
Abstract
Purpose
Peripheral autonomic neuropathy, including enteric neuropathy, may be subtle and unrecognized for several years. Diagnosis of enteric neuropathy demands complicated examinations such as full-thickness bowel biopsy. We hypothesized that knowledge about simultaneous occurrence of different types of neuropathy would lead to faster recognition and diagnosis of autonomic/enteric neuropathy. The aim of the present systematic review was to increase the awareness of disease groups causing autonomic and enteric neuropathy along with sensorimotor neuropathy.
Methods
A systematic search strategy was used in PubMed, Embase and Web of Science. First, 4978 articles were identified. Review of titles/abstracts rendered exclusion of animal studies, articles not written in English or full-length, case reports, conference abstracts and duplicates until 357 articles remained. The full-length evaluation resulted in 35 studies (27 non-systematic reviews) which described objectively verified peripheral autonomic, enteric and sensorimotor neuropathy within the same disease.
Results
Diabetes is the most common disease in society rendering generalized peripheral neuropathy. Accumulation of tissue deposits in amyloidosis, Lewy body disorders and sarcoidosis lead to widespread peripheral neuropathy. Several autoimmune disorders such as systemic sclerosis and primary Sjögren’s syndrome present themselves with neuropathy. Paraneoplastic neuropathy may appear prior to symptoms from the malignancy. Both the infection per se, as well as the autoimmune response to the infection, i.e., Guillain–Barré syndrome, may lead to widespread peripheral neuropathy. Hereditary disorders with disturbed metabolism lead to intermittent attacks of neuropathy.
Conclusions
The major causes of generalized peripheral neuropathy are diabetes, diseases with tissue deposits, autoimmunity, infections, malignancy and metabolic diseases.
Introduction
The nervous system consists of several parts responsible for various tasks [Citation1], represented at a central and a peripheral level [Citation2,Citation3]. Approximately, 90% of the autonomous nervous system, including the enteric nervous system, is composed of postganglionic small nerves in the form of thin, myelinated A-delta fibers and unmyelinated C fibers [Citation4]. Small sensory nerves transmit pain, itch and temperature, while the autonomic efferent fibers transmit complex signals from the autonomic ganglia to blood vessels and internal organs to maintain homeostasis. The small fibers release electrical and paracrine chemical signals, which are essential for regulating inflammation and healing in the tissue [Citation5]. Thus, autonomic dysfunction leads to widespread effects in the body [Citation4,Citation6].
The development of neuropathy may have many different causes, where some diseases lead to generalized and other to isolated peripheral neuropathy. Normal aging leads to some form of neuropathy, and definite sensorimotor polyneuropathy diagnosed by neurological screening is present in around 5–13% of the population [Citation7]. We have recently described how a patient with drug-induced intestinal pseudo-obstruction and a patient with Ehlers-Danlos syndrome both exhibited damages to the autonomic, enteric and sensorimotor nervous system when systematically examined [Citation8–10]. On the other hand, some toxic agents mainly affect the peripheral sensorimotor system leading to pain and paresthesia [Citation11,Citation12]. Malnutrition predominately affects the central nervous system, with slow mentation, memory impairment, attention deficits and dementia, and peripheral sensorimotor neuropathy [Citation2,Citation13], and may explain neuropathy observed after bariatric surgery [Citation14]. The neuropathic changes observed in celiac disease and inflammatory bowel disease (IBD) may be caused by concomitant malnutrition, but an autoimmune genesis to the sensorimotor neuropathy or drug effects cannot be ruled out [Citation15,Citation16].
In many cases, patients with enteric neuropathy are undiagnosed for decades before being properly examined by the healthcare system [Citation17]. Some function tests, as well as full-thickness bowel biopsies, are rather complicated methods not used in all hospitals [Citation17,Citation18].
We hypothesized that more knowledge about potential causes of neuropathy, enhanced awareness of simultaneous damages in different parts of the peripheral nervous system, and more simple examinations to diagnose enteric neuropathy, which is situated in less accessible organs, would improve patient outcome. Therefore, autonomic and sensorimotor neuropathy could, in some circumstances, possibly lead to a reasonable assumption of damage in the enteric nervous system as well, without having to perform a full-thickness bowel biopsy or antroduodenal manometry [Citation17].
The present systemic review aimed to increase the awareness of disease groups and etiologies that can cause autonomic and enteric neuropathy along with sensorimotor neuropathy.
Methods
This review was written according to the PRISMA (preferred reporting items for systematic reviews and meta-analysis) statement [Citation19], with the help of the PRISMA E & E (explanation and elaboration) document [Citation20]. According to the review protocol, human original and review articles, written in English, that described diseases which exhibited concomitant peripheral autonomic, enteric and sensorimotor neuropathy should be included. An objective assessment should verify the neuropathy; reports of sole symptoms of any neuropathy were excluded. Systematic reviews and cohort studies with an appropriate control cohort were intended to be studied. Autonomic neuropathy was defined as any form of autonomic dysfunction from visceral organs, excluding the gastrointestinal tract, which was separated in this study, or skin dysfunction such as disturbances in sweat gland function and thermoregulation [Citation6]. Enteric neuropathy was defined as pathological transit time or emptying tests, pathological manometry or histopathological signs of neuropathy in the enteric nervous system [Citation21]. Sensorimotor neuropathy was defined as chronic mononeuropathy or polyneuropathy according to nerve conduction tests and skin biopsies [Citation7,Citation8]. Publications that described signs of inflammation or vasculitis in the tissue without the direct involvement of the nerves were not included.
Information sources
A systematic search strategy was used in three databases: PubMed, Embase (Elsevier) and Web of Science (Clarivate Analytics), on 11 September 2020. We used keywords and relevant thesaurus terms when applicable. No language or publication date restrictions were applied. In Embase, conference abstracts were excluded. The search strategy was designed and executed in collaboration with an information specialist.
PubMed
The search terms in PubMed were: (Intestinal Pseudo-Obstruction [MeSH] OR ((enteric OR gastrointestinal) AND (neuropathy OR nervous system OR dysmotility OR pseudo-obstruction)) AND peripheral neuropathy. The search identified 2343 records.
Embase
The search terms in Embase were: 'intestine pseudoobstruction'/exp OR 'pseudoobstruction'/exp OR 'enteric neuropathy'/exp OR enteric OR gastrointestinal AND 'neuropathy'/exp OR neuropathy OR 'nervous system'/exp OR 'nervous system' OR (nervous AND system) OR dysmotility OR 'pseudoobstruction'/exp OR pseudoobstruction OR (pseudo AND ('obstruction'/exp OR obstruction)) OR 'pseudo obstruction'/exp OR 'pseudo obstruction' AND 'peripheral neuropathy'/exp OR 'peripheral neuropathy' OR (peripheral AND ('neuropathy'/exp OR neuropathy)) AND [embase]/lim NOT ([embase]/lim AND [medline]/lim) NOT 'conference abstract':it. The search strategy identified 1674 records.
Web of science
The following topics were used: TOPIC: (Intestinal Pseudo-Obstruction OR enteric OR gastrointestinal) AND TOPIC: (neuropathy OR nervous system OR dysmotility OR pseudo-obstruction) ANDTOPIC: (peripheral neuropathy). These topics rendered 1660 records.
Quality assessment
The screening of all articles was performed by one author (BO). Assessments of full-length articles intended to be included were performed independently of all authors. In case of discordance, the articles were reread, and after discussion, a consensus was made.
Results
The combined searches resulted in 5677 records, and after deduplication in EndNote, 4977 records remained. These were screened of titles and abstracts along with one article from another source. Animal studies, articles not written in English, articles not available in full-length, case reports, conference abstracts and further duplicates were excluded (). After this screening, 357 unique articles remained, which were studied in full-length. Further articles were then excluded due to different reasons, most often because the neuropathy was assessed only by symptom reports and no objective examinations were performed (). Only four reviews were identified that had tried to perform systematic literature reviews, although only one database was used to search for articles [Citation22–25]. One of the original articles that examined a cohort of patients used a control cohort to compare the prevalence of neuropathy [Citation26] (). Therefore, the initial intention to include only systematic reviews and original studies with a control cohort had to be repudiated. Finally, 35 articles remained (27 reviews), which could be included in the present systematic review (). No additional articles were included after the evaluation of the reference lists of the chosen original articles. No meta-analysis could be executed due to a lack of control material and information about ratios for the presence of neuropathy.
Figure 1. PRISMA flowchart over the systemic review.
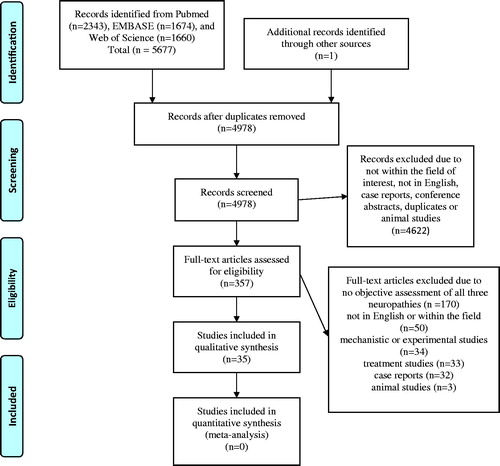
Table 1. Full-length articles describing concomitant peripheral autonomic, enteric and sensorimotor neuropathy.
Nomenclature
Neuropathy refers to damage across the whole neuronal system [Citation6], whereas neuronopathy is the degeneration of the neuronal cell body. Ganglionopathy, which is a form of neuronopathy, is damage to the autonomic or sensory ganglionic cells. The different types of autonomic disorders, which typically include gastrointestinal and urogenital dysfunction, can be divided into several classifications, with orthostatic, non-orthostatic and diffuse symptoms being one classification from a clinical point of view [Citation6]. There are several forms of peripheral sensorimotor neuropathy, such as large-fiber neuropathy, sensory ganglionopathy, polyradiculopathy and small fiber neuropathy (SFN) [Citation27].
Chronic polyneuropathy refers to distal symmetric neuropathy, which is widespread damage of peripheral nerves [Citation5,Citation7]. The most common damage on the nerves is at the axons’ distal ends, which are most vulnerable to disruption in the supply of energy and nutrients. A description of small or unmyelinated fibers is presented in . SFN mainly involves unmyelinated somatic afferents and autonomic efferents [Citation28], which are often located together [Citation6]. In typical cases, SFN presents with pain in the feet and/or symptoms of autonomic neuropathy, most often in the form of anhidrosis/hyperhidrosis, cardiovascular neuropathy or gastrointestinal dysmotility. Cardiovascular symptoms include orthostatic hypotension and tachycardia, as well as impaired cognitive function, headache and exercise intolerance due to circulatory insufficiency with secondary ischemia. Gastrointestinal symptoms may involve the entire gastrointestinal tract and may reflect both dysmotility and insufficient splanchnic circulation [Citation5,Citation6,Citation29]. SFN may co-exist with large-fiber neuropathy and ganglionopathy and can be idiopathic (73%) or secondary (10% diabetic) to an underlying disease [Citation6,Citation28].
Table 2. Classification of nerve fibers.
We herein divide the peripheral neuropathies into diabetic neuropathy, neuropathy due to excessive tissue deposits, autoimmune or inflammation-related neuropathy, infection-related neuropathy and hereditary neuropathy.
Diabetic neuropathy
The prevalence of diabetes mellitus is steadily increasing and is estimated to affect 425 million people worldwide [Citation30]. Diabetic neuropathy occurs in 20–30% of patients with type 1 diabetes after 20 years of disease duration and 50–60% of patients with type 2 diabetes after 10–15 years [Citation31,Citation32]. There are conflicting results on whether sex has any effect on the development of neuropathy. The neuropathy may appear two years after the debut of type 1 diabetes and before or within one year of type 2 diabetes [Citation29]. Objective signs of neuropathy, e.g., pathological autonomic function tests or reduced intra-epidermal nerve fiber density, only lead to symptoms in 20–50% of these patients. The genesis of neuropathy is multifactorial and not fully explained, but hyperglycemia, dyslipidemia, inflammation and immune responses, and macro-and microvascular diseases have been proposed [Citation28,Citation32]. The mechanisms of hyperglycemia and vasculopathy involve polyol pathway activation, oxidative stress, protein kinase C (PKC) activation, and the formation of advanced glycation end products (AGEs). Elevated triacylglycerol levels can induce an accumulation of toxic acylcarnitine, and elevated low-density lipoproteins (LDL) can lead to oxidized LDLs and toll-like receptor 4 (TLR4). Altered sphingolipid metabolism may result in neurotoxic deoxysphingolipids [Citation30]. No causality between the presence of autoantibodies and diabetic neuropathy has been demonstrated. Optimizing metabolic control is essential to prevent induction and progression of neuropathy [Citation29,Citation32]. Also, the nutrient supply is essential since neuropathy may be associated with low levels of cobalamins and vitamin D [Citation29,Citation30].
Diabetic neuropathy is categorized as diffuse when multiple nerves are affected, including both autonomic neuropathy and sensorimotor polyneuropathy, and is categorized as focal when only single nerves are affected. The longest nerves are affected first [Citation28,Citation29,Citation33]. Neuropathy in the vagus nerve renders widespread effects since it has multiple innervations and accounts for 75% of all parasympathetic activity [Citation29].
Distal symmetric sensorimotor neuropathy affects 72% of patients who are diagnosed with diabetic neuropathy [Citation33]. Sensory symptoms include numbness, tingling, pain and paresthesia [Citation33]. The motor symptoms are not clinically obvious but can be identified on electrophysiological examinations [Citation28].
Diabetic autonomic neuropathy may present itself with resting tachycardia, orthostatic hypotension, silent myocardial ischemia, exercise intolerance, sudomotor neuropathy, gastrointestinal dysmotility, incontinence, neurogenic bladder and hypoglycemia, among many other symptoms [Citation29,Citation33]. The cardiovascular symptoms depend on reduced reflexes in the hemodynamic homeostasis, thereby leading to failure of autonomic compensations in response to, e.g., standing and resting/exercise. Hypoglycemia is assumed to depend on an impaired epinephrine response, which is essential for the body to counteract hypoglycemia [Citation29].
Gastrointestinal dysmotility involves vagal and intrinsic neural denervation and reduced numbers of interstitial cells of Cajal. Both abnormal muscle contraction and muscle inhibition, as well as reduced sensation of pressure changes, may explain the symptomatology [Citation28]. Constipation is the most common gastrointestinal symptom and is present in 60% of all diabetes patients, and diarrhea is observed in 20% of the patients [Citation29]. Although neuropathy is evident in many of these patients, other etiologies to the gastrointestinal symptoms may be considered, e.g., pancreatic insufficiency, adverse drug effects and colitis. Esophageal dysmotility (50%) and gastroparesis (40%) are the most common signs of enteric neuropathy in long-standing diabetes [Citation31]. Gastrointestinal dysmotility further impairs metabolic control, which may accelerate the development of neuropathy [Citation28,Citation29,Citation32]. Modern drug treatment with glucagon-like peptide-1 (GLP1) and dipeptidyl peptidase-4 (DPP4) may further impair gastrointestinal motility, at least during the initial treatment, through their effects to reduce postprandial glucose levels partly by a delay of the gastric emptying [Citation30].
Neuropathy due to excessive tissue deposits
Amyloidosis
Amyloidosis is a group of diseases characterized by the deposition of amyloid fibrils in various tissues throughout the body leading to organ dysfunction. All parts of the peripheral nervous system may be involved, such as the nerve trunks, plexuses or ganglia. In the nerves, the amyloid deposits are situated in the epi-, peri- or endoneurium [Citation34]. Gastrointestinal involvement exists with a prevalence of around 30% in patients with amyloidosis, and the small intestine is the most common segment to be affected [Citation22].
Primary (amyloidosis light chain, AL) amyloidosis with an accumulation of light-chain amyloid fibrils is the most common form of systemic amyloidosis. This form is caused by plasma cell dyscrasia and has the most prominent gastrointestinal involvement. The symptoms debut in the middle-age and are more frequent in men than in women. Peripheral sensorimotor neuropathy is commonly the first and most prominent symptom, followed by autonomic and enteric neuropathy leading to constipation and diarrhea [Citation35,Citation36].
Secondary (AA) amyloidosis means deposition of the acute-phase reactant protein ‘serum amyloid A protein,’ which is produced during chronic inflammatory or infectious diseases. The other forms are classified by the precursor protein involved, such as transthyretin, β2-microglobulin (secondary to long-term dialysis) and lysozyme. Senile amyloidosis has been reported in 10–36% of subjects above 80 years of age [Citation22].
Transthyretin-associated neuropathic amyloidosis is an autosomal dominantly inherited mutation and has been called familial amyloid polyneuropathy type 1 (FAP-I), also known as the Portuguese-Swedish-Japanese type [Citation37]. This disease starts with sensorimotor polyneuropathy in early adulthood and progresses with autonomic and enteric neuropathy, with a mean survival of 5–15 years after diagnosis [Citation34]. The clinical features differ between the countries, with a higher debut age in Sweden and more gastric retention symptoms, whereas the Japanese population has more orthostatic hypotension symptoms [Citation37,Citation38].
Neurosarcoidosis
Sarcoidosis may generate granuloma accumulation of the peripheral nerves, mainly in the epi- and perineurium. Necrotizing vasculitis is observed in some cases. The prevalence of neurological symptoms in sarcoidosis is 5–13%. Patients who have involvement of the central nervous system have a much worse prognosis than those with sole involvement of the peripheral nervous system. Gastrointestinal symptoms include gastroparesis, constipation and diarrhea [Citation39].
Neurodegenerative synucleinopathies – Lewy body disorders
Abnormal aggregation of α-synuclein in the perikaryon, axons and dendrites of different brain regions represents the pathological hallmark of Parkinson’s disease and other Lewy body diseases. In recent years, these aggregates have been increasingly reported outside the central nervous system, and it is debated whether they may occur in the peripheral nervous system before they occur in the brain [Citation27]. Aggregates of α-synuclein have been found in several cortical areas, the brainstem, the spinal cord, paravertebral sympathetic ganglia, along the vagus nerve, myenteric ganglia, nerve fibers crossing the striated muscles and skin nerves [Citation27,Citation40,Citation41]. These histopathological aggregations are the basis of nerve atrophy and loss of epidermal nerves, reduced salivary secretion and gastrointestinal dysfunction. Constipation is the most prominent symptom of the gastrointestinal tract, but esophageal dysmotility and gastroparesis are also common [Citation40].
Autoimmune or inflammation-related neuropathy
Autoimmune peripheral neuropathy may be due to a systemic autoimmune disease, an autoimmune disorder specifically targeting peripheral nerves or ganglia or malignancy [Citation42]. The autoantibodies may be directed against membrane receptors, directed against cell surface glycoproteins (gangliosides) or paraneoplastic and directed against intracellular onconeural antigens. Autoantibodies directed against ganglionic neuronal acetylcholine receptors (AChRs; i.e., nicotine receptors) are of particular interest since these are biomarkers of an antibody-mediated autonomic dysfunction [Citation43], indicating a potentially treatable autonomic dysfunction. The higher the antibody titer, the more widespread neuropathy [Citation42]. The paraneoplastic antibodies are more specific to a unique cancer form than to a specific neuropathy form, and their titers do not correlate with the degree of neuropathy [Citation43,Citation44].
Antiphospholipid syndrome
Antiphospholipid syndrome is a systemic autoimmune disease with the presence of antiphospholipid antibodies and an increased risk of thromboses in arterial, venous and small vessels. A substantial portion of these patients may have an underlying joint hypermobility syndrome and other concomitant autoimmune systemic disorders, such as thyroiditis, autoimmune hepatitis, primary Sjögren’s syndrome and vasculitis. Skin biopsies have revealed reduced small sensory nerve fiber density, and the patients showed numerous autonomic disorders and gastrointestinal dysmotilities [Citation4].
Antineutrophil cytoplasmic antibody-associated vasculitis
A worldwide study recruited all cases (n = 955) with antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis to identify the prevalence of vasculitic neuropathy in this disease. The cohort represented granulomatosis with polyangiitis (n = 572), eosinophilic granulomatosis with polyangiitis (n = 165) or microscopic polyangiitis (n = 218). Vasculitic neuropathy was present in 28% of the cases, with sensory or sensorimotor neuropathy being the most prevalent, whereas autonomic and enteric neuropathy were less common [Citation45].
Primary Sjögren’s syndrome
Primary Sjögren’s syndrome is an autoimmune disease characterized by lymphocytic infiltration of various exocrine glands as well as circulating anti-Ro/SS-A and anti-La/SS-B antibodies targeting RNA-associated antigens [Citation18]. There have been reports of circulating antibodies against the human muscarinic AChR (situated on effector cells stimulated by postganglionic neurons) subtype 3 (m3AChR) [Citation56], which is the dominant receptor in lacrimal and salivary glands [Citation57,Citation58]. The muscarinic receptor subtype 3 is also a functionally important AChR in the gastrointestinal tract, where it mediates the contraction of stomach fundus smooth muscles [Citation59]. Animal studies have shown a role for autoantibodies in the loss of secretory function of exocrine tissue in Sjögren’s syndrome [Citation60]. This autoimmunity directed against important receptors may explain why patients with Sjögren’s syndrome also exhibited autonomic and enteric neuropathy with delayed gastric emptying, as well as reduced vibration perception [Citation46]. In accordance, Sjögren’s syndrome patients have shown an impaired microvascular response to cholinergic stimuli [Citation61].
Systemic sclerosis
Systemic sclerosis is a systemic microvascular disease that leads to fibroblast activation and excessive collagen production, primarily in the skin and internal organs. Signs of peripheral neuropathy may be found simultaneously or before the onset of the other symptoms. The exact pathophysiology of the neuropathy is unknown, but it is speculated to depend on autoimmunity with edema and collagen deposits in the epi-, peri- and endoneural vessels. These changes lead to deleterious ischemia and impaired removal of toxic metabolites. Furthermore, compressive conditions induce progressive demyelination. A third theory is that anti-myenteric neuronal antibodies are directed against the nervous system [Citation47]. Neuropathy is present in both localized and systematic sclerosis, as well as in morphea. Previously, peripheral sensorimotor neuropathy was considered an uncommon feature of this disease, but recent research has found a prevalence of sensorimotor neuropathy in about 14% of patients. Autonomic neuropathy is due to injury at the ganglion level and higher autonomic centers and is observed in up to 80% of patients with sclerosis [Citation23]. Enteric neuropathy is common in sclerosis. The earliest esophageal neural dysfunction is located at the lowest two-thirds of the esophagus, composed of smooth muscles innervated by autonomic nerves, and is associated with engagement of the entire gastrointestinal tract [Citation47].
Paraneoplastic neuropathy
Paraneoplastic neuropathy is rare and occurs in less than 0.01% of patients with malignancy [Citation44]. The paraneoplastic condition often presents sub-acutely [Citation61]. In most cases, the neuropathy precedes the symptoms and diagnosis of malignancy [Citation43], which is prevalent in 5–60% of patients with potential paraneoplastic neuropathy, depending on the disorder [Citation44]. It has been proposed that cellular mechanisms have been involved in the pathophysiology and include neuron damage mediated by CD8-positive cytotoxic T cells [Citation44,Citation48]. The histopathological findings resemble those of patients with primary Sjögren’s syndrome [Citation49]. Several antibodies such as antineuronal nuclear antibody-type 1 (anti-Hu/ANNA-1), collapsin response-mediator protein-5 antibodies (anti-CV2/CRMP-5), anti-ganglionic AChR antibodies and voltage-gated calcium channels (VGCCs) antibodies are recognized in about 50% of cases, and the specificity of these antibodies to diagnose paraneoplastic neuropathy is more than 90% [Citation43,Citation44,Citation48,Citation49]. Only the VGCC antibodies have been shown to be directly involved in the pathogenesis, whereas the other antibodies, which are directed against intracellular antigens, may be markers of autoimmunity rather than a cause of the neuropathy. The most common forms of paraneoplastic neuropathy are sensory neuropathy and cerebellar degeneration. Sensorimotor diseases (Guillain-Barré syndrome), vasculitic neuropathy, autonomic ganglionopathy and gastrointestinal hypomobility leading to esophageal dysmotility, gastroparesis, constipation and chronic intestinal pseudo-obstruction have been observed [Citation43,Citation44,Citation48].
Acute autonomic and sensory neuropathy
Acute autonomic and sensory neuropathy is a rare disorder with autonomic neuropathy and sensory impairment due to severe neuron death in sympathetic ganglia, dorsal root ganglia and myenteric plexuses. The condition is triggered by a respiratory or gastrointestinal infection in two-thirds of the patients. Gastrointestinal symptoms are more prominent than other autonomic symptoms and may be presented as paralytic ileus [Citation27].
Infection-related neuropathy
Varicella-zoster virus
Varicella-zoster virus belongs to the human herpesvirus group that causes varicella as a primary infection. After the acute infection, the virus resides latently in the peripheral ganglia. Several decades later, the virus may be reactivated, spontaneously or after a triggering event, to cause herpes zoster. Triggering factors are other diseases, drugs, infections, stress, trauma and malignancy. Post-herpetic neuralgia is the most important complication of herpes zoster, but segmental motor weakness may also occur due to the involvement of the spinal roots, spinal horns and brachial plexuses. Guillain-Barré syndrome is more common after varicella than after herpes zoster. Autonomic and enteric ganglia may also harbor latent varicella-zoster virus that can be reactivated in those ganglia as well as in sensory ganglia and lead to autonomic dysfunction with severe abdominal pain, ulcers and intestinal pseudo-obstruction, so-called ‘enteric zoster’ [Citation50].
Other viruses
Epstein-Barr virus has been reported to develop acute autonomic neuropathy with paresthesia, orthostatic intolerance and gastrointestinal symptoms in the form of intestinal pseudo-obstruction. The acute phase can occasionally show prolonged dysfunction [Citation25].
Chagas disease
Chagas disease is caused by the hemoflagellate parasite Trypanosoma cruzi through bites from Triatominae bugs. The disease is endemic in Central and South America. Initially, the infective trypomastigote circulates in the blood. During the active parasite multiplication, there is an intense interstitial inflammatory reaction. When this acute phase declines, the infected subject may be asymptomatic for several years. In the chronic phase, destruction of ganglion cells occurs in the central nervous system as well as the peripheral autonomic, enteric and sensorimotor nervous systems. This destruction leads to impaired heart rate regulation; loss of gastrointestinal motility, preferentially in the esophagus, descending colon, sigmoid colon and rectum; and muscle denervation [Citation25,Citation51].
Lyme disease
Lyme disease is caused by the spirochete Borrelia burgdorferi, which is a disseminated disease involving the musculoskeletal, cardiovascular, enteric and central nervous systems [Citation25]. Many of these patients do not have a previous history of chronic erythema migrans and may not remember being bitten by ticks. Liquor analysis may reveal anti-Borrelia IgM and IgG and lymphocytosis. Pathological studies have shown lymphocellular infiltrates in the autonomic ganglia [Citation62]. Symptoms of chronic intestinal pseudo-obstruction may improve with antibiotic therapy, which suggests a reversible autonomic neuropathy. However, long-term symptoms like chronic fatigue, musculoskeletal symptoms and postural orthostatic tachycardia syndrome (POTS) may occur [Citation25].
Guillain-Barré syndrome
Guillain-Barré syndrome is characterized by the acute and rapid progress of symmetric weakness, paresthesia, numbness, neuropathic pain and hypo- or areflexia. Later, arterial hypotension or hypertension, abnormal sweating and gastrointestinal dysmotility with constipation and ileus may develop. Although the exact mechanism is unknown, 50–70% of cases appear 1–2 weeks after a respiratory infection, gastrointestinal infection or another immune stimulus that induces autoimmune responses against peripheral nerves and their spinal roots. Campylobacter jejuni is the single most reported bacteria to cause the syndrome. The annual incidence of the disease is 0.5–2 cases/100,000. Guillain–Barré syndrome is not associated with other autoimmune diseases and is thought to be mainly humoral-mediated (anti-ganglioside antibodies) rather than T-cell-mediated. Most cases are caused by acute inflammatory demyelinating polyradiculoneuropathy. The mortality rate is 3–7% [Citation24].
Hereditary neuropathy
Acute intermittent porphyria
Acute intermittent porphyria is the most common and severe form of acute hepatic porphyria. The disease is an autosomal dominant condition with lower porphobilinogen deaminase levels, which causes an accumulation of HEM precursors. Abdominal pain, neurological dysfunction and psychiatric disturbances are the classic triad of the disease. Diverse kinds of autonomic neuropathy in the form of symptoms of the cardiovascular system and excessive sweating have been observed, as well as paralytic ileus, muscle weakness, painful flaccid paralysis and sensory involvement. The reason for the neurological damage is poorly understood, but axonal degeneration of sensorimotor and autonomic nerve fibers, rather than demyelination, has been cited as a cause of the dysfunction. Although the neurological effects are reversible, a mortality rate of 20–50% has been reported [Citation52].
Tyrosinemia
Tyrosinemia is an autosomal recessive disorder of the amino acid metabolism caused by a deficiency of fumarylacetoacetate hydrolase, which is the final step in the tyrosine breakdown pathway. The accumulation of tyrosine metabolites causes liver and renal failure, but intermittent neurologic crises have also been observed. Histopathological examinations have shown evidence of axonal degeneration and secondary demyelination. The neurological findings are most often hypertension, pain in the legs and abdomen, vomiting and ileus [Citation53].
Mitochondrial depletion syndromes
Mitochondrial DNA (mtDNA) depletion syndromes (MDS) are autosomal recessive disorders with a broad genetic and clinical spectrum due to a severe reduction of mtDNA, which is essential to provide respiratory chain components for energy production. Mutation of several genes may cause mtDNA depletion. MDS is classified into four clinical forms, where the neurogastrointestinal form is associated with mutations in TYMP and the clinical presentation of mitochondrial neurogastrointestinal encephalopathy (MNGIE). It is a progressive chronic disease that presents before the age of 20 in most cases, with weight loss, progressive gastrointestinal dysmotility and peripheral demyelinating motor and sensory neuropathy. Orthostatic hypotension is one of the most common autonomic dysfunctions. The other forms of MDS can present with encephalomyopathic symptoms, spinal muscular atrophy-like symptoms and gastrointestinal dysmotility in the form of abdominal pain, diarrhea, feeding difficulties and gastroesophageal reflux [Citation54]. In a case series of eight patients with complex regional pain syndrome type I (CRPS-I), in addition to their underlying mitochondrial disease, one out of three siblings presented with predominant symptoms from enteric neuropathy, one with predominant symptoms from autonomic neuropathy, and the third with predominant symptoms from sensorimotor neuropathy [Citation26]. In other cases, all three types of neuropathy were present in the same subject [Citation26].
Fabry disease
Fabry disease is an X-linked lysosomal storage disorder caused by deficient activity of α-galactosidase A. This deficiency predominantly leads to the accumulation of globotriaosylceramide (GL-3) in vascular endothelial cells and neural cells, amongst other cell types in the body. The earliest manifestation may be hypohidrosis, whereas other expressions of neuropathy develop later in life with different phenotypes. Some cases present with mild symptoms and slow progress, where only symptoms from a few organs are present. Gastrointestinal manifestations are found as postprandial abdominal pain and bloating followed by increased motility with early satiety, vomiting and diarrhea [Citation55].
Discussion
This is, to our knowledge, the first systematic review performed within this area of objectively verified, generalized peripheral neuropathy, including searches in three different databases. The primary causes of generalized peripheral neuropathy found in the present review were diabetes, diseases with an accumulation of tissue deposits, autoimmunity, infections, malignancy and metabolic diseases.
Clinical investigations of neuropathy
The findings in this review suggest that concomitant peripheral autonomic and sensorimotor neuropathy is not uncommon. Despite this, these patients are handled by separate specialists in the healthcare system. In order to improve the diagnosis of gastrointestinal symptoms, future studies need to compare the presence of enteric neuropathy in the same patient when objective findings of autonomic and sensorimotor neuropathy are described.
To objectively diagnose neuropathy, a panel of functional tests and circulating autoantibodies need to be performed and analyzed [Citation18,Citation29,Citation43]. Roughly, the autonomic function tests can be divided into tests of cardiac parasympathetic nervous system function (heart-rate variability and response tests), sympathetic adrenergic function (blood pressure response tests) and sympathetic cholinergic function (thermoregulation, sweat, sudomotor and sympathetic skin response tests) [Citation18,Citation29]. Tissue biopsies are relevant to diagnose deposits such as amyloid fibrils [Citation35,Citation36] and α-synuclein [Citation27,Citation40,Citation41]. Skin biopsies are relevant to define intra-epidermal nerve fiber density, i.e., SNF [Citation8], whereas nerve conduction velocity studies demonstrate large-fiber neuropathy [Citation30]. A full-thickness biopsy and measurement of intestinal motility are necessary to diagnose enteric dysmotility [Citation17]. Magnetic resonance imaging (MRI) may describe alterations in the central nervous system [Citation23] but is of no use to describe damages in the enteric nervous system [Citation63].
The awareness of the close relation between small sensory and autonomic nerve fibers may change the examination methods in daily clinical praxis [Citation6]. Skin biopsies should perhaps be a more prevalent method when diagnosing diseases with subtle autonomic dysfunction and gastrointestinal symptoms [Citation64]. Autonomic function tests and skin biopsies are associated with considerably fewer risks and inconvenience for the patients than full-thickness bowel biopsies and should be considered more frequently. Further development of new techniques to examine gastrointestinal motility is warranted. The recently reported 3D modality is a promising technique to verify neuropathy [Citation9], not only in the enteric nervous system but also in other tissues [Citation10].
Clinical consequences of neuropathy
Many cases of neuropathy end up as idiopathic neuropathy without any identified etiology. Still, several patients report that the symptoms debuted after an infection or a vaccination episode. Several common viruses have been discussed to evoke neuropathy [Citation25]. Thus, peripheral neuropathy secondary to viral infections may be much more frequent than hitherto assumed.
Diagnosis and treatment of autonomic neuropathy are important to prevent secondary complications. Cardiovascular neuropathy may impair the possibility for the subjects to exercise or live an active life. Since both impaired microcirculation and sudomotor function lead to dry skin and loss of sweating, these changes can contribute to the development of ulcers and gangrenes. Furthermore, there is a strong association between reduced heart rate variability and silent myocardial ischemia and mortality, with an increased mortality risk of 2.14 (95% confidence interval of 1.83–2.51) in patients with cardiovascular autonomic neuropathy [Citation29]. Enteric neuropathy impairs the possibility to eat regularly and optimally deliver food and drugs to the small intestine. All these factors have a negative impact on the metabolic control of diabetes, which enhances the development of neuropathy [Citation29,Citation30,Citation32]. Thus, neuropathy has a considerable relevance on both quality of life and survival. Gastrointestinal symptoms without objective findings on endoscopy are called irritable bowel syndrome (IBS). These patients are treated with dietary advice, psychological treatment and drugs to reduce gastrointestinal symptoms [Citation65]. A thorough examination of the gastrointestinal tract and its motility may lead to a diagnosis of gastrointestinal dysmotility instead of IBS [Citation17,Citation21], with a different treatment to improve motility with enhanced metabolic control and reduced symptoms [Citation31]. Nevertheless, it is important to recognize that the gastrointestinal symptoms in Sjögren’s syndrome or other disorders may not only depend on enteric neuropathy [Citation17,Citation46]. The finding of a correlation between levels of feces calprotectin, organic gastrointestinal disease and inflammatory rheumatic activity suggests that Sjögren’s syndrome may be situated in the entire gastrointestinal mucosa and not only in the mouth [Citation66].
Strengths and limitations
This review is the first systematic review in the field, with objectively verified peripheral neuropathy in different disease groups. There are several limitations, where the lack of systematic reviews and original studies with an appropriate control group is the most significant limitation. However, since this seems not to have been examined scientifically, we cannot influence these issues. Furthermore, several studies have only assessed the gastrointestinal symptoms but have not performed any objective examinations of transit time, peristalsis or sphincter pressures. Several more diseases may exhibit gastric dysmotility if properly examined. Improving the diagnosis of peripheral neuropathy is essential to improve the patients’ healthcare in many different aspects, from the quality of life to improved metabolic control and increased survival.
To conclude, several common diseases may cause peripheral neuropathy in multiple organs, where the major causes of generalized neuropathy identified in the present systematic review are diabetes, diseases with an accumulation of tissue deposits, autoimmunity, infections, malignancy and metabolic diseases.
Acknowledgements
We want to thank Maria Björklund, information specialist at the library of Lund University, for kindly help with the search strategy.
Disclosure statement
The authors declare that there is no conflict of interest or competition.
Additional information
Funding
References
- Spencer NJ, Hu H. Enteric nervous system: sensory transduction, neural circuits and gastrointestinal motility. Nat Rev Gastroenterol Hepatol. 2020;17(6):338–351.
- Maxwell PJ, Montgomery SC, Cavallazzi R, et al. What micronutrient deficiencies should be considered in distinct neurological disorders? Curr Gastroenterol Rep. 2013;15(7):331.
- Coon EA, Cutsforth-Gregory JK, Benarroch EE. Neuropathology of autonomic dysfunction in synucleinopathies. Mov Disord. 2018;33(3):349–358.
- Schofield JR. Autonomic neuropathy—in its many guises—as the initial manifestation of the antiphospholipid syndrome. Immunol Res. 2017;65(2):532–542.
- Oaklander AL. Immunotherapy prospects for painful small-fiber sensory neuropathies and ganglionopathies. Neurotherapeutics. 2016;13(1):108–117.
- Novak P. Autonomic disorders. Am J Med. 2019;132(4):420–436.
- Hanewinckel R, Drenthen J, van Oijen M, et al. Prevalence of polyneuropathy in the middle-aged and elder population. Neurology. 2016;87(18):1892–1898.
- Ohlsson B, Dahlin LB, Englund E, et al. Autonomic and peripheral neuropathy with reduced intraepidermal nerve fiber density can be observed in patients with gastrointestinal dysmotility. Clin Case Rep. 2020;8(1):142–148.
- Peruzzi N, Veress B, Dahlin L, et al. 3D analysis of the myenteric plexus of the human bowel by x-ray phase-contrast tomography – a future method? Scand J Gastroenterol. 2020;55(10):1261–1267.
- Eckermann M, Peruzzi N, Frohn J, et al. 3D nanotomography of unstained human skin biopsies may differentiate morphological structures in the dermis and epidermis between subjects. Skin Res Tech. 2020.
- Gebremedhn EG, Shortland PJ, Mahns DA. The incidence of acute oxaliplatin-induced neuropathy and its impact on treatment in the first cycle: a systematic review. BMC Cancer. 2018;18(1):410.
- Eltobgy M, Oweira H, Petrausch U, et al. Immune-related neurological toxicities among solid tumor patients treated with immune checkpoint inhibitors: a systematic review. Expert Rev Neurother. 2017;17(7):725–736.
- Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521–4539.
- Frantz DJ. Neurologic complications of bariatric surgery: involvement of central, peripheral, and enteric nervous systems. Curr Gastroenterol Rep. 2012;14(4):367–372.
- Moris G. Inflammatory bowel disease: an increased risk factor for neurological complications. World J Gastroenterol. 2014;20:1228–1237.
- Jericho H, Guandalini S. Extra-intestinal manifestation of celiac disease in children. Nutrients. 2018;10:755.
- Bengtsson M, Hammar O, Mandl T, et al. Evaluation of gastrointestinal symptoms in different patient groups using the visual analogue scale for irritable bowel syndrome (VAS-IBS). BMC Gastroenterol. 2011;11(1):122.
- Freeman R. Autonomic peripheral neuropathy. Neurol Clin. 2007;25(1):277–301.
- Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097.
- Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700.
- Wingate D, Hongo M, Kellow J, et al. Disorders of gastrointestinal motility: towards a new classification. J Gastroenterol Hepatol. 2002;17:S1–S14.
- Ebert EC, Nagar M. Gastrointestinal manifestations of amyloidosis. Am J Gastroenterol. 2008;103(3):776–787.
- Amaral TN, Peres FA, Lapa AT, et al. Neurologic involvement in scleroderma: a systematic review. Semin Arthritis Rheum. 2013;43(3):335–347.
- Esposito S, Longo MR. Guillain-Barré syndrome. Autoimmun Rev. 2017;16(1):96–101.
- Carod-Artal FJ. Infectious diseases causing autonomic dysfunction. Clin Auton Res. 2018;28(1):67–81.
- Higashimoto T, Baldwin EE, Gold JI, et al. Reflex sympathetic dystrophy: complex regional pain syndrome type I in children with mitochondrial disease and maternal inheritance. Arch Dis Child. 2008;93(5):390–397.
- Benarroch EE. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol. 2014;10(7):396–407.
- Sasaki H, Kawamura N, Dyck PJ, et al. Spectrum of diabetic neuropathies. Diabetol Int. 2020;11(2):87–96.
- Vinik AI, Maser RE, Mitchell BD, et al. Diabetic autonomic neuropathy. Diabetes Care. 2003;26(5):1553–1579.
- Sharma JK, Rohatgi A, Sharma D. Diabetic autonomic neuropathy: a clinical update. J R Coll Physicians Edinb. 2020;50(3):269–273.
- Tesfaye S, Boulton AJM, Dyck PJ, et al. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care. 2010;33(10):2285–2293.
- Pop-Busui R, Boulton AJM, Feldman EL, et al. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care. 2017;40(1):136–154.
- Sharma V, Verma P, Guleria R, et al. Diabetic pandemonium. Int J Pharm Sci Rev Res. 2012;17:65–72.
- Hund E, Linke RP, Willig F, et al. Transthyretin-associated neuropathic amyloidosis. Pathogenesis and treatment. Neurology. 2001;56(4):431–435.
- Kelly JJ Jr., Kyle RA, O'Brien PC, et al. The natural history of peripheral neuropathy in primary systemic amyloidosis. Ann Neurol. 1979;6(1):1–7.
- Lim AY, Lee JH, Jung KS, et al. Clinical features and outcomes of systemic amyloidosis with gastrointestinal involvement: a single-center experience. Korean J Intern Med. 2015;30(4):496–505.
- Suhr OB, Svendsen IH, Andersson R, et al. Hereditary transthyretin amyloidosis from a Scandinavian perspective. J Intern Med. 2003;254(3):225–235.
- Sekijima Y, Ueda M, Koike H, et al. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. 2018;13(1):6.
- Nozaki K, Judson MA. Neurosarcoidosis. Curr Treat Options Neurol. 2013;15(4):492–504.
- Wakabayashi K, Mori F, Tanji K, et al. Involvement of the peripheral nervous system in synucleinopathies, tauopathies and other neurodegenerative proteinopathies of the brain. Acta Neuropathol. 2010;120(1):1–12.
- Gelpi E, Navarro-Otano J, Tolosa E, et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord. 2014;29(8):1010–1018.
- Dineen J, Freeman R. Autonomic neuropathy. Semin Neurol. 2015;35(4):458–468.
- Vernino S. Antibody testing as a diagnostic tool in autonomic disorders. Clin Auton Res. 2009;19(1):13–19.
- Honnorat J, Antoine JC. Paraneoplastic neurological syndromes. Orphanet J Rare Dis. 2007;2:22.
- Bischof A, Jaeger VK, Hadden RDM, et al. Peripheral neuropathy in antineutrophil cytoplasmic antibody-associated vasculitides: insights from the DCVAS study. Neurol Neuroimmunol Neuroinflamm. 2019;6(6):e615.
- Kovács L, Papós M, Takács R, et al. Autonomic nervous system dysfunction involving the gastrointestinal and the urinary tracts in primary Sjögren's syndrome. Clin Exp Rheumatol. 2003;21:697–703.
- Cerinic MM, Generini S, Pignone A, et al. The nervous system in systemic sclerosis (scleroderma). Clinical features and pathogenetic mechanisms. Rheum Dis Clin North Am. 1996;22(4):879–892.
- Lorusso L, Hart IK, Ferrari D, et al. Autonomic paraneoplastic neurological syndromes. Autoimmun Rev. 2007;6(3):162–168.
- Koike H, Tanaka F, Sobue G. Paraneoplastic neuropathy: wide-ranging clinicopathological manifestations. Curr Opin Neurol. 2011;24(5):504–510.
- Kennedy PGE, Gershon AA. Clinical features of varicella-zoster virus infection. Viruses. 2018;10:609.
- Fernandez A, Hontebeyrie M, Said G. Autonomic neuropathy and immunological abnormalities in Chagas' disease. Clin Auton Res. 1992;2(6):409–412.
- Mehta M, Rath GP, Padhy UP, et al. Intensive care management of patients with acute intermittent porphyria: clinical report of four cases and review of literature. Indian J Crit Care Med. 2010;14(2):88–91.
- Mitchell G, Larochelle J, Lambert M, et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322(7):432–437.
- El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013;10(2):186–198.
- Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8(9):539–548.
- Sumida T, Tsuboi H, Iizuka M, et al. Anti-M3 muscarinic acetylcholine receptor antibodies in patients with Sjögren's syndrome. Mod Rheumatol. 2013;23(5):841–845.
- Mauduit P, Jammes H, Rossignol B. M3 muscarinic acetylcholine receptor coupling to PLC in rat exorbital lacrimal acinar cells. Am J Physiol. 1993;264(6 Pt 1):C1550–C1560.
- Proctor GB, Carpenter GH. Regulation of salivary gland function by autonomic nerves. Auton Neurosci. 2007;133:3–18.
- Stengel PW, Yamada M, Wess J, et al. M(3)-receptor knockout mice: muscarinic receptor function in atria, stomach fundus, urinary bladder, and trachea. Am J Physiol Regul Integr Comp Physiol. 2002;282(5):R1443–R1449.
- Robinson CP, Brayer J, Yamachika S, et al. Transfer of human serum IgG to nonobese diabetic Igmu null mice reveals a role for autoantibodies in the loss of secretory function of exocrine tissues in Sjögren's syndrome. Proc Natl Acad Sci U S A. 1998;95(13):7538–7543.
- Kovács L, Török T, Bari F, et al. Impaired microvascular response to cholinergic stimuli in primary Sjögren's syndrome. Ann Rheum Dis. 2000;59:48–53.
- Duray PH. Histopathology of clinical phases of human Lyme disease. Rheum Dis Clin N Am. 1989;15:691–710.
- Månsson S, Ekberg O, Ohlsson B. Motility index measured by magnetic resonance enterography is associated with sex and mural thickness. World J Gastroenterol. 2020;26(36):5484–5497.
- Fedorowski A. Postural orthostatic tachycardia syndrome: clinical presentation, aetiology and management. J Intern Med. 2019;285(4):352–366.
- Lacy BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology. 2016;150(6):1393–1407.
- Andréasson K, Ohlsson B, Mandl T. Elevated levels of faecal calprotectin in primary Sjögren's syndrome is common and associated with concomitant organic gastrointestinal disease. Arthritis Res Ther. 2016;18:9.