously described [Citation12,Citation13]. Transmucosal potential difference (PD), short circuit current (Isc) and transmucosal electrical resistance (TMER) were assessed for tissue viability. Live E. coli HM427 were added to the mucosal side at a final concentration of 1 × 108 CFU/ml. Samples from the serosal compartment were collected after 120 min in 2% PFA, and the number of transported bacteria was quantified by flow cytometry (FACscan, BD, Franklin Lakes, NJ, USA). Paracellular permeability was assessed using the 384 Da inert probe 51Cr-EDTA (Perkin Elmer), which was added to the mucosal side to a final concentration of 34 μCi/ml according established protocols [Citation12,Citation13]. Permeability was calculated during the 30–90 min period and given as Papp (apparent permeability coefficient; cm/s × 10−6).
Preparation of bacteria
The bacterial passage across the endoscopic biopsies and Caco-2 cell monolayers was studied using the CD-associated colonic AIEC strain HM427 [Citation10]. AIEC was transformed with a plasmid, pEGFP (BD Biosciences), for expression of enhanced green fluorescent protein (EGFP), as previously described [Citation14], and cultured for experimental use according to standard operating procedures. Moreover, the internalization of another strain of live EGFP incorporated E. coli HB101 (One Shots TOP10 Competent Cells, Invitrogen) was studied in the in vitro model. Bacteria were transfected as described previously [Citation13].
Study of internalization of HM427 E. coli by caco-2 epithelial cell monolayer
Caco-2 cells (1 × 106 cells/ml) were seeded in 12 well plates and cultured for 5–10 days until ∼80% confluent according to standard operating procedures. The cells were then randomly divided into six groups (see Supplementary Table 1 for overview): Group 1 – cells were treated with media only, vehicle group. Group 2 – cells were treated with TNFα (TNF) (R&D Systems) (10ng/ml) for 24 h. Group 3 – cells were treated first with TNF for 24 h and then with the endocytosis inhibitors, either colchicine (10μM) (Sigma-Aldrich) to disrupt tubulin polymerization essential in membrane ruffling [Citation15], or methyl-β-cyclodextrine (mβcd; 2.5 mM) (Sigma-Aldrich) which chelates cholesterol and thus inhibits lipid raft formation by impeding clathrin-coated vesicle formation [Citation16], each for 20 min prior to infection. Group 4 – cells were treated with endocytosis inhibitors alone for 20 min prior to infection. Group 5 – cells were treated with infliximab (1 μg/ml) for 24 h. Group 6 – cells were treated with a combination of TNF (10ng/ml) and infliximab (1 μg/ml) for 24 h.
After the stimulation, Caco-2 cells were exposed to EGFP-expressing E. coli HM427 (108 cfu/ml) for 3 h. The gentamicin protection assay was used to quantify internalized bacteria. Fluorescent intensity was measured using Victor3V multilabel reader (Perkin Elmer, Waltham, MA, USA).
Study of bacterial transport through caco-2 epithelial cell monolayer
Caco-2 epithelial cells (1 × 106 cells/ml) were seeded on 3 μm transwell filter inserts (Corning, Fisher Scientific), according to standard operating procedures. Upon reaching confluence, the cells were divided into six groups and were treated as described above. TNF and infliximab were administered to the basolateral side of the monolayers and the endocytosis inhibitors were added on both apical and basolateral sides. Media was changed to HBSS supplemented with 10% fetal bovine serum prior to administration of endocytosis inhibitors. After stimulation the Caco-2 cells were exposed to EGFP-expressing E. coli HM427 (108 CFU/ml) on the apical side for 3 h. After the 3 h infection period, changes in transepithelial electrical resistance (TEER) was measured using a voltmeter (Millipore,Burlington, MA, USA) and media samples were collected on the basolateral side to assess bacterial transport. Fluorescent intensity of the bacteria in the media was measured using Victor3V multilabel reader. Caco-2 epithelial cells on Transwell filter inserts were fixed in 4% PFA solution for further confocal microscopy studies. Experiments were performed in triplicates, number of experiments were between 5 and 9.
Bacteria-lipid raft interaction in the in vitro model studied by co-localization
Caco-2 epithelial cells were seeded on 3 μm transwell filter inserts and cultured and stimulated with TNF and infliximab on the basolateral side. Cells were pretreated with mβcd (2.5 mM) during 20 min followed by 3 h exposure to E. coli HM427 (108 CFU/ml on the apical side). After infection cells were washed in warm PBS, fixed with 4% PFA and permeabilized in 0.2% Triton X-100 in PBS. F-actin was labeled by incubating cells with Alexa Fluor 633-conjugated phalloidin (Molecular Probes) diluted 1:50 for 40 min in dark chamber at room temperature. After washing cells were stained with Alexa Fluor 594-conjugated cholera toxin subunit B (Molecular Probes) 1:1000 for 45 min at 4 °C. After washing, filters were mounted with 4', 6-diamidino-2-phenylindole dihydrochloride (ProLong-DAPI; Cell Signalling Technology, MA). Images were acquired with Zeiss LSM 710 laser scanning confocal microscope (Jena, Germany) using 60× oil objective. Z stack images were obtained and co-localization was studied using Huygens Professional software (Scientific volume imaging BV, Hilversum, The Netherlands).
Data presentation and statistical analysis
All data were analyzed using the GraphPad Prism 5 Software (GraphPad Software Inc. La Jolla, CA). Data are reported as median and 25th–75th percentile or as box plots unless otherwise stated. Differences between data obtained from the different groups were analyzed, for multiple comparisons, by Kruskal–Wallis test followed by Mann–Whitney U-test. Values of p < .05 were considered statistically significant. Degree of significance was indicated for datasets obtained as follows: p-value < .05 (*), p-value < .01 (**), or ns (non-significant).
Results
Effects of infliximab treatment on colonic barrier function in colonic CD mucosa
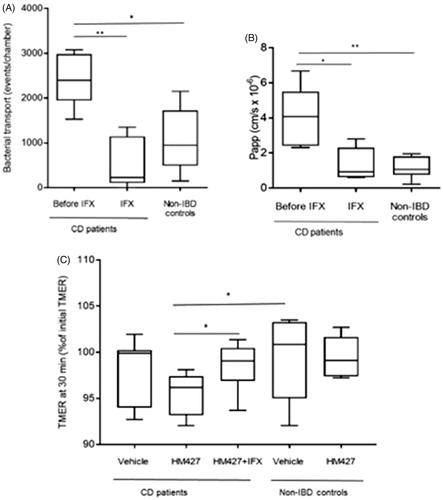
Mucosal barrier function was studied in biopsies of the sigmoid colon in CD patients with colonic involvement before infliximab treatment and four to 6 weeks after initiation of treatment, respectively. All seven patients had a reduction in their CDAI scores, and 2 of 3 with increased CRP at baseline normalized their CRP (). Light microscopy assessment by a clinical pathologist confirmed no active inflammation before or after infliximab treatment.
Biopsies of all groups showed stable and similar PD values (), signifying good viability during the Ussing chamber experiments [Citation12,Citation13]. In line with previous studies of biopsies in patients with colonic inflammation [Citation17], biopsies of CD before infliximab had higher TMER (135 (112–167)% of control values; p < .05) than biopsies after treatment and in controls (), probably due to slight tissue edema.
Table 2. The electrophysiological parameters, potential difference (PD), short circuit current (Isc) and transmucosal epithelial resistance (TMER) at start of experiments patients with biopsies of Crohn’s disease (CD) and controls.
Infliximab restores mucosal barrier function in colonic CD
Transmucosal passage of AIEC HM427 was increased in biopsies from patients with active disease. After infliximab treatment, bacterial passage was significantly reduced, and at similar levels as in the control group (). Correspondingly, the increase in paracellular permeability, as assessed by 51Cr-EDTA flux, was significantly reduced by infliximab treatment (). To assess mucosal response to AIEC, the change in TMER during 30 min of mucosal exposure to HM427 was studied. Colonic CD mucosa before infliximab treatment demonstrated a significant decrease in TMER (to 96 (92–98)% of median in biopsies of controls) during bacterial exposure (p < .05 versus control tissue ± bacteria). Infliximab treatment restored the TMER response to HM427 to control levels ().
Figure 1. (A–C) Assessment of intestinal barrier function in colonic biopsies. Intestinal barrier function was assessed in Ussing Chambers before and after infliximab (IFX) for CD patients (n = 7) and in non-IBD controls (n = 8). Panels show (A) Bacterial transport, displayed as number of fluorescent events in FACS. (B) Paracellular permeability expressed as apparent permeability coefficient (Papp). (C) Transmucosal electrical resistance (TMER). Changes in TMER were measured during 30 minutes of exposure to E. coli HM427 or vehicle. TMER at 30 min is given as % of initial TMER. All data are presented in box plots showing median, 25th–75th percentile for each group. (*p < .05 and **p < .01, calculated by Mann–Whitney U-test).
Effects of TNF and infliximab on TEER and bacterial uptake in caco-2 epithelial monolayers
To study effects of TNF and infliximab at the epithelial level, avoiding confounding from sub-epithelial cell populations, experiments were also performed in Caco-2 monolayers.
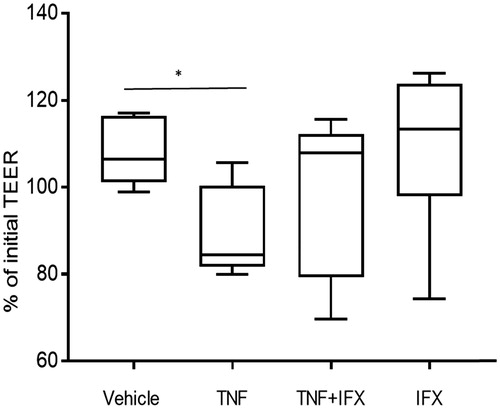
Infliximab normalizes TNF-induced TEER decrease in caco-2 monolayers
TNF (10 ng/ml, 24 h) significantly decreased the transepithelial electrical resistance (TEER) in Caco-2 monolayers compared to TEER during control experiments (vehicle) (p < .05) (). When monolayers were exposed to a combination of TNF and infliximab there was no significant change in TEER. Infliximab alone did not significantly alter TEER.
Figure 2. Effects of TNF and infliximab on transepithelial electrical resistance (TEER) of Caco-2 monolayers. Caco-2 cells were grown on transwell filters and exposed to vehicle, 10ng/ml TNF, TNF +1µg/ml infliximab (IFX) or IFX alone for 24 h (n = 9–10 from three separate experiments). TEER is presented as percentage of initial (time 0 min) and given as box plots showing median, 25th–75th percentile in each group (*p < .05, calculated by Mann–Whitney).
Infliximab reduces TNF-induced uptake of HM427 in caco-2 monolayers
Monolayers were pretreated as above with TNF for 24 h or with a combination of TNF and infliximab. Transepithelial transport of HM427 in monolayers that received TNF + infliximab was 21% lower than those exposed to TNF only (p = .01; ). In addition, TNF-induced internalization of E. coli HM427 in epithelial cells of Caco-2 monolayers was reduced by 10% by infliximab (p < .01; ).
Table 3. Transepithelial uptake and epithelial cell internalization of AIEC HM427 in TNF-exposed Caco-2 monolayers.
TNF and infliximab affect epithelial uptake of E. coli via the lipid raft pathway
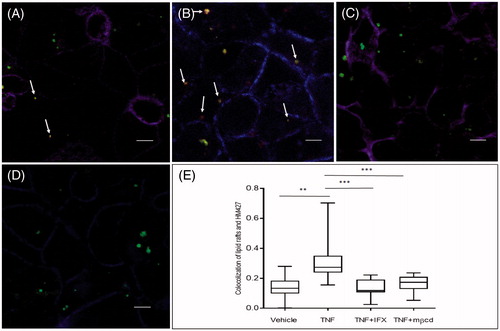
Colchicine (blocks microtubule rearrangement) had no significant effect on TNF-induced transport of E. coli HM427 (). However, TNF-induced transepithelial transport of bacteria was reduced by 50% when monolayers were exposed to mβcd (inhibitor of lipid raft formation) (p = .001; ). Similarly, colchicine had no effect on bacterial internalization (), whereas treatment with mβcd significantly decreased TNF-induced internalization of E. coli HM427 by Caco-2 cells (p = .001; ). The effects of TNF and mβcd were also repeated and confirmed using E. coli HB101 (Supplementary figure 1).
In line with transport and internalization experiments, TNF and infliximab affected co-localization of lipid rafts and HM427 in immunofluorescence experiments in Caco-2 monolayers (). Co-localization of HM427 and lipid rafts increased when monolayers were pretreated with TNF, whereas in the group that received a combination of TNF and infliximab, co-localization of HM427 and lipid rafts were at control (vehicle) levels ().
Figure 3. Effects of TNF, infliximab and methyl-β-cyclodextrin on co-localisation of E. coli HM427 and lipid rafts in Caco-2 monolayers. Caco-2 cells were cultured on transwells filters and exposed to TNF, infliximab (IFX) and methyl-β-cyclodextrin (mβcd) on the basolateral side. Panels show monolayers exposed to fluorescent HM427 E. coli (green) on the apical side in (A) Control; non exposed Caco-2 cells (vehicle) and Caco-2 cells exposed to (B) TNF (10ng/ml), (C) TNF + IFX (1µg/ml). (D) TNF + mβcd (2,5mM). Arrows indicate co-localisation of HM427 with lipid rafts (i.e. yellow (merge green and red) bacteria). (E) Quantitative data of co-localisation coefficient of HM427 (green) and lipid rafts (red) in the photomicrographs of the various groups. Data is presented as boxplots showing median, 25th–75th percentile in each group. (*p < .05 and **p < .01, calculated by Mann–Whitney U-test).
Discussion
The present study focused on the effects of infliximab on colonic barrier function in CD patients with clinical indications for anti-TNF therapy. Collectively, the novel data of the present study show that infliximab restores the barrier to CD-associated colonic AIEC, and suggest that TNF, in addition to unlocking the paracellular leak pathway, can affect epithelial barrier by promoting uptake of bacteria via lipid rafts; an event inhibited by infliximab. Using macroscopically non-inflamed colonic biopsies mounted in Ussing chambers, we demonstrated that patients with active CD have significantly increased paracellular permeability and increased transmucosal passage of AIEC HM427 compared to healthy controls, which we assumed was driven by elevated mucosal TNF production in the CD patients [Citation9]. To test this hypothesis, intestinal permeability and bacterial transcytosis were studied again in the same patients after infliximab treatment. We found that paracellular permeability and bacterial transcytosis were significantly decreased compared to prior to the treatment of infliximab, reaching similar levels as biopsies of controls. These data support the view that at least one of the factors behind increased permeability in CD was in fact TNF.
A correlation between loosening of tight junction integrity and high secretion of pro-inflammatory cytokines has been associated with an increased bacterial penetration through the barrier in in vitro [Citation18] and in vivo studies [Citation19]. The findings of decreased paracellular permeability following infliximab treatment are in accordance with studies on the restoration of intestinal permeability by infliximab, as measured by 51Cr-EDTA permeability [Citation20]. In addition, Zeissig et al have shown that altered epithelial resistance and apoptotic index in biopsies of CD patients were equal to the control group after 2 weeks of infliximab therapy [Citation4]. In order to deepen the understanding of the mechanisms behind these processes, and to study direct effects of TNF and infliximab at the epithelial level, we used an in vitro model of Caco-2 cell monolayers, known to express receptors for TNF, TNFR1 and TNFR2 [Citation21]. Our results showed that treatment with TNF for 24 h significantly reduced the TEER of epithelial monolayers, suggesting increased permeability. Infliximab normalized the TNF-induced fall in TEER, supporting a report on the anti-human TNF antibody adalimumab and its antagonism of TNF-induced disruption of tight junction function in Caco-2 monolayers [Citation5]. Our data suggest that infliximab exerts direct effects on intestinal epithelia in vitro and corroborate the effects on intestinal barrier function in CD biopsies in our ex vivo studies. It should, however, be noted that the results presented here have been obtained using a colonic AIEC only and further experiments needs to be performed in order to elucidate if the mechanisms observed are applicable to other AIECs associated with CD.
Numerous studies have shown that TNF weakens the tight junctional barrier [Citation2]. TNF affects the tight junction leak pathway via MLCK activation, probably mainly via TNFR2 [Citation22]. This could mean that bacteria can then exploit the paracellular pathway to penetrate the epithelial barrier. It has also been shown that AIEC such as LF82 is able to increase paracellular permeability and decrease TEER by re-distribution of the tight junctional proteins in an in vitro model [Citation23]. In previous studies we demonstrated E. coli uptake via both the paracellular and transcellualar pathways in ileum of CD [Citation24], potentially mediated via TNF [Citation25]. To further explore the mechanisms by which bacteria could go across colonic epithelium, internalization assays, as a surrogate of transcellular transport, were performed in Caco-2 monolayers. Infliximab significantly decreased HM427 internalization in TNF-exposed monolayers, thus suggesting that TNF does not only exploit the tight junctional complexes, but also affects transcellular pathways to increase bacterial uptake by Caco-2 cells. Enterocytes rarely internalize bacteria under normal conditions, but there has been growing evidence that epithelial cell-bacterial interaction is a complex process that involves more than one pathway. During these interactions bacteria can modulate cell pathways that are involved in inflammatory process [Citation26,Citation27] and cell growth [Citation28], and may thus affect both the paracellular pathway and endocytosis. Given the reduction in HM427 internalization after treatment it is tempting to speculate that infliximab might be more effective in the subgroup of patients displaying an increased number of AIEC in the colonic mucosa, however, no data exit today on how the presence of AIEC strains correlate to disease severity in IBD.
To study potential transcellular pathways for colonic AIEC we used two endocytosis inhibitors, mβcd and colchicine, and found that TNF and infliximab may affect epithelial cell function via lipid rafts, whereas microtubules seem to be less affected. The microtubule pathway has previously been shown to be involved in uptake of the AIEC LF82 in HEp-2 and prostate RWPE-1 monolayers, but uptake routes have not been fully reported in intestinal cell lines, and not for colonic-type AIEC, such as HM427. Lipid rafts are specialized microdomains, enriched in cholesterol and glycosphingolipids, and found in the plasma membrane of mammalian cells. These microdomain formations are involved in a great number of cellular functions [Citation29]. For example, a recent study showed that lipid rafts regulate differentiation and sodium absorption via the phosphatidylinositol 3-kinase/Akt signaling pathway in Caco-2 cells [Citation30]. In vitro studies have also shown that pathogens may exploit lipid rafts in order to gain entry into the cells [Citation31]. E. coli was one of the first bacteria shown to invade cells via the lipid raft pathway [Citation32]. This is further supported by in vitro studies that have shown that E. coli C25 use the transcellular pathway via lipid rafts structures in order to pass the epithelial barrier under the influence of a combination of IFNγ and TNF [Citation33]. We found that monolayers pretreated with mβcd before TNF had a significantly lower internalization and transport of HM427. In addition, infliximab decreased the co-localization of lipid rafts and HM427. This is in line with previous studies showing that lipid rafts play an important role in bacterial uptake and transport through epithelial cell monolayers [Citation18] and adds the novel findings of infliximab having beneficial effects on barrier function by interacting at the epithelial level with TNF-induced effects on AIEC uptake via lipid rafts.
IGAS_Johan_et_al_Supplemental_Content.zip
Download Zip (26.3 KB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Salim SY, Söderholm JD. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:362–381.
- Ma TY, Iwamoto GK, Hoa NT, et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376.
- Söderholm JD, Streutker C, Yang P-C, et al. Increased epithelial uptake of protein antigens in the ileum of Crohn’s disease mediated by tumour necrosis factor alpha. Gut. 2004;53:1817–1824.
- Zeissig S, Bojarski C, Buergel N, et al. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut. 2004;53:1295–1302.
- Fischer A, Gluth M, Pape U-F, et al. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2013;304:G970–G979.
- Li Z, Arijs I, De Hertogh G, et al. Reciprocal changes of Foxp3 expression in blood and intestinal mucosa in IBD patients responding to infliximab. Inflamm Bowel Dis. 2010;16:1299–1310.
- Söderholm JD, Olaison G, Peterson KH, et al. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn's disease. Gut. 2002;50:307–313.
- MacDonald TT, Hutchings P, Choy MY, et al. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol. 1990;81:301–305.
- Darfeuille-Michaud A, Neut C, Barnich N, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterology 1998;115:1405–1413.
- Martin HM, Campbell BJ, Hart CA, et al. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology. 2004;127:80–93.
- Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124.
- Keita AV, Gullberg E, Ericson A-C, et al. Characterization of antigen and bacterial transport in the follicle-associated epithelium of human ileum. Lab Invest. 2006;86:504–516.
- Wallon C, Braaf Y, Wolving M, et al. Endoscopic biopsies in Ussing chambers evaluated for studies of macromolecular permeability in the human colon. Scand J Gastroenterol. 2005;40:586–595.
- Moussata D, Goetz M, Gloeckner A, et al. Confocal laser endomicroscopy is a new imaging modality for recognition of intramucosal bacteria in inflammatory bowel disease in vivo. Gut. 2011;60:26–33.
- Herd H, Daum N, Jones AT, et al. Nanoparticle geometry and surface orientation influence mode of cellular uptake. ACS Nano. 2013;7:1961–1973.
- Rodal SK, Skretting G, Garred O, et al. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol Biol Cell. 1999;10:961–974.
- Wallon C, Persborn M, Jönsson M, et al. Eosinophils express muscarinic receptors and corticotropin-releasing factor to disrupt the mucosal barrier in ulcerative colitis. Gastroenterology. 2011;140:1597–1607.
- Clark EC, Patel SD, Chadwick PR, et al. Glutamine deprivation facilitates tumour necrosis factor induced bacterial translocation in Caco-2 cells by depletion of enterocyte fuel substrate. Gut. 2003;52:224–230.
- Söderholm JD, Yang P-C, Ceponis P, et al. Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology. 2002;123:1099–1108.
- Suenaert P, Bulteel V, Vermeire S, et al. Hyperresponsiveness of the mucosal barrier in Crohn's disease is not tumor necrosis factor-dependent. Inflamm Bowel Dis. 2005;11:667–673.
- Mochizuki T, Satsu H, Shimizu M. Signaling pathways involved in tumor necrosis factor alpha-induced upregulation of the taurine transporter in Caco-2 cells. FEBS Lett. 2005;579:3069–3074.
- Su L, Nalle SC, Shen L, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145:407–415.
- Wine E, Ossa JC, Gray-Owen SD, et al. Adherent-invasive Escherichia coli, strain LF82 disrupts apical junctional complexes in polarized epithelia. BMC Microbiol. 2009;9:180.
- Keita AV, Salim SY, Jiang T, et al. Increased uptake of non-pathogenic E. coli via the follicle-associated epithelium in longstanding ileal Crohn's disease. J Pathol. 2008;215:135–144.
- Salim SY, Silva MA, Keita AV, et al. CD83 + CCR7- dendritic cells accumulate in the subepithelial dome and internalize translocated Escherichia coli HB101 in the Peyer’s patches of ileal Crohn’s disease. Am. J. Pathol. 2009;174:82–90.
- Haller D, Russo MP, Sartor RB, et al. IKK beta and phosphatidylinositol 3-kinase/Akt participate in non-pathogenic Gram-negative enteric bacteria-induced RelA phosphorylation and NF-kappa B activation in both primary and intestinal epithelial cell lines. J Biol Chem. 2002;277:38168–38178.
- Fries W, Renda MC, Lo Presti MA, et al. Intestinal permeability and genetic determinants in patients, first-degree relatives, and controls in a high-incidence area of Crohn’s disease in Southern Italy. Am J Gastroenterol. 2005;100:2730–2736.
- Sun J, Hobert ME, Rao AS, et al. Bacterial activation of beta-catenin signaling in human epithelia. Am J Physiol Gastrointest Liver Physiol. 2004;287:G220–G227.
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603.
- Li X, Leu S, Cheong A, et al. Akt2, phosphatidylinositol 3-kinase, and PTEN are in lipid rafts of intestinal cells: role in absorption and differentiation. Gastroenterology 2004;126:122–135.
- Wooldridge KG, Williams PH, Ketley JM. Host signal transduction and endocytosis of Campylobacter jejuni. Microb. Pathog. 1996;21:299–305.
- Baorto DM, Gao Z, Malaviya R, et al. Survival of FimH-expressing enterobacteria in macrophages relies on glycolipid traffic. Nature 1997;389:636–639.
- Bowie RV, Donatello S, Lyes C, et al. Lipid rafts are disrupted in mildly inflamed intestinal microenvironments without overt disruption of the epithelial barrier. Am J Physiol Gastrointest Liver Physiol. 2012;302:G781–G793.