
Abstract
Background: Intestinal degenerative neuropathy without extra-intestinal involvement occurs as familial forms (FIDN) but the genetics behind is unknown. We studied a Swedish family with autosomal dominant disease and several cases of chronic intestinal pseudo-obstruction (CIP).
Methods: We included 33 members of a family sharing a male ancestor. Chronic intestinal symptoms including diarrhoea occurred in 11, four had severe CIP. DNA was analysed with SNP-microarray (Affymetrix), linkage (Allegro Software) and gene dosage (CNAG 3.0).
Results: Genetic linkage was found to the short arm of Ch9 to a 9.7 Mb region with 45 protein-coding genes, 22 of which were duplicated (1.2 Mb duplication) (dup(9)(p21.3) with breaking point in the FOCAD-gene. Lod score for the region was 3.4. Fourteen subjects were duplication carriers including all 11 subjects having severe chronic symptoms/CIP. Nineteen subjects had no duplication. The occurrence of gastrointestinal symptoms in the family was strongly linked to duplication carrier-ship (p = .0005). The two branches of the family had separate maternal ancestors (A and B). Including the previous generation, severe disease (overt CIP and/or death from intestinal failure) was assessed to occur in 100% (5/5) of duplication carriers in branch A and in 21% (3/14) in branch B (p = .005). In branch B the onset of symptoms was later (median 38 vs. 24 yrs) and three duplication carriers were symptom-free.
Conclusions: In this family with autosomal dominant hereditary intestinal neuropathy, the disorder is linked to a 9.7 Mb region in Ch9 including a 1.2 Mb duplication. There is a significant difference in disease expressivity between family branches, seemingly related to separate maternal ancestors.
Introduction
Chronic idiopathic intestinal pseudo-obstruction (CIP) occurs as neuropathic, myopathic or mesenchymatic forms [Citation1–3]. For the myopathic forms much interest has been directed to genetic variants of actin in the last decade [Citation4–6]. For the neuropathic diseases leading to CIP, both autosomal dominant and autosomal recessive forms have been identified [Citation1,Citation7] as well as a recessive X-linked chronic neuropathic form leading to CIP reportedly linked to Xq28 and the FLNA (filamine A) gene [Citation8,Citation9]. In general these variants of CIP occur together with dysfunctions in one or several other organs such as the central nervous system, cardiovascular system or the urinary tract.
Hereditary neuropathic disorders confined to the intestinal tract and leading to CIP are relatively rare but may be clinically challenging. Three families with an autosomal dominant form of familial intestinal degenerative neuropathy (FIDN) were reported from 1980 to 1991 [Citation10–12]. In 2011 we reported a large Swedish family with such characteristics, i.e., severe intestinal derangement in both men and women but without oesophageal or extra-intestinal involvement [Citation13]. The onset of symptoms was at adult age, 18 to 70 years, with a medium around 30 years.
The clinical picture in patients with FIDN is similar to several other diseases leading to intestinal failure. Chronic diarrhoea is the most common symptom [Citation10,Citation11,Citation13], but abdominal pain, constipation, nausea and vomiting are also frequent. The final stage of the disease is chronic intestinal pseudo-obstruction, a life-threatening condition. There is no cure for the disease. An important issue is that, at young age, members of FIDN families do not know whether they are carriers of the risk to convey the disease to their children because the genetic background is unknown.
The aim of the present study was to investigate genomic loci associated with disease in a family with FIDN and to assess the relation between chromosomal aberration and the occurrence of gut symptoms.
Material and methods
The study was approved by the Regional Ethics Committee at the University of Gothenburg. Written informed consent was obtained from all study subjects. The blood samples for DNA analysis were de-identified and kept at Department of Laboratory Medicine while the identification code was kept at Institute of Medicine, University of Gothenburg.
Family material
The study subjects belonged to the Swedish kindred with an autosomal dominant form of FIDN previously reported [Citation13]. There was no known consanguinity. The intestinal disease had been demonstrated by histopathology on full-thickness material from three patients in the branches of the family tree (Subjects III:12; III:16; IV:2 in the Pedigree, ). In ten investigated subjects having symptoms compatible with intestinal failure all had chronic diarrhoea and various abnormalities in bowel motility, and six had overt CIP with dilated intestinal loops in the absence of mechanical obstruction [Citation13].
Figure 1. Pedigree showing the kindred with autosomal familial intestinal degenerative neuropathy (FIDN) with indications of clinical and genetic features. Filled symbols indicate chronic gut symptoms. Genetically tested subjects have coloured symbols: Red = Ch9 duplication; Green = no Ch9 duplication.
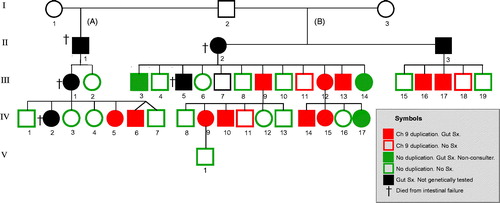
In the present study, DNA from 33 family members was analysed. They were all first-degree relatives to, or were themselves patients with verified CIP or histologically verified degenerative intestinal neuropathy. Eight of the ten previously reported patients with chronic symptoms [Citation13] could be included (two subjects with CIP, III:1 and IV:2, had died from intestinal failure). Moreover, three additional subjects with chronic diarrhoea for 15 to 23 years who also had consulted gastroenterologists were included. Three further subjects had mild abdominal pain (non-consulters) while 19 subjects had been symptom-free. Blood from one husband and one wife to family members was also collected for the linkage analysis. Family members below 18 years of age were not included. The participants’ disease history was explored and they were specifically asked about five groups of symptoms: abdominal pain, diarrhoea, constipation, dysphagia and nausea/vomiting, as well as age at onset of symptoms.
SNP array analyses
DNA was extracted from blood samples and DNA microarray experiments performed as described previously [Citation14]. Affymetrix 250 K SNP arrays (with 262,000 different SNP probes over the genome; Affymetrix, Santa Clara, CA, USA) were used. For all subjects genotypes based on 262,000 loci throughout the genome were compared. Analysis was performed using GDAS (GeneChip® DNA Analysis software) and GTYPE (Affymetrix) for extraction of genotype calls. Linkage was performed using the Allegro software and Z-values calculated; a Z-value ≥3.3 was considered significant. Analysis for gene dosage was also performed with CNAG 3.0 (Copy Number Analyzer for Affymetrix GeneChip Mapping arrays software, version 3.0; Genome Laboratory, Tokyo University). Analysis was also performed using the RFI model (regions free of incompatibilities) described by us earlier [Citation15].
Chi-square test of the association between the occurrence of chromosomal aberrations and the occurrence of intestinal symptoms was performed when appropriate using 2-sided Fischer’s Exact test.
Exome sequencing
DNA from a trio including one in-law healthy subject (III:9, IV:9, and the healthy spouse of III:9) and three additional symptomatic family members (individuals III:14, IV:6, and IV:14; ) were subjected to exome sequencing (Otogenetics corp. Atlanta, GA, USA), aiming for an average coverage of 50X. Bioinformatical handling of exome data (read trimming, mapping, variant calling and annotation) was done through the cloud platform DNAnexus (DNAnexus, Mountain View, CA, USA). Only high quality calls with a minimum of 10X coverage and located within coding sequence or canonical splice sites were kept for further analysis.
Results
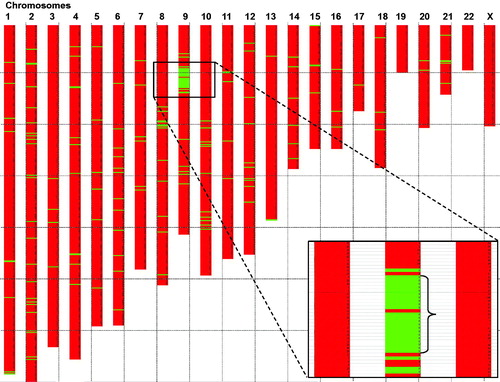
Linkage analysis (Allegro) of SNP array derived genotypes showed a statistically significant linkage between a region in the short arm of chromosome 9 and the chronic intestinal symptoms, Z = 3.4. All 11 patients with chronic intestinal symptoms including diarrhoea showed linkage to a 9.7 Mb region located 13.83 to 23.57 Mb from the p-terminal (pter) in chromosome 9 (). This region is covered by approximately 1060 SNP loci in the used microarray. Among the 19 subjects without intestinal symptoms, three individuals also displayed the linked haplotype while the remaining 16 did not carry the haplotype.
Figure 2. Mapping of a common disease haplotype among affected subjects in the large pedigree (). Genome wide analysis of all affected individuals from the family were analysed using the RFI model (region free of incompatibilities). SNP loci (using Affymetrix arrays with 262,000 individual SNPs) were grouped into 2620 groups of 100 consecutive loci. Regions with 100 consecutive loci showing three or more incompatibilities are shown as a red box in the chromosome plot. No incompatibilities in the group of 100 loci is indicated by a green horizontal box in the chromosome plot. Note that a region on chromosome region 9p showed a strong indication of a haplotype shared by the affected members of the family, thus indicating a common ancestral founder disease haplotype and a mutation. The common ancestral region on chromosome 9 constitute region 13.83 Mb–23.57 Mb from pter (9.74 Mb; 45 protein coding genes).
Gene dose analysis (CNAG) shows that all 14 subjects with the linked haplotype also carry a duplication of 1.2 Mb in Ch9, located 20.9–22.1 Mb from pter [dup(9)(p21.3)] (). The distal breakpoint of the duplication is located within the FOCAD (KIAA1797) gene while the proximal breakpoint is located between CDKN2B and DMRTA1 (). Nineteen subjects, 16 symptom-free and three with milder symptoms (non-consulters) had no duplication in the short arm of Ch9. The presence of Ch9 duplication among the 33 study subjects is shown in the pedigree (, red symbols). According to RefSeq (GCF_000001405.25_GRCh37.p13; 2017-04-19) there are 45 protein coding genes located in the genomic segment linked to FIDN whereof 22 are included in the duplicated region ().
Figure 3. Analysis of SNP array genome profile showed that affected family members in addition to the 9.74 Mb region described in , have a 1.2 Mb duplication in the region 20.9–22.1 Mb from pter.
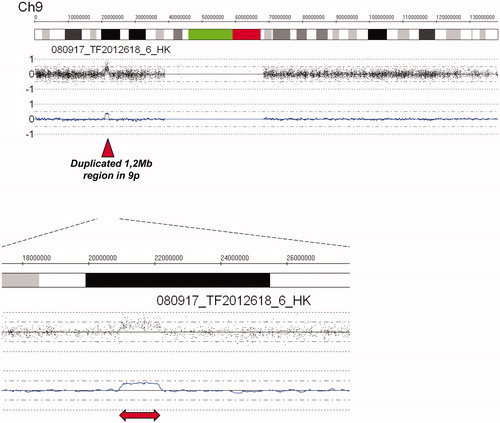
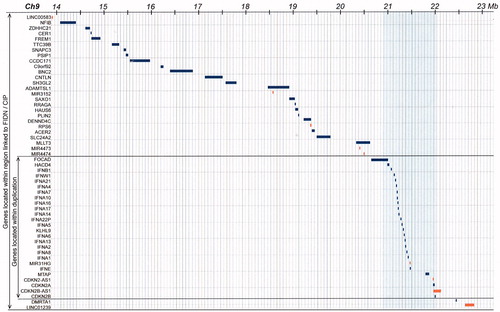
Figure 4. Detailed map of all denoted genes in the 9.7 Mb region of common ancestry and the 1.2 Mb region of duplication. Thus, 22 protein coding genes are within the duplicated gene region. Based on the extension of the duplicated region, one gene, the FOCAD gene (Focadhesin; HGNC: 23377), is predicted to be disrupted by one of the newly introduced breakpoints.
Exome sequencing analysis of five subjects with intestinal symptoms and one in-law showed only six single nucleotide variants in coding sequence of genes located within the linked Ch9 region (13.83-23.57 Mb) that segregated with disease. All six variants are present in dbSNP and include three heterozygous synonymous variants (FREM1, rs10738380; PSIP1, rs2821529; RRAGA, rs41268997) and three missense variants (TTC39B, rs1407977; DENND4C, rs17818730; FOCAD, rs150147497).
Symptoms in relation to the Ch9 duplication
The prevalence of gut symptoms in the 33 family members is shown in . Among the 14 carriers of the 1.2 Mb Ch9 duplication 11 subjects (79%) had chronic symptoms and had consulted gastroenterologists. Chronic diarrhoea was a major symptom in all of them. Three carriers of the duplication had no intestinal symptoms. In contrast to the duplication-carriers, only three out of 19 subjects (16%) without the duplication reported mild abdominal pain (upper abdominal 2, lower abdominal 1). They were non-consulters and none of them had diarrhoea. There was a strong association between the presence of the Ch9 duplication and the occurrence of intestinal symptoms (p = 0.0005; Chi-square test) (). In the eleven subjects with Ch9 duplication and chronic symptoms, the onset of symptoms was from the age of 20 to 70 years, median 31 years. The three duplication carriers without symptoms were 43, 60 and 69 years, respectively.
Table 1. Gastrointestinal symptoms and CIP in 33 family members with or without the 1.2 Mb duplication in chromosome 9 at genetic testing.
Table 2. The 26 duplicated genes (of which 22 are protein coding) in the 1.2 Mb duplication in Ch9 linked to FIDN [dup(9)(p21.3)].
Seven of the eleven duplication carriers with symptoms had done motility investigations. All seven showed jejunal hypomotility and abnormal phase III at manometry while delayed small bowel transit and jejunal enteropathogens were found in 6/7 [Citation13]. Four of the duplication carriers had severe CIP.
Disease severity in the family branches
As outlined in the Pedigree, the common male ancestor (I:2) in this family had children with two women, A and B. Given that the Ch9 duplication was transferred via his children to the present study subjects () and knowing their disease history we assessed the proportion of severe cases among duplication carriers in the two family branches A and B. Verified CIP and/or death from intestinal failure was regarded as the most severe disease stage and occurred in 100% (5/5) in branch A and in 21% (3/14) in branch B (p = .005). Furthermore, the age at onset of symptoms also differed between branches. Median age was 23 years (range 18–35) in branch A and 38 years (range 21–70) in branch B. The boy III:5 () born in 1932 was not included in the assessment; he died from ileus at 3 years age (Parish death register), but otherwise scant clinical information was available.
Discussion
The present study shows that the FIDN that causes disabling symptoms and CIP in this family is linked to a 9.7 Mb region including 1.2 Mb duplication in the short arm of chromosome 9. This is a novel finding since in the similar families with FIDN without oesophageal or extra-intestinal manifestations previously reported [Citation10–12] there is no information about associated chromosomal changes (Emeran Mayer; Michael Camilleri; personal communications).
The coupling between FIDN and Ch9 aberration could be firmly demonstrated, partly due to the size of the family and the high proportion of participating family members; 32 out of 33 subjects still alive in generation III–IV and born from 1927 could be tested ().
The high prevalence of gut symptoms in the tested family members was strongly linked to the Ch9 duplication. While 79% of the duplication carriers had chronic gut symptoms only 16% of subjects without Ch9 duplication had symptoms that were milder and compatible with functional gut disorder. This proportion is similar to that in the general population [Citation16] indicating that family members without Ch9 duplication are not at increased risk to have intestinal disease.
Our genealogy and disease history studies of this family [Citation13] allowed an overall assessment of the likely occurrence of Ch9 duplication in the family branches A and B and its relation to disease severity. With the 100% occurrence of severe disease (death from intestinal failure and/or severe CIP) in branch A compared to 21% in branch B (p = .005) there is apparently a significant difference in disease expressivity between the branches, seemingly related to different maternal ancestors. Furthermore, the disease expressivity seems to be paralleled by a difference in penetrance since, in contrast to branch A, three duplication carriers in branch B had no gut symptoms and onset of symptoms was significantly later in branch B.
The breakpoint of the Ch9 duplication was found to be located within the FOCAD gene and will most likely disrupt or alter gene function. It has been suggested that monoallelic germline FOCAD deletions could lead to colorectal cancer predisposition but this is not prevalent in this family.
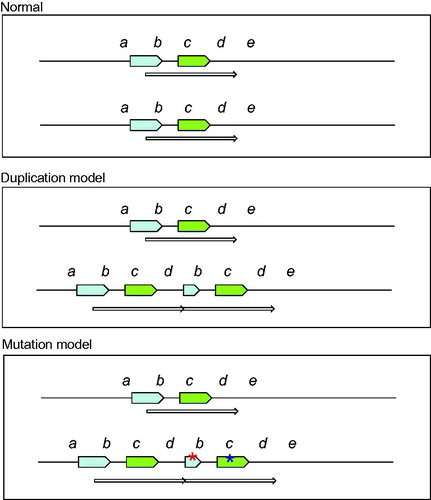
It is not exactly clear which gene is responsible for the enteric neuropathy in this family. One possibility to be considered is a gene dose effect, i.e., the occurrence of a dose sensitive gene or genes (, middle panel). Among the 22 protein-coding genes within the duplication (), there are several interferon genes and also the cell cycle regulating gene CDKN2A (cyclin-dependent kinase inhibitor 2A) (). Severe developmental abnormalities with neural involvement are well known to be associated with gene duplications. Examples are the RAI1 gene in Ch17 causing the Potocki–Lupski syndrome [Citation17] and duplication in Ch15 (15q11-12) [Citation18]. Interestingly, a duplication in Xq28 causing neuropathic CIP has been reported and proposed to be linked to the Filamin A (FLNA) gene [Citation19].
It can at present not be excluded that one of the genes in the Ch9 region, within or outside the duplication () is mutated and associated with the familial intestinal neuropathy described here (, lower panel). However, exome sequencing did not indicate any novel or deleterious alterations within coding sequence of the genes in the Ch9 region.
Figure 5. Tentative model of the chromosome region 9p, 1.2 Mb duplication. The green arrow represents the genes that are duplicated and thus present in 3 copies instead of the normal 2 copies. They include e.g., a group of interferon genes and the tumour suppressor genes CDKN2A/CDKN2B. The blue arrow represents the disrupted FOCAD gene where the 3’-end is present in three copies while the 5’-end is present in normal two copies.
The importance of the Ch9 duplication for the development of intestinal disease in this family may be elucidated by the two sisters III:1 and III:2 in the Pedigree (). While the likely duplication carrier III:1 who died from CIP had three children with severe, disabling CIP, her duplication free sister had two children and five grandchildren, all of them without gut symptoms. Hitherto, several younger family members have abstained from getting own biological children. From now, testing for the Ch9 duplication should offer a possibility to evaluate if younger family members are at risk to develop the disease and to transfer that risk to the next generation.
The present novel findings linking the neural function of the intestine to a gene locus in chromosome 9 seem to be important not only for the clinical handling and guiding in the affected family. Further research on the genetic mechanisms and on the genes involved may open up for a deeper understanding of the enteric nervous system and the pathophysiology of CIP.
| Abbreviations | ||
| FIDN | = | familial intestinal degenerative neuropathy |
| CIP | = | chronic intestinal pseudo-obstruction |
| SNP | = | single nucleotide polymorphism |
| Ch9 | = | chromosome 9 |
| pter | = | p-terminal |
| Mb | = | million base pairs |
Acknowledgements
The authors thank research nurses Gisela Ringström, Annette Ekblad and Patricia Ebbersten for excellent assistance and patient logistics.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Stanghellini V, Cogliandro RF, de Giorgio R, et al. Chronic intestinal pseudo-obstruction: manifestations, natural history and management. Neurogastroenterol Motil. 2007;19(6):440–452.
- De Giorgio R, Cogliandro RF, Barbara G, et al. Chronic intestinal pseudo-obstruction: clinical features, diagnosis and therapy. Gastroenterol Clin North Am. 2011;40(4):787–807.
- Gabbard SL, Lacy BE. Chronic intestinal pseudo-obstruction. Nutr Clin Pract. 2013;28(3):307–316.
- Sipponen T, Karikoski R, Nuutinen H, et al. Three-generation familial visceral myopathy with alpha-actin-positive inclusion bodies in intestinal smooth muscle. J Clin Gastroenterol. 2009;43(5):437–443.
- Lehtonen HJ, Sipponen T, Tojkander S, et al. Segregation of missense variant in smooth muscle actin gamma-2 with autosomal dominant visceral myopathy. Gastroenterology. 2012;143(6):1482–1491.
- Milunsky A, Baldwin C, Zhang X, et al. Diagnosis of chronic intestinal pseudo-obstruction and megacystis by sequencing the ACTG2 gene. J Pediatr Gastroenterol Nutr. 2017;65(4):384–387.
- Auricchio A, Brancolini V, Casari G, et al. The locus for a novel syndromic form of neuronal intestinal pseudo-obstruction maps to Xq28. Am J Hum Genet. 1996;58(4):743–748.
- Gargiulo A, Auricchio R, Barone M, et al. Filamin A is mutated in X-linked chronic intestinal pseudo-obstruction with central nervous involvement. Am J Hum Genet. 2007;80(4):751–758.
- Deglincerti A, De Giorgio R, Cefle K, et al. A novel locus for syndromic chronic idiopathic intestinal pseudo-obstruction maps to chromosome 8q23-q24. Eur J Hum Genet. 2007;15(8):889–897.
- Roy AD, Bharucha H, Nevin NC, et al. Idiopathic intestinal pseudo-obstruction: a familial visceral neuropathy. Clin Genet. 2008;18(4):291–297.
- Mayer EA, Schuffler MD, Rotter JI, et al. Familial visceral neuropathy with autosomal dominant transmission. Gastroenterology. 1986;91(6):1528–1535.
- Camilleri M, Carbone LD, Schuffler MD. Familial enteric neuropathy with pseudoobstruction. Digest Dis Sci. 1991;36(8):1168–1171.
- Ahlfors F, Linander H, Lindström M, et al. Familial intestinal degenerative neuropathy associated with chronic intestinal pseudo-obstruction. Neurogastroenterol Mot. 2011;23(4):347–355.
- Carén H, Erichsen J, Olsson L, et al. High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: four cases of homozygous deletions of the CDKN2A gene. BMC Genomics. 2008;9:353.
- Ohlsson M, Hedberg C, Brådvik B, et al. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain. 2012;135(6):1682–1694.
- Agréus L. The epidemiology of functional gastrointestinal disorders. Europ J Surg Suppl. 1998;583:60–66.
- Potocki L, Bi W, Treadwell-Deering D, et al. Characterization of Potocki–Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80(4):633–649.
- Depienne C, Moreno-De-Luca D, Heron D, et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry. 2009;66(4):349–359.
- Clayton-Smith J, Walters S, Hobson E, et al. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur J Hum Genet. 2009;17(4):434–443.