
ABSTRACT
Mechanisms contributing to unusually high cation exchange capacity and potassium ion selectivity in several halloysite-rich soils remain a topic of intense debate. In spite of the large number of studies, a unifying mechanism to explain the high charge and K+ selectivity has not been elucidated. High K+ selectivity occurs in several soils from northern California whose clay fraction is dominated by hydrated tubular halloysite (1.0 nm) and abundant Fe (hydr)oxides. To investigate the mechanism(s), we measured K+/Ca2+ selectivity and charge properties of the clay-size fraction of two California soils following various pretreatments, including organic matter removal by H2O2, Fe (hydr)oxide removal by citrate-dithionite (CD), and alteration of the basal spacing by dehydration (reduced-pressure drying). Transmission electron microscopy and X-ray diffraction indicated a predominance of tubular halloysite morphology with a 1.0-nm peak following Mg-saturation. In contrast, reduced pressure-dried samples showed a 0.7-nm peak without a 1.0-nm peak, indicating effective dehydration. The K+ selectivity was strongly linked to the interlayer spacing (1.0 vs 0.7 nm) of halloysite and the presence of Fe (hydr)oxides. The 0.3 nm larger interlayer spacing in hydrated halloysite appears to contribute to K+ selectivity as the weakly hydrated K+ can readily dehydrate and enter into the interlayer space while the strongly hydrated Ca2+ is too large to enter, a mechanism similar to K+ selectivity of ion channels in human nerve and muscle tissue. The Fe (hydr)oxides may reduce the permanent negative charge and enhance K+ selectivity via physically blocking interlayer exchange sites and repulsing cations, especially divalent cations, due to their positive charge. Results of this study suggest the occurrence of a high-charge, K+-selective halloysite for which hydration/dehydration and Fe (hydr)oxides strongly influence the charge and selectivity characteristics by altering ion access to interlayer exchange sites.
1. Introduction
Mechanisms contributing to unusually high cation exchange capacity (CEC) and K+/Ca2+ selectivity in several halloysite-rich soils remain a topic of intense debate (Joussein et al. Citation2005; Yang et al. Citation2016). Halloysite is a 1:1 aluminosilicate mineral, which is characterized by a diversity of morphologies, chemical composition, specific surface area, structural disorder, and physicochemical properties (e.g., CEC, ion selectivity) (Dahlgren et al. Citation2004; Joussein et al. Citation2005; Joussein Citation2016). Among the diversity of properties, CEC values for halloysite generally range between 2 and 10 cmolc kg−1, although values in excess 20 cmolc kg−1 clay have been reported (Bailey Citation1990; Norrish Citation1995). The larger CEC values may originate from permanent negative charges derived from Al3+ vacancies in the octahedral sheet, isomorphous substitution of Al3+ for Si4+ in the tetrahedral sheet, and/or isomorphous substitution of Fe2+ for Al3+ or nonstoichiometric substitution of Fe3+ for Al3+ (i.e., cation vacancies) in the octahedral sheet leading to a deficit of positive charge (Soma et al. Citation1992; Takahashi et al. Citation2001; Churchman et al. Citation2016; Joussein Citation2016; Yang et al. Citation2016).
There are several reports of soils dominated by halloysite in their clay fraction showing strong K+ or NH4+ retention (e.g., Okamura and Wada Citation1984; Quantin et al. Citation1988; Delvaux et al. Citation1989; Delvaux et al. Citation1990a; Delvaux et al. Citation1990b; Delvaux et al. Citation1992; Fontaine et al. Citation1989; Espino-Mesa and Hernandez-Moreno Citation1994; Escudey and Galindo Citation1988; Escudey et al. Citation1997). The high K+ (or NH4+) selectivity has been explained in terms of (i) K+- or NH4+-selective halloysite (Wada and Odahara Citation1993; Okamura and Wada Citation1984; Ndayiragije and Delvaux Citation2004), (ii) presence of a halloysite-smectite mixed-layer clay (Delvaux et al. Citation1990a; Delvaux et al. Citation1990b), (iii) presence of mica, vermiculite or zeolite impurities (Ming and Mumpton Citation1989; Parfitt Citation1992), (iv) preferential repulsion of divalent cations by Fe oxides surface coatings (Escudey and Galindo Citation1988; Escudey et al. Citation1997), and (v) whole salt (KCl) retention within halloysite tubes (Wada Citation1958; Wada Citation1959; Thomas Citation1960). However, no unique mechanism has been demonstrated to explain both the high charge and K+ selectivity of halloysitic clays (reviewed by Joussein et al. Citation2005).
Takahashi et al. (Citation2001) found high K+ selectivity in the <2-mm fraction of subsurface horizons in soils derived from andesitic basalt flows in northern California. The clay fraction was dominated by hydrated tubular halloysite (1.0 nm) with abundant free iron oxides (70 – 100 g Fe kg−1 by citrate-dithionite extraction). No detectable 2:1 layer silicates, 1:1–2:1 mixed-layer clays, or zeolites were identified in the clay-size fraction; however, trace levels of these constituents are very difficult to detect. The high K+ selectivity in the fine-earth fraction (<2 mm) was attributed to the existence of a K+-selective halloysite phase and preferential repulsion of divalent cations by Fe oxide surface coatings, although the contribution of 2:1 minerals detected in the silt fraction could not be ignored (Takahashi et al. Citation2001). The 27Al-NMR spectrum of the halloysite-rich clay indicated a poorly-ordered kaolin and a tetrahedral Al content of ~2% attributed to isomorphous substitution of Al3+ for Si4+ in the tetrahedral layer.
The primary objective of this paper was to further elucidate the mechanism(s) for high K+-selectivity of volcanic soils dominated by halloysite from northern California. Our previous study examined K+-selectively in the fine-earth fraction (<2-mm fraction) of these soils (Takahashi et al. Citation2001). This study builds on our previous research by rigorously examining the charge and K+/Ca2+ selectivity properties of the clay-size fraction (<2 µm) following various pretreatments. For high charge and K+-selective halloysite, it is considered that the halloysite should possess permanent negative charges that are selectively accessible to K+. Because permanent negative charge sites are considered to be located in interlayers, the basal spacing of halloysite (0.7 or 1.0 nm) was assessed (reviewed by Joussein et al. Citation2005; Joussein Citation2016). Thus, this study examined the permanent and variable charge characteristics and the changes in K+/Ca2+ selectivity of the clay fraction upon selective removal of organic matter (by H2O2) and free iron oxides (by citrate-dithionite treatment) and alteration of the basal spacing by dehydration (reduced-pressure drying). In addition, the <2-mm fraction from a comparably weathered soil formed in andesitic lahar that was similarly dominated by tubular halloysite, but with detectable 2:1 clay minerals, was included to gain insights on the potential role of 2:1 layer silicates on charge and K+/Ca2+ selectivity characteristics.
2. Material and methods
2.1. Experimental soils
Soils formed on extrusive volcanic materials were collected from two areas in northern California. Soils from two sites used in the original study of K+ selectivity (Takahashi et al. Citation2001) are located in the vicinity of Mt. Shasta volcano in the Cascade Range (LeTrab: 41°16ʹ0”N, 121°48ʹ10”W; Red Tank: 41°18ʹ59”N, 121°46ʹ46”W). These soils formed in andesitic basalt flows and were overlain by rhyolitic ash (25–40 cm) from Holocene eruptions (Takahashi et al. Citation1993; Takahashi et al. Citation2001). Based on Takahashi et al. (Citation2001), we selected the 2Bt3 horizons from the LeTrab (fine-loamy, halloysitic, mesic, Andic Haploxeralf) and Red Tank (fine, halloysitic, mesic, Andic Haploxeralf) pedons for more intensive study. These horizons have clay mineralogy dominated by halloysite and display considerable weathering as evidenced by high clay (33 and 51%) and free Fe oxide (93 and 45 g Fe kg−1) concentrations ().
Table 1. Selected physical and chemical properties of soil samples.
Soils from a second area located in the western Sierra Nevada (Aiken: 38°42ʹ38.2”N, 120°33ʹ04.0”W) were selected to include a soil whose clay mineralogy was dominated by tubular halloysite, but also contained detectable 2:1 layer silicates that may be expected to affect charge and K+ selectivity (Rasmussen et al. Citation2007). The parent material of the Aiken soil consists of Miocene- to Pliocene-aged andesitic lahar with evidence of late-Pleistocene to early-Holocene volcanic ash inputs in the surface horizons. The Aiken pedon was classified as a fine, halloysitic, mesic Andic Palehumult. The Bt3 and Bt4 horizons selected for this study have a clay mineralogy dominated by halloysite with minor 2:1 layer silicates and display appreciable weathering as evidenced by the high clay (39 and 51%) and free Fe oxide (22 and 23 g Fe kg−1) concentrations.
2.2. Clay preparation for charge and k+/ca2+ selectivity assessment
The clay-size fraction (<2 µm) from the LeTrab and Red Tank soils was isolated from air dry soil using the pipette method following dispersion with sonication (15 kHz, 150 W, 10 min) and pH adjustment to ~9 with NaOH. Five treatments were prepared from the collected clay specimens:
Air dried
Organic matter removal: Air-dried samples were treated with 5% H2O2.
Reduced-pressure drying: Air-dried clays were further dried under reduced pressure at about 15 Pa overnight.
Free iron oxide removal: Air-dried samples were treated with sodium citrate and dithionite (CD treated) for 16 hr (Holmgren Citation1967). After washing with 80% acetone, the clay samples were air dried.
Iron oxide removal/reduced-pressure drying: Following free iron oxide removal by CD (Treatment 4), the iron oxide free clays were dried under reduced pressure overnight.
A preliminary study demonstrated no difference on charge and K+/Ca2+ selectivity properties between field moist and air dried soils. Therefore, we elected to work with air-dried clays to maintain consistent moisture conditions among all treatments. We acknowledge that the clay pretreatment methods used in the present study may affect clay charge characteristics in several ways and therefore any conclusions must recognize the potential for artifacts.
2.3. Determination of permanent and variable charge characteristics
Permanent and variable charge characteristics of the pretreated clay samples were determined by the CsCl method of Anderson and Sposito (Citation1991). This method was developed for 2:1 type minerals that possess micaceous structure. Therefore, the values obtained in this study should be considered ‘apparent’ permanent negative charge because the clay samples did not possess any detectable micaceous minerals (K2O% <0.1%). Briefly, samples were saturated with Cs+ using CsCl solutions (0.5 M and then 0.05 M) and washed with ethanol. Then, the Cs-saturated samples were dried at 65°C for 48 h. The Cs+ was extracted from the dried samples with 0.01 M LiCl (variable charge) and then 1 M NH4OAc (permanent charge). Concentrations of Cs+ in the extracted solutions were measured by atomic absorption spectrophotometry (A-2000; Hitachi High Technologies Corp., Tokyo, Japan).
2.4. K+/ca2+ exchange equilibrium study
A K+-Ca2+ exchange equilibrium study was performed using the method of Wada and Odahara (Citation1993) on the five clay treatments from the LeTrab and Red Tank soils and also on the non-treated <2-mm fraction of the LeTrab, Red Tank and Aikens soils. Exchange equilibrium values were measured across a range of K adsorption ratios ([K+]/[Ca2+]1/2) from 0.1 to 4.5 mM1/2 (final values after equilibrium). Samples were washed five times with 0.5 M CaCl2 to saturate exchange sites with Ca2+, then washed six times to equilibrate the exchange sites with mixed KCl-CaCl2 solutions having a constant 0.02 M Cl− concentration. After the final decantation, adsorbed K+ and Ca2+ were extracted five times using 1 M NH4OAc solution. Concentrations of K+ and Ca2+ in the equilibrated and extracted solutions were measured by atomic absorption spectrophotometry. The equivalent fractions of adsorbed K+ (EK) and solution K+ (ĒK) were compared using the Vanselow (KV) selectivity coefficient:
where NK and NCa denote molar fractions of adsorbed K+ and Ca2+, and (K+) and (Ca2+) denote aqueous ion activities. Aqueous ion activities were calculated using activity coefficients computed from the Davies equation.
2.5. Transmission electron microscopy (TEM)
TEM observation was performed on the K+-saturated clay specimens (CD-air-dried and CD-reduced pressure-dried) spotted onto C-coated collodion films using a Hitachi H-7650 operating at 100 kV (Hitachi High Technologies Corp., Tokyo, Japan). TEM observation was also performed on the CD-treated and K+-saturated clay specimens isolated from the Akin soil horizons.
2.6. X-ray diffraction (XRD) analysis
The CD-air-dried samples and CD-reduced pressure-dried clay samples were saturated with Mg2+ (non-treated and solvated with formamide) and K+ (heated to 25, 300 and 550°C) for analysis with a MiniFlex XRD (Rigaku Corp., Tokyo, Japan) using Cu Kα radiation generated with a 30 kV accelerating potential and a 15 mA tube current. Samples were step-scanned for 2 s at a 0.02° 2θ step with the random orientation method. The CD-air-dried clay fraction (<2 µm) from the Aiken soil was also analyzed by XRD using the same XRD protocols.
3. Results
3.1. TEM and XRD analysis of LeTrab and Red Tank clay samples
Transmission electron microscopy showed a predominance of tubular morphology with lesser amounts of spheroidal morphology (), which are representative morphologies for halloysite (Joussein et al. Citation2005; Churchman et al. Citation2016; Yang et al. Citation2016). There were no morphological differences between the air-dried and reduced pressure-dried samples (data not shown). No identifiable 2:1 minerals, zeolites or allophane/imogolite phases were observed. We previously reported that concentrations of K (0.08–0.10% K2O), Fe (0.94–1.14% Fe2O3), and Mg (0.06–0.09% MgO) were low in the LeTrab and Red Tank CD-treated clay fractions indicating that they are only a minor structural component of the clay fraction (Takahashi et al. Citation2001).
Figure 1. Transmission electron micrographs of the citrate-dithionite-treated clay fractions of LeTrab 2Bt3 and Red Tank 2Bt3 samples. Reference scales are 0.5 µm.

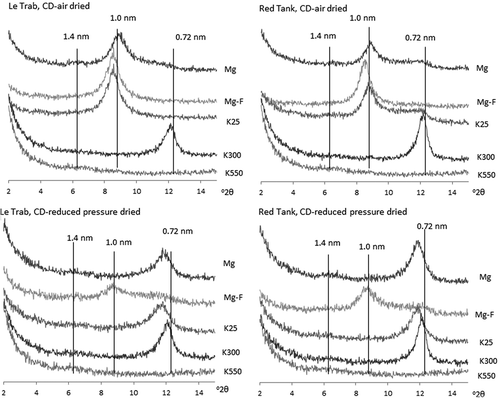
The CD-air dried clay samples from both LeTrab and Red Tank soils showed a prominent 1.0-nm peak with Mg-saturation (). Upon K-saturation and heating to 300°C, the 1.0-nm peak collapsed to 0.7 nm, and the peak disappeared after heating to 550°C. The combined TEM images and XRD patterns confirmed the dominance of halloysite (1.0 nm) in the clay fraction. The CD-reduced pressure-dried samples showed a 0.7-nm peak without a 1.0 nm peak, indicating that the interlayer water was effectively dehydrated by reduced-pressure drying. The 0.7-nm peak largely expanded to 1.0-nm with formamide treatment indicating that the mineral retained the fundamental nature of halloysite.
Figure 2. X-ray diffractograms of CD-air dried and CD-reduced pressure dried clay fractions of LeTrab 2Bt3 and Red Tank 2Bt3 samples.
3.2. Permanent and variable negative charge characteristics of LeTrab and Red Tank clay fractions
Permanent and variable negative charges determined by the method of Anderson and Sposito (Citation1991) are shown in . The permanent negative charge for air-dried LeTrab and Red Tank clays decreased by 16% (from 7.7 to 6.5 cmolc kg−1) and 27% (from 9.5 to 6.9 cmolc kg−1) upon reduced-pressure drying, respectively. In contrast, free iron oxide removal with CD increased the permanent charge by 63% (from 7.7 to 12.6 cmolc kg−1) and 80% (from 9.5 to 17.1 cmolc kg−1) for the LeTrab and Red Tank clay fractions, respectively. Reduced pressure-drying of CD treated clays decreased the permanent charge of the LeTrab clays from 12.6 to 8.4 cmolc kg−1 and the Red Tank clays from 17.1 to 10.4 cmolc kg−1.
Table 2. Permanent and variable negative charges of clay fractions after the treatments.
The variable charge component of both clay samples was also decreased by reduced pressure drying; from 13.6 to 8.3 cmolc kg−1 for LeTrab and from 10.4 to 7.7 cmolc kg−1 for Red Tank. In contrast to the permanent charge, the difference in variable charge values between air dried and CD-air dried samples was small.
3.3. Potassium ion selectivity of LeTrab and Red Tank clay fractions
The variation in the sum of extracted K+ and Ca2+ in the K+-Ca2+ exchange equilibrium study was small (i.e., small SD values) for all treatments (). This indicates that the preferential adsorption of CaCl+ was negligible and K+ fixation was not an important factor (Takahashi et al. Citation2001) because the sum of extracted K+ and Ca2+ values is nearly constant across the entire K+ vs. Ca2+ saturation range (Supplementary Fig.1); adsorption of CaCl+ would increase the K+ + Ca2+ values in the lower K+ saturation range and K+ fixation would decrease the K+ + Ca2+ values in the higher K+ saturation range. The sums of exchangeable K+ + Ca2+ were 11.7 and 13.3 cmolc kg−1 for air-dried LeTrab and Red Tank samples, respectively. These values were 1.4–1.5 times larger than the measured permanent charges, but 55–67% of the total negative charges (permanent + variable; ). The trend for changes in the exchangeable K+ + Ca2+ values across the various clay pretreatments was similar to changes in the permanent (r2 = 0.69 and 0.71 for LeTrab and Red Tank) and total negative charges (r2 = 0.33 and 0.83 for LeTrab and Red Tank) ( & ). Reduced pressure drying decreased the exchangeable K+ + Ca2+ values while the CD treatment increased the values.
Table 3. Average and standard deviation values of adsorbed Ca2++K+ in K+-Ca2+ exchange equilibrium study for clay fractions of LeTrab and Red Tank soils.
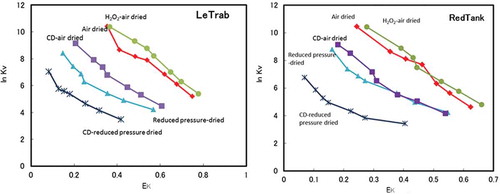
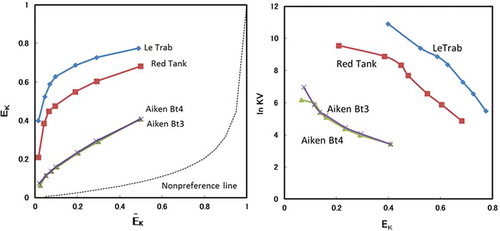
Plots of adsorbed K+ saturation vs. solution K+ saturation and Vanselow (KV) selectivity vs. K-saturation are shown in and , respectively. The air-dried LeTrab and Red Tank clays showed very high K+ selectivity as compared to the non-preferential K+-Ca2+ line. Following either reduced pressure drying or CD treatment, the K+ selectivity showed a large decrease for both clay samples. The decreased K+ selectivity was generally larger for the reduced pressure drying as compared to the CD treatment (). For example, at a given adsorbed K+ saturation level the ln KV for K+ selectivity decreased by 2–3, with reduced pressure drying being lower than the CD treatment by ~1, except for adsorbed K+ saturation levels greater than 0.3 for the Red Tank clays which had similar values between the reduced pressure drying and CD treatments (). Reduced pressure drying of the CD treatment further decreased the K+ selectivity, indicating a synergist effect of the combined treatments. The H2O2 treatment slightly increased the K+ selectivity, possibly due to removal of organic matter (2–3 g C kg−1) that would exhibit a preferential affinity for divalent cations such as Ca2+.
Figure 3. Equivalent fractions of adsorbed K+ (EK) vs. equivalent fraction of solution K+ (ĒK) for clay fractions with the treatments. Non-preference line (dashed line) calculated following Sposito et al. (Citation1983).
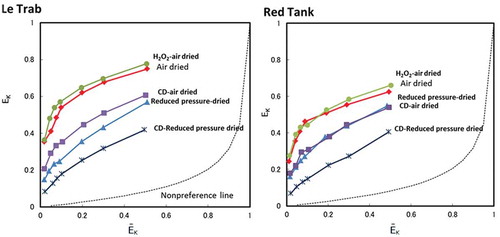
Figure 4. The Vanselow selectivity coefficient (ln KV) as a function of the equivalent adsorbed fraction of K+ (EK) for clay fractions with the treatments.
3.4. TEM images and XRD patterns for the clay fraction of Aiken soil
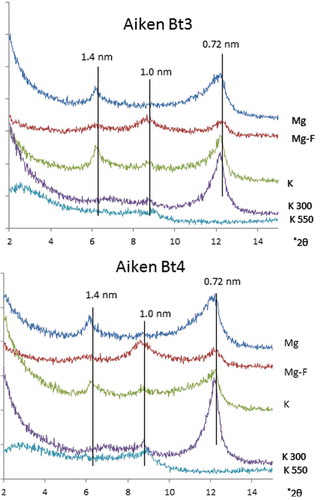
The TEM images of the clay fraction for the Aiken Bt3 and Bt4 horizons showed the predominance of tubular morphology similar to the clay fraction of the LeTrab and Red Tank samples (). The XRD patterns for the clay fractions of the Aiken soils showed distinct 0.7 nm- and 1.4 nm-peaks for Mg-saturation; there was no distinct peak at 1.0 nm as observed in LeTrab and Red Tank soils (). With formamide solvation, the 0.7 nm-peak partially expanded to 1.0 nm and then disappeared with 550°C heating, indicating the existence of an in situ dehydrated halloysite phase. The 1.4 nm-peak collapsed to 1.0 nm with K-saturation and heating (). These characteristics indicate the presence of 2:1 type minerals with appreciable hydroxy-Al interlayering.
Figure 5. Transmission electron micrographs of the citrate-dithionite-treated clay fractions of Aiken Bt3 and Aiken Bt4 soils.

Figure 6. X-ray diffractograms of CD-air dried clay fractions of Aiken Bt3 and Aiken Bt4 soils.
3.5. Potassium ion selectivity in the fine-earth fraction (<2 mm) of LeTrab, Red Tank and Aiken soils
The average values for the sum of extracted K+ and Ca2+ in the K+-Ca2+ exchange equilibrium study for the <2-mm fraction were 11.4 cmolc kg−1 for LeTrab soil and 10.6 cmolc kg−1 for Red Tank soil (). In comparison, the values for the Aiken soils were substantially lower (7.5 and 7.0 cmolc kg−1 for Bt3 and Bt4, respectively) in spite of their similar clay contents and dominance by tubular kaolin minerals. Plots of adsorbed K+ saturation vs. solution K+ saturation showed much higher K+ selectivity in the <2-mm fraction of the LeTrab and Red Tank soils in comparison to the Aiken soils ().
Table 4. Average and standard deviation values of adsorbed Ca2++K+ in K+-Ca2+ exchange equilibrium study for fine earth fractions (<2 mm) of LeTrab, Red Tank and Aiken soils.
Figure 7. Equivalent fractions of adsorbed K+ (EK) vs. equivalent fraction of solution K+ (ĒK) (left) and the Vanselow selectivity coefficient (ln KV) as a function of the equivalent adsorbed fractions of K+ (EK)(right) for fine earth fractions (<2 mm). Non-preference line (dashed line) calculated following Sposito et al. (Citation1983).
4. Discussion
To investigate possible mechanism(s) for the high K+ selectivity of halloysitic clays (1.0 nm) in the Bt horizons of the LeTrab and Red Tank pedons, we performed charge characterization and K+-Ca2+ ion selectivity experiments on the clay fractions following several pretreatments. Previous studies indicated that there were no detectable 2:1 minerals, halloysite-2:1 mixed layer minerals or zeolites in the clay fractions of the LeTrab and Red Tank soils (Takahashi et al. Citation1993; Takahashi et al. Citation2001). We also compared the K+ selectivity of these soils to a soil (Aiken Bt horizons) dominated by dehydrated tubular halloysite (0.7 nm expandable to 1.0 nm with formamide treatment) and containing a minor component of 2:1 layer silicates.
In contrast to the idealized halloysite structure, permanent negative charge measurements following several clay pretreatments indicated the presence of considerable permanent charge (7–17 cmolc kg−1) on the LeTrab and Red Tank clay fractions (). A previous 27Al-NMR characterization of the Red Tank 2Bt3 clay fraction indicated a poorly ordered halloysite with a tetrahedral Al content of about 2% (i.e., 2% Al3+ and 98% Si4+ in tetrahedral sheet) (Takahashi et al. Citation2001). This level of isomorphous substitution could account for ~15 cmolc kg−1 of permanent negative charge (assuming substitution of Al3+ for Si4+ in the tetrahedral layer) and is consistent with the value of 17.1 cmolc kg−1 measured in the CD-treated Red Tank Bt3 clay fraction in this study (). Similarly, Fe2+ substitution for Al3+ in the octahedral sheet (Churchman et al. Citation2016) could conceivably contribute 11.6 to 13.5 cmolc kg−1 of permanent charge, assuming that the Fe was associated with isomorphous substitution rather than Fe contaminants from Fe (hydr)oxides. Therefore, permanent charge associated with isomorphous substitution in either the tetrahedral or octahedral sheets or Al3+ vacancies in the octahedral sheet could contribute to the permanent charge measured in the LeTrab and Red Tank clay fractions.
Bailey (Citation1990) proposed that the interlayer water in halloysite (1.0 nm) is associated with exchangeable cations that balance the layer charge. The decrease in permanent charge following reduced pressure drying (of both the air dried and CD-air dried treatments) may be attributed to a decrease of ion accessibility to some ion-exchange sites because of interlayer collapse (1.0 → 0.7 nm). This is consistent with the findings that hydrated halloysite (1.0 nm) generally has a larger CEC than dehydrated forms (0.7 nm) (Grim Citation1968; Norrish Citation1995). In contrast, the near doubling of permanent charge upon free iron oxide removal was ascribed to an increase of ion accessibility to the interlayer by removal of prominent iron (hydr)oxide coatings that hinder ion diffusion to and from the interlayer (Shao and Wang Citation1991). Variable charge negative sites were not strongly affected by iron oxide removal, but did demonstrate a large reduction following reduced pressure drying. These variable charge sites are associated with broken bonds at particle edges. Changes in charge characteristics following strong soil drying are commonly reported and attributed to irreversible changes in the chemical (e.g., change in surface coordination) and physical (e.g., decrease in surface area and aggregation) surface properties (Dowding et al. Citation2005).
There are several studies of K+-Ca2+ selectivity for 2:1 clay minerals reported in the literature (Robbins and Carter Citation1983; Ogwada and Sparks Citation1986; Feigenbaum et al. Citation1991; Wada and Seki Citation1994; Bond Citation1995). In general, when the EK is 0.4 in the EK vs. ln KV diagram (), the ln KV values are in the range of 2–6 as shown in the literature cited above. In sharp contrast, the clay fractions of the LeTrab and Red Tank samples were much higher, about 11 for LeTrab and 9 for Red Tank (). This literature comparison is also consistent with the differences measured in the <2-mm fraction between the LeTrab/Red Tank soils and the Aiken soils that contained dehydrated halloysite and a minor 2:1 layer silicate component. However, the hydroxy-Al interlayering of the 2:1 layer silicates in the Aiken soil may change the K+-Ca2+ selectivity dynamics compared to 2:1 layer silicates having no hydroxy-Al interlayers. Given that the pH range of the LeTrab/Red Tank soils (5.9–6.2) were similar to the Aiken soil horizons (5.8–5.9), it may be expected that any 2:1 layer silicates present in the LeTrab/Red Tank soils would also contain hydroxy-Al interlayers as in the Aiken soil horizons. Thus, we interpret these results to suggest that the mechanism for high K+ selectivity in the LeTrab and Red Tank clay fractions is not solely due to 2:1 clay mineral impurities.
If the halloysite is the source of the high K+ selectivity, the halloysite should possess permanent negative charge and the basal spacing (0.7 nm or 1.0 nm) should be a key factor affecting the accessibility of K+ to the permanent charge sites. The evidence obtained in this study suggests the presence of permanent negative charge that increases following removal of free iron oxides. Following CD treatment, K+ selectivity decreased ( and ) and the sum of exchangeable Ca2++K+ increased. This indicates that the iron minerals may physically block the exchange sites of halloysite and repulse cations, especially divalent cations, due to their positive charges (Escudey and Galindo Citation1988; Takahashi et al. Citation2001). Iron (hydr)oxides, which possess a zero point of charge (ZPC) of ~8.8 (Sverjensky and Sahai Citation1996), have a net positive surface charge at the ambient pH of our soils (). Thus, charge-to-charge repulsion could make access of divalent cations (e.g., Ca2+) to the internal negative surface charge sites more difficult than for monovalent cations (e.g., K+).
With reduced-pressure drying and dehydration of interlayer waters, the K+ selectivity showed a large decrease ( and ). The selectivity for Aiken soils, whose halloysite was naturally dehydrated, was far lower compared to those of LeTrab and Red Tank soils, but similar to that of the reduced pressure-dried LeTrab and Red Tank clay fractions. These results provide strong evidence that the hydrated form of halloysite (1.0 nm-basal space) was an important factor affecting the high K+ selectivity in this study. The potassium ion has a comparatively lower hydration energy and easily dehydrates to monatomic K+ that can selectively enter the interlayer of halloysite (1.0 nm) relative to hydrated Ca2+. With the dehydration of the halloysite and the collapse of the interlayer, K+ appears to have a more difficult time accessing the permanent charge sites in the interlayer and the selectivity of K+ is correspondingly decreased. This mechanism contributing to K+ selectivity in hydrated halloysite has potential similarities to the K+ selectivity associated with ion channels in human nerve and muscle tissues (Degrève et al. Citation1996). The selectivity of these channels was related to the hydration structure of ions with the weakly hydrated K+ being selectively transported (as opposed to the larger strongly hydrated Na+) through ion channels with diameters of 0.3 nm. The diameter of these K+-selective ion channels is equivalent to the 0.3 nm difference in the interlayer distance between hydrated (1.0 nm) and dehydrated (0.7 nm) halloysite/kaolin phases. Thus, the ionic diameter of K+ relative to the hydrated halloysite interlayer spacing may play an important role in its preferential selectivity versus the much larger hydrated Ca2+.
TEM observation showed that the clay fractions of LeTrab and Red Tank soils were dominated by tubular halloysite. Tubular halloysite has the structure of several concentric layers of 1:1 aluminosilicates in a rolled internal configuration when viewed in cross-section (e.g., similar to a roll of paper). When the tubular halloysite (1.0 nm) dehydrates upon drying, parts of the layer structure may become disordered resulting in an irregular interlayer spacing. Dehydration was shown to expand the diameter of individual halloysite tubes, and layers within the rolls comprising the tubes became separated to form large voids between the individual sheets and slit-shaped pores along the tube axis (Kohyama et al. Citation1978; Kohyama et al. Citation1982; Churchman et al. Citation1995). Thus, some parts of the dehydrated halloysite interlayer may expands allowing access to hydrated Ca2+, and therefore result in a decrease in K+ selectivity. Similarly, some parts of the interlayer may become blocked due to the collapsed interlayer resulting in the loss of preferential access to K+ and also to the loss of accessible permanent charge to all ions. The much lower sum of extracted K+ and Ca2+ measured for the naturally dehydrated halloysite in the Aiken soil compared to the LeTrab/Red Tank soils () may also result in part from in situ dehydration and lack of access of cations to interlayer permanent charge sites. Thus, the concomitant decrease of K+ selectivity and permanent charge by the reduced-pressure drying is consistent with a high-charge, K+-selective halloysite (1.0 nm).
There have been previous reports indicating that halloysite with a basal spacing of 1.0 nm was responsible for high K+ selectivity of soils. Delvaux et al. (Citation1990a), (Citation1990b) and Ndayiragije and Delvaux (Citation2004) showed a close correlation between the amounts of halloysite and KV values and considered that the selectivity was derived from hydrated halloysite (1.0 nm). Some other studies have reported that soils with dehydrated halloysite (0.7 nm) also showed high K+ selectivity (Fontaine et al. Citation1989; Wada and Odahara Citation1993). However, in the study of Fontaine et al. (Citation1989), the existence of halloysite-smectite mixed layer clays was suggested and may contribute to the high K+ selectivity. Overall, there is paucity of evidence for high K+ selectivity in soils dominated by dehydrated halloysite (0.7 nm).
5. Conclusions
The results obtained in this study indicate the presence of a K+-selective halloysite phase having considerable permanent negative charge. The K+ selectivity of the halloysite was strongly linked to its interlayer spacing (1.0 vs. 0.7 nm). The 0.3 nm larger interlayer spacing in hydrated halloysite appears to contribute to K+ selectivity as the weakly hydrated K+ can readily dehydrate and enter into channels with a 0.3 nm diameter while the strongly hydrated Ca2+ is too large to enter, as has been demonstrated for K+ selective ionic channels in human muscle and nerve tissues. Thus, ion hydration energy plays an important role as the ions must lose their hydration shell to enter the halloysite interlayer space. The role of Fe (hydr)oxides was also shown to strongly affect the permanent charge and K+ selectivity. The Fe (hydr)oxides appear to physically block the exchange sites of halloysite and repulse cations, especially divalent cations, due to their positive charge. Verification of these mechanisms to explain high K+ selectivity for halloysite is required for a variety of other halloysite-rich soils experiencing high K+ selectivity.
EXCa_K_vs_EK_solution_.pptx
Download MS Power Point (153.9 KB)Related Research Data
References
- Anderson SJ, Sposito G 1991: Cesium-adsorption method for measuring accessible structural surface charge. Soil Sci. Soc. Am. J., 55, pp. 1569–1575. doi:10.2136/sssaj1991.03615995005500060011x
- Bailey SW 1990: Halloysite – a critical assessment. In eds. Farmer VC, Taedy Y, Proceedings 9th Int. Clay Conf., Strasbourg, France 1989, pp. 89–98. Science Géologiques, Mémoires No. 86.
- Blakemore LC, Searle PL, Daly BK 1981: Soil Bureau Laboratory Methods: A. Methods for Chemical Analysis of Soils, New Zealand, Soil Bureau Sci. Rep. 10A.
- Bond WJ 1995: On the Rothmund-Kornfeld description of cation exchange. Soil Sci. Soc. Am. J., 59, pp. 436–443. doi:10.2136/sssaj1995.03615995005900020024x
- Churchman GJ, Davy TJ, Aylmore LAG, Gilkes RJ, Self PG 1995: Characteristics of fine pores in some halloysites. Clay Miner., 30, pp. 89–98. doi:10.1180/claymin.1995.030.2.01
- Churchman GJ, Pasbakhsh P, Lowe DJ, Theng BKG 2016: Unique but diverse: some observations on the formation, structure and morphology of halloysite. Clay Miner., 51, pp. 395–416. doi:10.1180/claymin.2016.051.3.14
- Dahlgren RA, Saigusa M, Ugolini FC 2004: The nature, properties and management of volcanic soils. Adv. Agron., 82, pp. 113–182.
- Degrève L, Vechi SM, Junior CQ 1996: The hydration structure of the Na+ and K+ ions and the selectivity of their ionic channels. Biochim. Biophys. Acta, 1274, pp. 149–156. doi:10.1016/0005-2728(96)00019-9
- Delvaux B, Dufey JE, Vielvoye L, Herbillon AJ 1989: Potassium exchange behavior in a weathering sequence of volcanic ash soils. Soil Sci. Soc. Am. J., 53, pp. 1679–1684. doi:10.2136/sssaj1989.03615995005300060011x
- Delvaux B, Herbillon AJ, Dufey JE, Vielvoye L 1990a: Surface properties and clay mineralogy of hydrated halloysitic soil clays. I: existence of interlayer K+ specific sites. Clay Miner., 25, pp. 129–139. doi:10.1180/claymin.1990.025.2.01
- Delvaux B, Herbillon AJ, Vielvoye L, Mestdagh MM 1990b: Surface properties and clay mineralogy of hydrated halloysitic soil clays. II. Evidence for the presence of halloysite/smectite (H/Sm) mixed-layer clays. Clay Miner., 25, pp. 141–160. doi:10.1180/claymin.1990.025.2.02
- Delvaux B, Tessier D, Herbillon AJ, Burtin G, Jaunet AM, Vielvoye L 1992: Morphology, texture, and microstructure of halloysitic soil clay as related to weathering and exchangeable cation. Clays Clay Miner., 40, pp. 446–456. doi:10.1346/CCMN.1992.0400409
- Dowding CE, Borda MJ, Fey MV, Sparks DL 2005: A new method for gaining insight into the chemistry of drying mineral surfaces using ATR-FTIR. J. Colloid Interface Sci., 292, pp. 148–151. doi:10.1016/j.jcis.2005.05.075
- Escudey M, Diaz P, Foerster JE, Galindo G 1997: Adsorbed ion activity coefficients in K-Ca exchange on soil fractions derived from volcanic materials. Aust. J. Soil Res., 35, pp. 123–130. doi:10.1071/S96051
- Escudey M, Galindo G 1988: Potassium-calcium exchange on inorganic clay fractions of Chilean Andepts. Geoderma, 41, pp. 275–285. doi:10.1016/0016-7061(88)90065-1
- Espino-Mesa M, Hernandez-Moreno JM 1994: Potassium selectivity in andic soils in relation to induced acidity, sulphate status and layer silicates. Geoderma, 61, pp. 191–201. doi:10.1016/0016-7061(94)90047-7
- Feigenbaum S, Bar-Tal A, Portnoy R, Sparks DL 1991: Binary and ternary exchange of potassium on calcareous montmorillonitic soils. Soil Sci. Soc. Am. J., 55, pp. 49–56. doi:10.2136/sssaj1991.03615995005500010008x
- Fontaine S, Delvaux B, Dufey JE, Herbillon AJ 1989: Potassium exchange behavior in Caribbean volcanic ash soils under banana cultivation. Plant Soil, 120, pp. 283–290. doi:10.1007/BF02377078
- Grim RE 1968: Clay Mineralogy. McGraw-Hill, p. 596. New York.
- Holmgren GG 1967: A rapid citrate-dithionite extractable iron procedure. Soil Sci. Soc. Am. Proceed., 31, pp. 210–211. doi:10.2136/sssaj1967.03615995003100020020x
- Joussein E 2016: Geology and mineralogy of nanosized tubular halloysite. In eds. Yang P, Thill A, Bergaya F, Nanosized Tubular Clay Minerals. Halloysite and Imogolite, Development in Clay Science, Vol. 7, pp. 12–48 Elsevier, Amsterdam.
- Joussein E, Petit S, Churchman J, Theng B, Righi D, Delvaux B 2005: Halloysite clay minerals - A review. Clay Miner., 40, pp. 383–426. doi:10.1180/0009855054040180
- Kohyama N, Fukushima K, Fukami A 1978: Observation of hydrated form tubular halloysite by an electron microscope equipped with an environmental cell. Clays Clay Miner., 26, pp. 25–40. doi:10.1346/CCMN.1978.0260103
- Kohyama N, Fukushima K, Fukami A 1982: Interlayer hydrates and complexes of clay minerals observed by electron microscopy using an environmental cell. In eds. Van Olphen H, Veniale F, Proceedings of the 7th International Clay Conference 1981. Developments in Sedimentology, Vol. 35, pp. 373–384. Elsevier, Amsterdam.
- Ming DW, Mumpton FA 1989: Zeolites in soils. In eds. Dixon JB, Weed SB, Minerals in Soil Environments, 2nd. pp. 873–911. SSSA Book Ser. no. 1, Madison, WI. USA.
- Ndayiragije S, Delvaux B 2004: Selective sorption of potassium in a weathering sequence of volcanic ash soils from Guadeloupe, French West Indies. Catena, 56, pp. 185–198. doi:10.1016/j.catena.2003.10.010
- Norrish K 1995: An unusual fibrous halloysite. In eds. Churchman GJ, Fitzpatrick RW, Eggleton RA, Clays – controlling the Environment, Proceeding 10th Int. Clay Conf. Adelaide, Australia 1993, pp. 275–284. CSIRO Publ., Melbourne, Australia.
- Ogwada RA, Sparks DL 1986: Use of mole or equivalent fractions in determining thermodynamic parameters for potassium exchange in soils. Soil Sci., 141, pp. 268–273. doi:10.1097/00010694-198604000-00003
- Okamura Y, Wada K 1984: Ammonium-calcium exchange equilibria in soils and weathered pumices that differ in cation exchange materials. J. Soil Sci., 35, pp. 387–396. doi:10.1111/j.1365-2389.1984.tb00295.x
- Parfitt RL 1992: Potassium-calcium exchange in some New Zealand soils. Aust. J. Soil Res., 30, pp. 145–158. doi:10.1071/SR9920145
- Quantin P, Gautheyrou J, Lorenzoni P 1988: Halloysite formation through in situ weathering of volcanic glass from trachytic pumices, Vico’s Volcano, Italy. Clay Miner., 23, pp. 423–437. doi:10.1180/claymin.1988.023.4.09
- Rasmussen C, Matsuyama N, Dahlgren RA, Southard RJ, Brauer N 2007: Soil genesis and mineral transformation across an environmental gradient on andesitic lahar. Soil Sci. Soc. Am. J., 71, pp. 225–237. doi:10.2136/sssaj2006.0100
- Robbins CW, Carter DL 1983: Selectivity coefficient for calcium-magnesium-sodium-potassium exchange in eight soils. Irrigation Sci., 4, pp. 95–102.
- Shao ZC, Wang WJ 1991: Relationship between iron oxides and surface charge characteristics in soils. Pedosphere, 1, pp. 29—39.
- Soma M, Churchman GJ, Theng BKG 1992: X-ray photoelectron spectroscopic analysis of halloysites with different composition and particle morphology. Clay Miner., 27, pp. 413–421. doi:10.1180/claymin.1992.027.4.02
- Sposito G, Holtsclaw KM, Charlet M, Journey C, Page AL 1983: Sodium-calcium and sodium-magnesium exchange on Wyoming bentonite in perchlorate and chloride background ionic media. Soil Sci. Soc. Am. J., 53, pp. 52–57.
- Sverjensky DA, Sahai N 1996: Theoretical prediction of single-site surface-protonation equilibrium constants for oxides and silicates in water. Geochim. Cosmochim. Acta, 60, pp. 3773–3797. doi:10.1016/0016-7037(96)00207-4
- Takahashi T, Dahlgren R, Van Susteren P 1993: Clay mineralogy and chemistry of soils formed in volcanic materials in the xeric moisture regime of northern California. Geoderma, 59, pp. 131–150. doi:10.1016/0016-7061(93)90066-T
- Takahashi T, Dahlgren RA, Theng BKG, Whitton JS, Soma M 2001: Potassium-selective, halloysite-rich soils formed in volcanic materials from northern California. Soil Sci. Soc. Am. J., 65, pp. 516–526. doi:10.2136/sssaj2001.652516x
- Thomas GW 1960: Factors affecting the removal of salts from halloysite. Soil Sci., 90, pp. 344–347. doi:10.1097/00010694-196012000-00005
- Wada K 1958: Adsorption of alkali chloride and ammonium halide on halloysite. Soil Plant Food, 4, pp. 137–144. doi:10.1080/00380768.1958.10430876
- Wada K 1959: Oriented penetration of ionic compounds between the silicate layers of halloysite. Am. Mineral., 44, pp. 153–165.
- Wada S–I, Odahara K 1993: Potassium-calcium exchange in five Ap soils from paddy fields and its effect on potassium concentration in soil solution. Soil Sci. Plant Nutri., 39, pp. 129–138. doi:10.1080/00380768.1993.10416982
- Wada S–I, Seki H 1994: Ca-K-Na exchange equilibria on a smectitic soil: modeling the variation of selectivity coefficient. Soil Sci. Plant Nutr., 40, pp. 629–636. doi:10.1080/00380768.1994.10414302
- Yang H, Zhang Y, Ouyang I 2016: Physicochemical properties of halloysite. In eds. Yang P, Thill A, Bergaya F, Nanosized Tubular Clay Minerals. Halloysite and Imogolite. Development in Clay Science, Vol. 7, pp. 67–91. Elsevier, Amsterdam.