
ABSTRACT
Under abiotic stress conditions, plants actively modify the transcriptome either for activation of stress tolerance mechanisms or adjustment of biological processes for adaptation to the stress. Comparative analysis of transcriptome data under different stressors and stress intensities is useful to understand how plants balance growth and survival under stress conditions. In this study, the transcriptome of Arabidopsis roots was investigated in response to four rhizotoxic stressors, namely aluminum (Al3+), cadmium (Cd2+), and copper (Cu2+) ions and sodium chloride (NaCl), under mild stress (50% inhibition of root elongation compared with the control) and severe stress (>90% inhibition of root elongation). Only Al treatment showed similar transcriptome responses between the stress intensities. The shared Al-specific up-regulated genes included known Al-tolerance genes and downstream genes of SENSITIVE TO PROTON1 (STOP1) such as ALUMINUM ACTIVATED MALATE TRANSPORTER1 and malic enzymes, suggesting that Al-defense mechanisms regulated by STOP1 might be activated over a broader range of Al stress intensities. The sulfur metabolism pathway was activated only under severe Al stress, suggesting that rapid activation of sulfur metabolism is crucial for survival of root cells in the early stage of severe Al stress. A large number of genes associated with the response mechanism to reactive oxygen species production were up-regulated by Cd and Cu treatment under both severe and mild stress, but the members of the genes sets differed between the stress severities. The results indicated that Cd and Cu stress trigger common transcriptional responses, but the responses are likely associated with the stress severity. The transcriptional response to mild NaCl stress was relatively smaller compared with those to the other stressors, suggesting that in Arabidopsis different mechanisms other than rapid transcriptomic responses are involved in the response to NaCl stress. Comparison between Al and the other stressors indicated that the STOP1 regulation system and central metabolic pathways contribute to the similarity of the transcriptional response to different intensities of Al stress. The results suggest that robustness of transcriptionally regulated Al tolerance systems to different stress intensities is important for both of growth and survival in plant roots under Al stress.
1. Introduction
As sedentary organisms, plants must adapt to environmental stresses, but are required to maintain a balance between growth and survival in agricultural lands and natural areas (Claeys and Inze Citation2013). The root system is important for maintaining growth because it is essential for acquisition of water and nutrients, and thus it sometimes determines growth of the whole plant. Rhizotoxicity of cadmium (Cd) and copper (Cu) ions may be encountered and enhanced by man-made activities, such as soil contamination resulting from mining activities, whereas aluminum (Al) and NaCl rhizotoxicities occur naturally (Wu et al. Citation1996; Kobayashi et al. Citation2016). In particular, Al rhizotoxicity, which disrupts root growth of diverse crops, is common in acid soils of pH < 5.5 and affects 40% of arable lands worldwide. Identification of molecular mechanisms for tolerance to rhizotoxicity in plants, especially for Al tolerance, would contribute to improvement of crop productivity globally in stressed soil conditions.
Although many Al tolerance genes have been identified by forward genetic approaches, such as fine mapping of mutants (see reviews by Liu et al. Citation2014; Daspute et al. Citation2017), additional studies using genome-wide approaches, including transcriptome analyses, are required to understand the complex molecular physiological mechanisms of Al tolerance (Yokosho et al. Citation2014; Li et al. Citation2017; Xu et al. Citation2017). Comparative transcriptomic analysis that includes several types of rhizotoxic stressors is one experimental approach to investigate complex stress tolerance mechanisms, such as that of Al tolerance, because the responsive genes and biological pathways can be classified into common and specific groups. In a previous study, we analyzed the root transcriptome response to relatively strong rhizotoxic stresses, namely Al, Cd, and Cu ions and NaCl, in Arabidopsis (Zhao et al. Citation2009). The transcriptome response to rhizotoxic treatments using concentrations that caused almost complete cessation of growth (i.e. > 90% growth inhibition) indicated that the transcriptional response to reactive oxygen species (ROS) and calcium (Ca) homeostasis were shared with all tested ions for survival rather than growth. By contrast, several known Al tolerance genes, such as ALUMINUM ACTIVATED MALATE TRANSPORTER1 (AtALMT1; Hoekenga et al. Citation2006), are relatively specifically inducible by Al rather than other rhizotoxic stressors (Zhao et al. Citation2009). Additional comparative analyses of responses to different stress conditions, such as milder stress conditions (e.g. 50% growth inhibition), may provide molecular information on Al tolerance mechanisms that operate with maintenance of root growth. In addition, comparative transcriptomic analyses between Al and other rhizotoxic stressors may help to reveal differential tolerance strategies of plants to other rhizotoxic ions and highlight the growth/survival strategies against Al toxicity by considering commonality and specificity among the stressors.
Previous transcriptome analyses of responses to Al stress usually compared Al treatment and the control condition and dose-response transcriptional information is limited at the transcriptome level. These analyses identified inducible expression of several major Al tolerance genes, which individually contribute to Al tolerance, such as AtALMT1 and ALUMINUM SENSITIVE 3 (ALS3) in Arabidopsis (Sawaki et al. Citation2009). However, the pH and Al concentration varies with soil depth and fluctuates with fertilizer application (You et al. Citation2015). It is well known that different defense mechanisms are employed for different intensities of drought stress. Severe drought reduces maximum Rubisco carboxylation and ribulose 1,5-bisphosphate regeneration results in decreased photosynthesis (Flexas and Medrano Citation2002; Chaves et al. Citation2009), whereas stomatal closure limits photosynthesis under milder drought stress (Flexas et al. Citation2014). The increased isoprene emission and zeaxanthin concentration observed under mild drought stress may protect the phytosynthetic apparatus against stresses (Tattini et al. Citation2015). These differences imply that plants have multiple physiological and transcriptional mechanisms that are employed to overcome a particular stress with adjustment of the balance between growth and survival. Clarification of common and different transcriptional mechanisms will contribute to the establishment of advanced breeding strategies. In addition, it remains unclear whether Al alters transcription in other biological processes in accordance with the balance between maintenance of growth and survival, such as metabolic pathways, hormone metabolism, signaling pathways that regulate development, and cell division. It also unknown whether the overall pattern of transcriptomic adaptation to Al stress is similar to that for other ions.
In this study, we adopted a comparative transcriptomic approach for (1) determination of similarities and differences in the transcriptional response to mild stress compared with that for severe stress, (2) characterization of growth/survival strategies against Al stress by comparison with multiple ion stresses, and (3) improvement in understanding how plants adapt to different rhizotoxic stress conditions through maintaining a balance between growth and survival. The root transcriptome of Arabidopsis thaliana plants grown under mild stress (50% inhibition of root elongation compared with root growth under the control condition) induced by the rhizotoxic stressors Al, Cd, and Cu ions and NaCl was compared with previously published root transcriptome data for the same rhizotoxic stressors under severe stress (>90% root growth inhibition). The results show that distinct pattern of transcriptomic adaptation by each rhizotoxic ion, including greater similarity of inducible genes group by Al at different stress intensities. This greater similarity of transcriptomic pattern of Al than other ions, in other words robustness of transcriptomic adaptation pattern at different stress intensities of Al, would be explained by the involvement of STOP1 regulated Al-inducible tolerance system, including upregulation of AtALMT1 with changes in gene expression for malate metabolism.
2. Methods
2.1. Rhizotoxic stressor treatments
Rhizotoxic stressor treatments and root sampling were performed as described by Zhao et al. (Citation2009). Arabidopsis thaliana accession Col-4 (N933, NASC; Nottingham Arabidopsis Stock Center, Nottingham, UK) seedlings were hydroponically pre-grown in modified 1/50 MGRL medium (Fujiwara et al. Citation1992) with 200 μM of CaCl2 and inorganic phosphorus at pH 5.0 for 10 days and transferred to the 1/50 MGRL solution with 200 μM of CaCl2 except inorganic phosphorus containing either 6 μM AlCl3, 3 μM CdCl2, 1.3 μM CuSO4, or 10 mM NaCl for 24 h. Concentrations of each ion and NaCl were adjusted to achieve 50% root growth inhibition compared with root growth under the control condition during a 1-week growth test. As a control, pre-grown seedlings were transferred to modified 1/50 MGRL medium lacking rhizotoxic stressors. Al free activity was calculated by GEOCHEM-EZ software version 1.0 (Shaff et al. Citation2010). Free Al ion at the plasma membrane (PM) surface was calculated by a SGCS electrostatic model (Kinraide and Wang Citation2010).
2.2. RNA isolation
Total RNA was isolated using the method described by Suzuki et al. (Citation2004), then quantified at A260 using a NanoDrop™ spectrophotometer (ND-1000, NanoDrop Technologies, Wilmington, DE, USA). The quality of RNA used for microarray analysis was measured using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) in accordance with the manufacturer’s instructions.
2.3. Microarray experiment
Microarray analyses were performed using the Agilent microarray system (Agilent Technologies, Palo Alto, CA, USA) as described by Zhao et al. (Citation2009). All procedures were carried out in accordance with the manufacturer’s protocols. Briefly, 1 μg total RNA from each sample was used to synthesize cRNA and was labeled with cyanine-5 (Cy5)- or cyanine-3 (Cy3)-labeled CTP (Perkin Elmer/NEN Life Sciences, Tokyo, Japan). The labeled cRNAs (including a treatment and a control sample) were competitively hybridized to the Arabidopsis 3 Oligo Microarray and then washed. The hybridized slides were scanned using Agilent DNA Microarray Scanner version 6.1 software and data points were extracted using Agilent Feature Extraction version 8.1 software. Three comparisons, including one dye-flip, were made between biologically independent samples. All microarray data have been deposited in the Gene Expression Omnibus of the National Center for Biotechnology Information under the accession number GSE108751.
2.4. Data analysis
All statistical analyses of microarray data were performed using R version 3.4.3 available at CRAN (http://cran.r-project.org/). Previously studied microarray data for the severe rhizotoxic ion stress condition (E-MEXP-1907) and the stop1 mutant under the Al stress condition (E-MEXP-1908) were downloaded from the ARRAYEXPRESS database to compare transcriptional similarities and differences between different stress intensities with consideration of STOP1 regulation. Quality control of the microarray data was performed using the default settings of GeneSpring GX version 12.6.1 software (Agilent, Tokyo, Japan). Probes for the mild and severe stress conditions were connected by their AGI codes. A total of 21,399 genes that passed the quality control in all 24 microarray experiments were used for statistical analyses and gene selection. Up- and down-regulated genes in response to an individual stressor were defined if their fold change values were in the upper or lower 2.5% (i.e. upper or lower 534 genes) in all three replications. Genes up-regulated by all mild stressors and unique gene groups were identified by Venn diagrams using the R package VennDiagram (Chen and Boutros Citation2011). Functional categorization of differentially expressed genes was performed using PANTHER (Thomas et al. Citation2003). Pathway enrichment analysis for up-regulated genes was performed using the BioAnalytic Resource Classification SuperViewer (Provart et al. Citation2003) and MapMan version 3.5.1 software (Thimm et al. Citation2004). Co-expression gene networks of up-regulated genes were prepared using NetworkDrawer implemented in ATTED-II (Aoki et al. Citation2016) with the ‘add a few genes’ option. Network density of individual gene modules was calculated using the NetWorkAnalyzer function implemented in Cytoscape version 3.2.0 software (Shannon et al. Citation2003). Enrichment analysis of octamers in the AtALMT1 promoter was performed using the method described by Yamamoto et al. (Citation2011).
3. Results
3.1. Global analysis of transcriptomic response to mild and severe rhizotoxic stress treatments
Rhizotoxic stress treatments were applied using a hydroponic culture system to study the transcriptomic response of four rhizotoxic stressors at concentrations that induce 50% root growth inhibition (see Kobayashi et al. Citation2007). Either of 6 μM AlCl3, 3 μM CdCl2, 1.3 μM CuCl2, and 10 μM NaCl was applied in the treatments. The transcriptome was obtained after incubation of seedlings for 24 h in each rhizotoxic solution. Transcriptomic responses to the severe stress condition (i.e. medium containing the rhizotoxic stressor at the concentration that induced > 90% root growth inhibition; Zhao et al. Citation2009), which were obtained in the previous study, were reanalyzed with the current data to compare transcriptomic responses at different levels of rhizotoxic stressors. It was predicted that Al-free activity and PM surface Al ion activity were 1.4 μM and 13.2 μM under the mild Al stress treatment, respectively. Al toxicity at the root plasma membrane was occurred even at moderate acidic condition when relatively lower Ca concentration used in this study (Kobayashi et al. Citation2013).
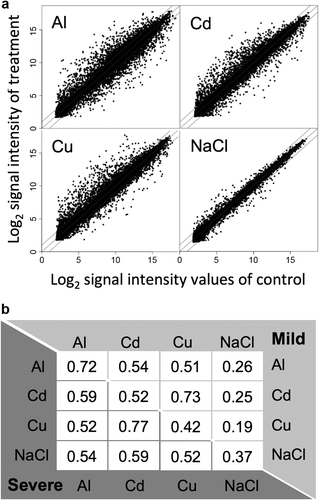
A total of 21,399 genes that passed the quality filters of all 24 microarray chips (two stress intensities, four stressors, three replications) were used for analyses. Scatter plots showed that NaCl altered the transcription of fewer genes than the other tested stressors (; fold change > 2 or < 0.5). The correlation matrix () showed that similar transcriptomic responses between the mild and severe stress conditions were observed for Al ions only (r = 0.72), in contrast to the lower Pearson correlation coefficients observed for the other stressors (r = 0.37–0.52). This result suggests that the transcriptomic response to Al stress is broadly occurred against different stress intensities and degrees of root growth inhibition. Similarities in transcriptomic responses between Cd and Cu ions were observed both under mild and severe stress. Pearson correlation coefficients between Cd and Cu were 0.73 and 0.77 under mild and severe stress, respectively, whereas the correlation coefficients between mild and severe stress were 0.52 and 0.42 for Cd and Cu, respectively. This result suggests that the heavy metals Cd and Cu induce a similar transcriptional response to that of Al and NaCl. In addition, different transcriptional systems might operate for the different stress intensities under Cd and Cu stress.
Figure 1. Whole transcriptome response to rhizotoxic stressor treatment. (A) Scatter plot of competitive microarray data from roots of Arabidopsis subjected to mild stress of rhizotoxic stressors. Roots of hydroponically grown seedlings were treated by control (pH 5.0, no toxicant) and rhizotoxic solutions containing AlCl3 (6 μM), CdCl2 (3 μM), CuCl2 (1.3 μM), or NaCl (10 mM) at pH 5.0 for 24 h. X and Y axes indicate signal intensities in the control and rhizotoxic treatments, respectively. The mean signal intensities from three biologically independent replications are plotted. The slope of the lines in each panel represent 2, 1, and 1/2 fold changes, respectively. (B) Pearson correlation matrix for the whole transcriptome in response to four rhizotoxic stressors. Correlation coefficients of 21,399 genes are shown for different stressors (Al, Cd, and Cu ions and NaCl) compared with the mild (upper matrix) or severe (lower matrix) stress conditions.
4. Stressor-specific and common responsive genes
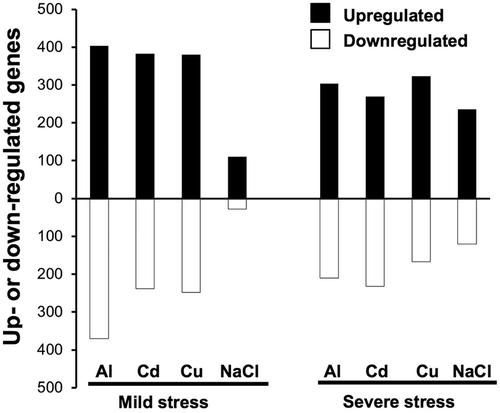
To accentuate the responsive genes for each stressor, the most up-regulated or down-regulated genes were grouped by ranking the fold change of the genes on the basis of whether the fold change value was in the upper and lower 2.5% in each microarray experiment (Zhao et al. Citation2009). The current gene selection methodology using ranking-based method has an advantage to compare transcriptome profiles with different dynamic range of fold change (i.e. NaCl induced much less genes than the other ions; and Supplementary Figure S1) and to extract the outstanding biological processes activated by individual stresses Additionally, our ranking-based criterion placed importance on reproducibility of gene lists in the three replicates (Guo et al. Citation2006; MAQC Consortium Citation2006) rather than collecting all of possible induced/suppressed genes. Of the 21,399 genes, 403, 382, 380, and 110 genes were classified as up-regulated by Al, Cd, Cu, or NaCl, respectively (). On the other hand, 370, 239, 248, and 28 genes were classified as down-regulated by Al, Cd, Cu, or NaCl, respectively. Those genes were overlapped for the most part with up- or down-regulated genes selected by a conventional gene selection method using statistical test based on t-statistic and fold change (Supplementary Figure S1). Salt stress induced and suppressed fewer genes under mild stress intensity than those under severe stress intensity, although a similar number of genes were up- or down-regulated in response to the other stressors. These results indicated that the transcriptional response to NaCl was relatively mild or slow compared with that to the other stressors, despite inducing the same degree of root growth inhibition.
Figure 2. Number of differentially expressed (up- and down-regulated) genes in Arabidopsis roots after treatment with rhizotoxic stressors for 24 h. Up- and down-regulated genes under the mild and severe stress conditions for Al, Cd, and Cu ions and NaCl. The genes were defined as those with fold change (treatment/control) values in the upper and lower 2.5% in all three replicates.
Venn diagrams identified gene groups most specifically responsive to each stressor, and the overlap between the most up- and down-regulated genes of each stressor at the different stress intensities (). These gene groups reflect the specificity of the transcriptional response to the four tested rhizotoxic stressors. The highest ratio was observed in the overlapped genes between Cd and Cu treatment (), which corresponded to the high correlation of the whole transcriptome response between Cd and Cu. Thirty-one genes were up-regulated in response to all four stressors under mild stress, whereas 58 genes were up-regulated in common in response to severe stress. No genes were suppressed by all four rhizotoxic stressors, although 13 genes were down-regulated in common under the severe stress intensity. Although fewer genes were up-regulated or down-regulated by all four stressors under the mild stress intensity because significantly fewer genes were selected in the mild NaCl stress treatment, many genes associated with ROS scavenging, such as GLUTATHIONE S-TRANSFERASE F2, GLUTATHIONE S-TRANSFERASE F3, PEROXIDASE 34, PEROXIDASE 52, PEROXIDASE 58, and a Ca-homeostasis associated gene that encodes a putative calcium-binding protein CML47, were in the common up-regulated gene group (). Although most of the up-regulated genes in common under mild stress were up-regulated in at least one severe stress condition, only three genes, namely NODULIN MTN21-LIKE TRANSPORTER UMAMIT36, ankyrin (At3g13950), and LACCASE 12, responded only to milder stress conditions and thus might be associated with stress tolerance without growth retardation.
Table 1. Upregulated genes by all four stressors (Al, Cd, Cu, and NaCl) under milder stress intensity.
Figure 3. Venn diagram of differentially expressed genes in response to individual rhizotoxic stressors. (A) The top 2.5% genes whose expression was most increased or decreased by each stressor drawn using the VennDiagram R package. The number of genes that was specific to each stressor is indicated in the small circle for comparison between the severe and mild stress intensities. (B) The proportion of genes overlapped between each two upregulated gene sets. Different letters indicate a significant difference in the ratio (Fisher’s exact test; p < 0.05). (C) Evaluation of specificity in individual stressor-dependent gene expression. Specifically expressed genes were defined as individual stressor-specific expressed genes in the Venn diagram without up-regulation by the other stressors (FC <2). Asterisks indicate significant difference in the ratio of specifically expressed genes (Fisher’s exact test; p < 0.05).
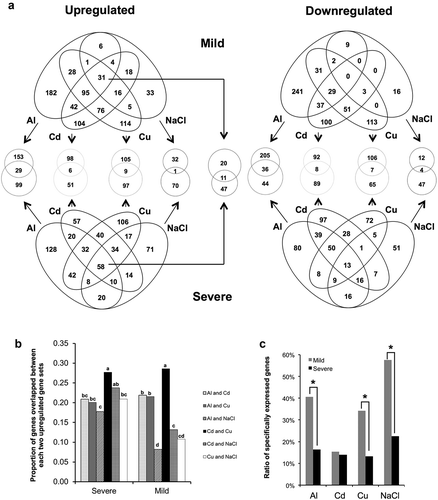
Venn diagrams also indicated that 182, 104, 114, and 33 genes showed stress-specific up-regulation under mild Al, Cd, Cu, and NaCl stress, respectively (; Supplementary Tables S1–S4). A higher proportion of genes were specifically responsive under mild stress than those under severe stress (). Typical Al tolerance genes, such as MULTIDRUG AND TOXIC COMPOUND EXTRUSION (MATE; Magalhaes et al. Citation2007) and ALUMINIUM-SENSITIVE 3 (ALS3; Larsen et al. Citation2005), were among the up-regulated genes under mild Al stress (Supplementary Table S1), whereas AtALMT1 was identified in the severe stress intensity. Mild Al stress specifically induced five genes, which were previously identified as expression level polymorphic genes in a RNA-Seq analysis employing six Arabidopsis thaliana accessions with contrasting degrees of Al tolerance: MYB112, LACCASE 7, NAC74, CHITINASE C (ChiC), and PLASMODESMATA-LOCATED PROTEIN 1 (PDLP1) (Kusunoki et al. Citation2017). These results suggested that several Al tolerance genes are highly specifically inducible by Al compared with the other tested stressors. Up-regulated genes under mild Cd stress included NAD(P)-binding Rossmann-fold superfamily protein (At1g07440) and MITOCHONDRION-LOCALIZED SMALL HEAT SHOCK PROTEIN 23.6 (ATHSP23.6-MITO), which were detected by proteome analysis in response to Cd stress in A. thaliana (Brahim et al. Citation2010; Sarry et al. Citation2006). Genes specifically responsive to NaCl included C2-DOMAIN ABA-RELATED 5 (CAR5), which is a positive regulator of abscisic acid-activated signaling pathways (Rodriguez et al. Citation2014). Enriched GO-BP terms were identified for the genes up-regulated in response to mild Cu stress, including ethylene-activated signaling pathways consisting of six ethylene response factors (ERF11, ERF016, ERF094, ERF096, ERF114, and ERF115) (Supplementary Table S5).
5. Possible mechanisms for the induction of the genes groups in response to both mild and severe Al stress
To investigate the possible mechanism for the up-regulated genes in response to different intensities of Al stress, we examined Al-inducible genes that overlapped between the mild and severe stress intensities and performed a co-expression network analysis. When the specific genes were compared between the stress intensities, a relatively larger overlap was identified for Al (29 genes) than for the other stressors (; ). Genes regulated by the STOP1 transcription factor (i.e. fold change (stop1/wild type) < 0.5) were significantly enriched in the 29 genes shared in both stress conditions (p = 5.257E-05 in Fisher’s Exact Test), including six genes such as an NADP-dependent malic enzyme (ME2) and a sulfate transporter (SURTR3;1). In addition, we performed a co-expression network analysis of Al-specific up-regulated genes under mild and severe stress intensities. Eleven small co-expression genes networks were identified by the ATTED-II co-expression database for the severe Al stress condition. Two (of which one was large) co-expression networks were identified for mild Al stress. Three of the co-expression networks identified for the severe Al stress condition were completely or partially included in the large network identified for mild Al stress. Two of the shared networks for severe Al stress contained four STOP1-regulated genes, whereas 11 STOP1-regulated genes were included in the large network derived for mild Al stress. These results suggested that a larger number of STOP1-regulated genes is one factor in the transcriptomic similarity to different stress intensities. Pearson’s correlation coefficient value of whole transcriptome between severe and mild Al stress was slightly decreased when it was calculated excluding STOP1-regulated genes, while it was not observed in the other stressors (Supplementary Figure S2). This hypothesis is further supported by higher relative appearance of part of STOP1-binidng sequence in the AtALMT1 promoter under both Al stress intensities (Supplementary Figure S3; Tokizawa et al. Citation2015). Pathway enrichment analysis of the Al-specific up-regulated genes identified three enriched MapMan-pathways (‘TCA/org transformation’, ‘development’, and ‘transport’) shared by the two stress conditions (Supplementary Table S6). When the up-regulated genes were mapped on the MapMan metabolic map, malate-related pathways, such as malic acid and malate dehydrogenase, were enriched (Supplementary Figure S4), which suggested that malate metabolic processes operated to support malate release by ALMT1.
Table 2. Al-specific upregulatd genes under both severe and mild stress intensities.
Enrichment of the GO-BP terms ‘glutathione metabolic process’ and ‘response to salicylic acid’ was observed in both Al stress treatments (; Supplementary Table S7), which suggested that a similar transcriptional response to ROS is an additional factor in the transcriptomic similarity to different stress intensities. Five and eight ROS-responsive genes up-regulated by REDOX RESPONSIVE TRANSCRIPTION FACTOR1 (RRTF1) overexpression (Matsuo et al. Citation2015) also were included in the co-expression gene networks constructed for the Al-specific up-regulated genes under severe and mild stress intensity, respectively (). Several different GO-BP terms were observed between the mild and severe Al stress conditions; ‘sulfate transmembrane transport’, ‘suberin biosynthetic process’, and ‘lateral root formation’ were only observed for severe Al stress, whereas ‘response to aluminum ion’ and ‘calcium ion transmembrane transport’ were only observed for mild Al stress. The KaPPA metabolic process viewer showed up-regulation of genes associated with the sulfur metabolism pathway for severe Al stress (Supplementary Figure S5). These results suggested that specific metabolic pathways were responsive to severe Al stress, although most of pathways were similar between the mild and severe Al stress conditions.
Figure 4. Co-expression gene network of Al-specific up-regulated genes under severe (A) and mild (B) stress. A co-expression gene network was constructed, using Al-specific up-regulated genes under severe and mild stress intensity as query genes, with the ATTED-II database. Closed and opened circles indicate query and co-expressed genes added according to co-expression relationships. Triangles and rectangles indicate genes regulated by SENSITIVE TO PROTON RHIZOTOXICITY1 (STOP1) and REDOX RESPONSIVE TRANSCRIPTION FACTOR1 (RRTF1), respectively. Dashed circles and ellipses indicate overlapped co-expression gene networks between the severe and mild stress intensities.
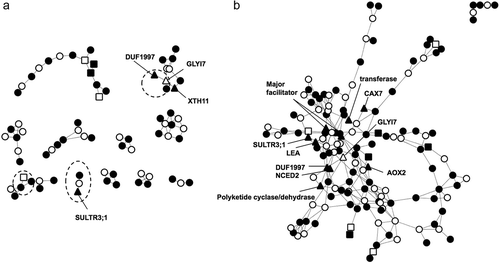
Figure 5. Common and specific biological pathways responsive to severe and mild Al stress intensity. Representative genes are listed in parentheses for each gene ontology (GO) term. Representative enriched GO terms in the biological process category for genes up-regulated under severe or mild Al stress. GO analysis was performed using the PANTHER classification system. All enriched GO-BP terms are listed in Supplementary Table S7.

6. Comparative analysis of genes up-regulated in response to both Cd and Cu
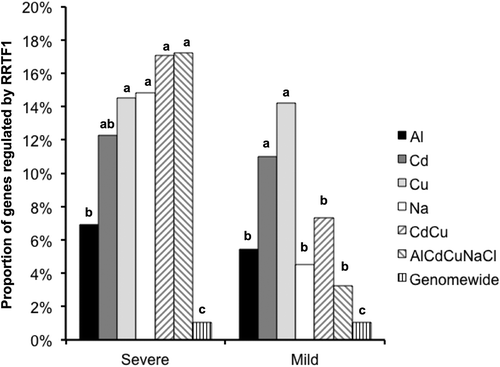
Under severe stress intensity, 164 genes were up-regulated in response to both Cd and Cu, whereas 218 up-regulated genes overlapped under mild Cd or Cu stress (). Responsive genes that overlapped between Cd and Cu stresses were compared between the severe and mild stress conditions to clarify the similarities and differences in the transcriptional response to severe and mild stress intensity (). Many similarities were observed in the transcriptional responses to Cd and Cu stress, that is transcription factors (C2H2, WRKY), peroxidase, ethylene responsive transcription factors, auxin responsive proteins, laccases, germin-like proteins, gluthathione S-transferases, and ABC transporters were up-regulated in response to both Cd and Cu under severe or mild stress intensity. Many GO groups overlapped between Cd and Cu for each stress intensity, and contained ROS response-related GO terms such as ‘toxin catabolic process’, ‘hydrogen peroxide catabolic process’, and ‘response to reactive oxygen species’ including PEROXIDASE 15/49/54/58, GSTF2/3, and GSTU4. It is suggested that both Cd and Cu induce similar ROS-mediated signaling pathways. Up-regulated gene groups contained several hydrogen peroxide (H2O2) inducible genes, which were up-regulated in H2O2-accumulating transgenic Arabidopsis that overexpresses RRTF1 ().
Figure 6. Comparison of enriched GO-BP terms between Cd and Cu stresses under severe and mild stress intensity. Venn diagram showing overlap in genes up-regulated by Cd and Cu stress. The number in parentheses indicates genes regulated by REDOX RESPONSIVE TRANSCRIPTION FACTOR1 (RRTF1; Matsuo et al. Citation2015). Representative enriched GO terms in the biological process category were classified into three groups. GO analysis was performed using the PANTHER classification system.
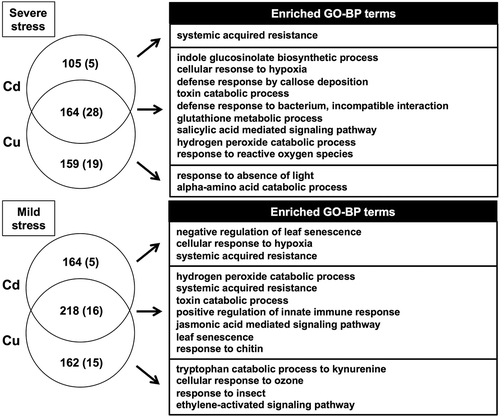
Figure 7. Proportion of genes regulated by RRTF1 among the up-regulated genes by individual stressors and transcriptome wide. Proportions are out of 222 genes upregulated in transgenic Arabidopsis overexpressing REDOX RESPONSIVE TRANSCRIPTION FACTOR1 (RRTF1; Matsuo et al. Citation2015). Different letters indicate a significant difference in the ratio (Fisher’s exact test; p < 0.05).
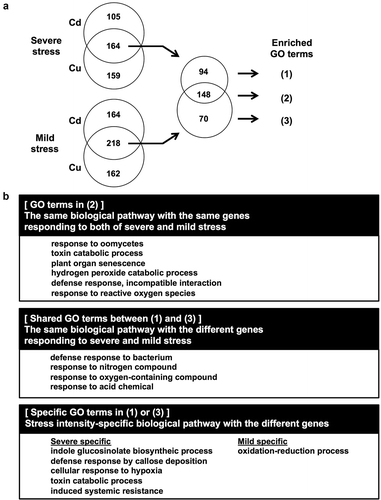
By contrast, only a few GO terms were enriched only under severe or mild stress conditions. For example, genes for ‘salicylic acid mediated signaling pathway’ were enriched in response to severe stress treatment, whereas genes for ‘jasmonic acid mediated signaling pathway’ were enriched under the mild stress condition. Only one GO term (systemin acquired resistance in Cd-specific genes) was shared by Cd- or Cu-specific gene groups under the mild and severe stress conditions (). The GO terms ‘Indole glucosinolate biosynthetic process’ and ‘callose deposition’ were enriched only in the severe stress-specific gene group, including NSL1, MYB51, and CYP81F2. In the mild stress-specific gene group, ‘oxidation-reduction process’ included LACCASE 12, L-ASPARTATE OXIDASE, PEROXIDASE 28/34/39, CYP71B3, and CYP81F1 (; Supplementary Tables S8–S9).
Figure 8. Common and specific biological pathways between severe and mild stress intensities in the overlapped genes up-regulated by Cd and Cu stress. (A) Venn diagram of genes up-regulated in response to Cd and Cu stress. (B) Representative enriched GO terms in the biological process category were classified into three groups. GO analysis was performed using the PANTHER classification system. All enriched GO-BP terms are listed in Supplementary Table S9.
7. Discussion
Transcriptional regulation plays a major role in stress tolerance of plants during adaptation to stress conditions. In a previous comparative transcriptomic analysis of rhizotoxic ions in Arabidopsis, we identified both ion-specific responses and common transcriptional responses, including activation of several stress tolerance genes. In the previous study, we applied severe stress of Al, Cd, Cu, and NaCl using concentrations of the rhizotoxic stressors that almost completely inhibited root growth. In the present study, we obtained the transcriptome under milder stress conditions that maintained 50% root growth relative to the control condition. The present analysis further identified different transcriptional responses to the same four rhizotoxic stressors under the mild stress condition, and examined differences in the transcriptomic responses between the two stress intensities.
Our previous study demonstrated that ROS-scavenging enzymes and Ca-binding proteins play an important role in the response to exposure to Al, Cd, Cu, and NaCl under severe stress (Zhao et al. Citation2009). It is notable that the ROS response to the four rhizotoxic stressors under severe stress was also observed under the mild stress intensity, although fewer ROS-responsive genes were up-regulated (). This finding suggested that protection against ROS and maintenance of Ca-homeostasis is a basic defense response to any rhizotoxic stressor, even under the mild stress intensity. On the other hand, stressor-specific gene responses were expanded under the mild stress intensity for all of the tested rhizotoxic stressors (). These results suggested that the stressor-specific gene response is dominant for stress tolerance while maintaining growth under the mild stress condition. Thus, utilization of mild stress intensities might be suitable for candidate gene selection in transcriptome experiments.
Comparative analysis between the severe and mild stress conditions showed that the transcriptomic response patterns differed between stress intensities. The four tested rhizotoxic stressors were classified into three groups on the basis of the transcriptomic similarity/difference between the severe and mild stress intensities (). The transcriptional response to Al stress was relatively similar between the severe and mild stress intensities in both the correlation analysis at the transcriptome level and in the overlap of Al-specific up-regulated genes compared with the other stressors ( and ). These results suggested that a similar transcriptional tolerance mechanism is triggered by different Al concentrations. This similarity can be partly explained by the Al-specific activation of genes regulated by the STOP1 transcription factor (Iuchi et al. Citation2007; ; ; Supplementary Figure S2–3). The mild Al stress intensity induced transcription of not only ALMT1 but also MATE and ALS3, which are representative Al-tolerance genes. Given that STOP1 is likely to be a ‘master switch’ for plant stress responses, recent studies indicate that it may regulate different gene sets, such as activation of genes responsive to phosphate deficiency (Balzergue et al. Citation2017; Mora-Macías et al. Citation2017). It suggests that several STOP1-regulated genes may require Al-specific activation, including other mechanisms such as up-regulation of the co-activator of target genes, or an Al-specific mechanism of post-translational activation of STOP1. Additionally, part of the Al-activated co-expression gene networks did not contain STOP1-regulated genes (). These gene groups may be regulated by other critical transcription factors, which show greater specificity to Al than to other ions, such as WRKY46 in Arabidopsis (Ding et al. Citation2013) and ABSCISIC ACID, STRESS AND RIPENING (ASR5; Arenhart et al. Citation2013) in rice. Up-regulation of ALMT1 and enrichment of tricarboxylic acid cycle-related genes, including malic enzymes, were observed under both severe and mild Al stress (; ; Supplementary Figure S3). Rapid and robust activation of malate release mechanism against different Al stress intensities also likely contributed to the similar transcriptome between the severe and mild Al stress conditions, because Al detoxification by malate chelation is a foremost defense system against Al ions. In addition, up-regulation of transcripts for malate metabolism under different Al stress intensities suggested activation of operation of biochemical pH-stat pathway under both Al stress conditions can also contribute to the transcriptomic similarity (Sakano Citation1998). By contrast, GO terms associated with ROS response, such as ‘glutathione metabolic process’ and ‘response to salicylic acid’, overlapped between the severe and mild Al stress conditions, which showed that ROS response is another important response regardless of stress intensity. Conversely, genes associated with sulfate transport and metabolism were activated only under severe Al stress (; Supplementary Figure S4). Sulfur fertilizer alleviates Al toxicity in barley, citrus, wheat, and oilseed rape via improvement in antioxidant protection and decrease in ROS concentrations (Chen et al. Citation2013; Guo et al. Citation2017; Qian et al. Citation2014). Activation of sulfur transport and metabolism is likely involved in scavenging the high quantities of ROS produced under severe Al stress. The commonality of shared tolerance mechanisms (i.e. organic acid excretion, pH regulation, and sulfur-mediated metabolisms) at different stress intensities would explain the similar transcriptomic adaptation pattern between the different Al stress intensities. Greater transcriptomic similarity between different stress intensities of Al could be also explained by the limited molecular target of Al than that of Cd/Cu, then the simplicity of phytotoxicity of Al compared to that of the other stressors. For example, Cd and Cu competitively bind to the metal binding domains in the metal binding proteins regulating metal homeostasis such as heavy metal transporting ATPases and metal chaperons (Dudev and Lim Citation2014).
Figure 9. Schematic illustration of the proposed model for the difference in transcriptional response between mild and severe stress intensities.
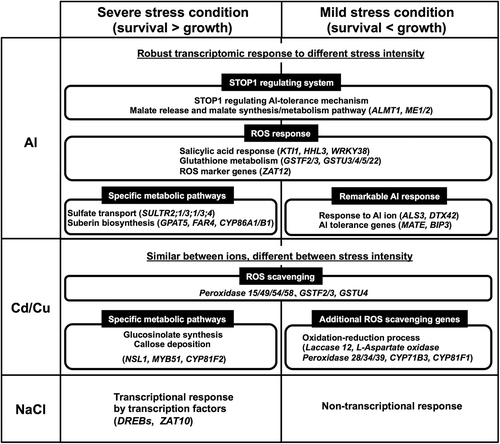
In contrast to the other tested rhizotoxic stressors, under mild NaCl stress transcriptomic change was much smaller than that observed under severe NaCl stress and in response to the other stressors. Significantly fewer genes were up-regulated under mild NaCl stress (), which suggested that NaCl tolerance is regulated by non-transcriptional responses under mild NaCl stress. Several NaCl tolerance genes, such as SALT OVERLAY SENSITIVE 1, which encodes a protein kinase (CIPK24), were not induced in response to NaCl treatment. By contrast, the short-term (24 h) exposure to severe NaCl stress rapidly induced the drought/salt responsive pathway regulated by DEHYDRATION RESPONSIVE ELEMENT BINDING PROTEIN 1A (DREB1A), DREB subfamily A-5 (At1g22810 and At5g21960), and ZAT10, and drastically altered the root transcriptome for defense (Zhao et al. Citation2009). Overexpression of DREB1A results in severe growth retardation under normal growing conditions (Kasuga et al. Citation1999). These results suggested that the non-transcriptional defense system might be employed for root growth under moderate and weak salinity stress conditions.
The current analysis also detected a high correlation between the responses to Cd and Cu ions, both in the entire transcriptome and among up-regulated genes ( and ; Supplementary Tables S8–S9). Both Cd and Cu are known to induce oxidative stress at the cellular level (Smeets et al. Citation2009; Cuypers et al. Citation2011). Overlapped up-regulated genes between the Cd and Cu stress treatments were mainly associated with ROS response/scavenging (–). Interestingly, glucosinolate biosynthesis and callose deposition genes were notable only under severe stress, which suggested that activation of these biological pathways is involved with survival under cessation of root growth. Mild stress also induced ROS response but in different gene members from those induced under severe stress.
In summary, we characterized differences in the transcriptional response to different stress intensities of four representative rhizotoxic stressors in relation to maintenance of the balance between growth and survival. The results suggested that the STOP1 regulation system and central metabolic pathways contributed to the similarity of the transcriptional response to different intensities of Al stress, and that robustness of Al tolerance systems to different stress intensities is more important for both of growth and survival in plant roots under Al stress than under the other tested rhizotoxic stressors. Commonality and specificity of the transcriptional response among the individual stressors clarified the biological processes involved in the tolerance to common and specific stressors. Stressor-specific up-regulated genes identified in this study can be investigated in future studies as candidate genes and physiological processes in relation to survival under severe stress and maintenance of plant growth under moderate stress, which are frequently encountered in agriculture. Combination of a genome-wide approach with the current approach may be required for efficient isolation of stress tolerance genes. For example, we recently identified several Al tolerance genes by studying expression level polymorphism between accession groups with contrasting Al tolerance (Kusunoki et al. Citation2017). In addition, integration of co-expression gene network analysis and genome-wide association studies for NaCl tolerance revealed previously unidentified NaCl tolerance mechanisms (e.g., involvement of the cell wall in NaCl tolerance; Kobayashi et al. Citation2016). A similar integrated approach may accelerate identification of genes involved in regulation of root growth under stress conditions.
Supplementary_Table_S9.pdf
Download PDF (20.6 KB)Supplementary_Table_S8.pdf
Download PDF (271.8 KB)Supplementary_Table_S7.pdf
Download PDF (18.2 KB)Supplementary_Table_S6.pdf
Download PDF (10.4 KB)Supplementary_Table_S5.pdf
Download PDF (14.1 KB)Supplementary_Table_S4.pdf
Download PDF (33.7 KB)Supplementary_Table_S3.pdf
Download PDF (103 KB)Supplementary_Table_S2.pdf
Download PDF (94.6 KB)Supplementary_Table_S1.pdf
Download PDF (160.9 KB)FigS5.jpg
Download JPEG Image (2.8 MB){kind=link}
FigS4.jpg
Download JPEG Image (2.5 MB){kind=link}
FigS3.jpg
Download JPEG Image (1.2 MB){kind=link}
FigS2.jpg
Download JPEG Image (914.3 KB){kind=link}
FigS1.jpg
Download JPEG Image (1.7 MB){kind=link}
Acknowledgments
We thank Robert McKenzie, PhD, from Edanz Group (www.edanzediting.com/ac), for editing a draft of this manuscript.
Disclosure statement
The authors have no conflicts of interest to declare.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Aoki Y, Okamura Y, Tadaka S, Kinoshita K, Obayashi T 2016: ATTED-II in 2016: a plant coexpression database towards lineage-specific coexpression. Plant Cell Physiol., 57, e5 (1–9). doi:10.1093/pcp/pcv165
- Arenhart RA, Lima JCDE, Pedron M, et al. 2013: Involvement of ASR genes in aluminium tolerance mechanisms in rice. 52–67. doi:10.1111/j.1365-3040.2012.02553.x
- Balzergue C, Dartevelle T, Godon C, et al. 2017: Low phosphate activates STOP1-ALMT1 to rapidly inhibit root cell elongation. Nat. Commun., doi:10.1038/ncomms15300
- Brahim S, Joke D, Ann C, Jean-Paul N, Marjo T, Arja T, Sirpa K, Frank VB, Karen S, Jaco V 2010: Leaf proteome responses of Arabidopsis thaliana exposed to mild cadmium stress. J. Plant Physiol., 167, 247–254. doi:10.1016/j.jplph.2009.09.015
- Chaves MM, Flexas J, Pinheiro C 2009: Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann. Bot., 103, 551–560. doi:10.1093/aob/mcn125
- Chen H, Boutros PC 2011: VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics, 12, 35. doi:10.1186/1471-2105-12-35
- Chen J, Wang WH, Wu FH, You CY, Liu TW, Dong XJ, He JX, Zheng HL 2013: Hydrogen sulfide alleviates aluminum toxicity in barley seedlings. Plant Soil, 362, 301–318. doi:10.1111/j.1744-7909.2010.00946.x
- Claeys H, Inze D 2013: The agony of choice: how plants balance growth and survival under water-limiting conditions. Plant Physiol., 162, 1768–1779. doi:10.1104/pp.113.220921
- Cuypers A, Karen S, Jos R, et al. 2011: The cellular redox state as a modulator in cadmium and copper responses in Arabidopsis thaliana seedlings. J. Plant Physiol., 168. 309–316. doi:10.1016/j.jplph.2010.07.010
- Daspute AA, Sadhukhan A, Tokizawa M, Kobayashi Y, Panda SK, Koyama H 2017: Transcriptional regulation of aluminum-tolerance genes in higher plants: clarifying the underlying molecular mechanisms. Front. Plant Sci., 8, 1–12. doi:10.3389/fpls.2017.01358
- Ding ZJ, Yan JY, Xu XY, Li GX, Zheng SJ 2013: WRKY46 functions as a transcriptional repressor of ALMT1, regulating aluminum-induced malate secretion in Arabidopsis. Plant J., 76, 825–835. doi:10.1111/tpj.12337
- Dudev T, Lim C 2014: Competition among metal ions for protein binding sites: determinants of metal ion selectivity in proteins. Chem. Rev., 114, 538–556. doi:10.1021/cr4004665
- Flexas J, Diaz-Espejo A, Gago J, Gallé A, Galmés J, Gulías J, Medrano H 2014: Photosynthetic limitations in Mediterranean plants: a review. Environ. Exp. Bot., 103, 12–23. doi:10.1016/j.envexpbot.2013.09.002
- Flexas J, Medrano H 2002: Drought-inhibition of photosynthesis in C3plants: stomatal and non-stomatal limitations revisited. Ann. Bot., 89, 183–189. doi:10.1093/aob/mcf027
- Fujiwara T, Hirai MY, Chino M, Komeda Y, Naito S 1992: Effects of sulfur nutrition on expression of the soybean seed storage protein genes in transgenic Petunia. Plant Physiol., 99, 263–268. doi:10.1104/pp.99.1.263
- Guo L, Lobenhofer EK, Wang C, et al. 2006: Rat toxicogenomic study reveals analytical consistency across microarray platforms. Nat. Biotechnol., 24, 1162–1169. doi:10.1038/nbt1238
- Guo P, Li Q, Qi Y-P, Yang L-T, Ye X, Chen -H-H, Chen L-S 2017: Sulfur-mediated-alleviation of aluminum-toxicity in Citrus grandis seedlings. Int. J. Mol. Sci., 18, 2570. http://www.mdpi.com/1422-0067/18/12/2570 doi:10.3390/ijms18122570
- Hoekenga OA, Maron LG, Piñeros MA, et al. 2006: AtALMT1, which encodes a malate transporter, is identified as one of several genes critical for aluminum tolerance in Arabidopsis. Proc. Natl. Acad. Sci. USA, 103, 9738–9743. doi:10.1073/pnas.0602868103
- Kasuga M, Liu Q, Miura S, Yamaguchi-Shinozaki K, Shinozaki K 1999: Improving plant drought, salt, and freezing tolerance by gene transfer of a single stress-inducible transcription factor. Nat. Biotechnol., 17, 287–291. doi:10.1038/7036
- Kinraide TB, Wang P 2010: The surface charge density of plant cell membranes (σ): an attempt to resolve conflicting values for intrinsic σ. J. Exp. Bot., 61, 2507–2518. doi:10.1093/jxb/erq082
- Kobayashi Y, Hoekenga OA, Itoh H, Nakashima M, Saito S, Shaff JE, Maron LG, Piñeros MA, Kochian LV, Koyama H 2007: Characterization of AtALMT1 expression in aluminum-inducible malate release and its role for rhizotoxic stress tolerance in Arabidopsis. Plant Physiol., 145, 843–852. doi:10.1073/pnas.0700117104
- Kobayashi Y, Kobayashi Y, Watanabe T, Shaff JE, Ohta H, Kochian L, Wagatsuma T, Kinraide TB, Koyama H 2013: Molecular and physiological analysis of Al3+ and H+ rhizotoxicities at moderately acidic conditions. Plant Physiol., 163, 180–192. doi:10.1104/pp.113.222893
- Kobayashi Y, Sadhukhan A, Tazib T, et al. 2016: Joint genetic and network analyses identify loci associated with root growth under NaCl stress in Arabidopsis thaliana. Plant, Cell Environ., 39. 918–934. doi:10.1111/pce.12691
- Kusunoki K, Nakano Y, Tanaka K, Sakata Y, Koyama H, Kobayashi Y 2017: Transcriptomic variation among six Arabidopsis thaliana accessions identified several novel genes controlling aluminium tolerance. Plant. Cell Environ., 40, 249–263. doi:10.1111/pce.12866
- Iuchi S, Koyama H, Iuchi A, Kobayashi Y, Kitabayashi S, Kobayashi Y, Ikka T, Hirayama T, Shinozaki K, Kobayashi M 2007: Zinc finger protein STOP1 is critical for proton tolerance in Arabidopsis and coregulates a key gene in aluminum tolerance. Proc. Natl. Acad. Sci. USA, 104, 9900–9905. doi:10.1073/pnas.0700117104
- Larsen PB, Geisler MJB, Jones CA, Williams KM, Cancel JD 2005: ALS3 encodes a phloem-localized ABC transporter-like protein that is required for aluminum tolerance in Arabidopsis. Plant J., 41, 353–363. doi:10.1111/j.1365-313X.2004.02306.x
- Li Y, Huang J, Song X, Zhang Z, Jiang Y, Zhu Y, Zhao H, Ni D 2017: An RNA-Seq transcriptome analysis revealing novel insights into aluminum tolerance and accumulation in tea plant. Planta, 246, 91–103. doi:10.1007/s00425-017-2688-6
- Liu J, Piñeros MA, Kochian LV 2014: The role of aluminum sensing and signaling in plant aluminum resistance. J. Integr. Plant Biol, 56, 221–230. doi:10.1111/jipb.12162
- MAQC Consortium 2006: The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol., 24. 1151–1161. doi:10.1038/nbt1239
- Magalhaes JV, Liu J, Guimarães CT, et al. 2007: A gene in the multidrug and toxic compound extrusion (MATE) family confers aluminum tolerance in sorghum. Nat. Genet., 39. 1156–1161. doi:10.1038/ng2074
- Matsuo M, Johnson JM, Hieno A, et al. 2015: High redox responsive transcription factor1 levels result in accumulation of reactive oxygen species in Arabidopsis thaliana shoots and roots. Mol. Plant, 8. 1253–1273. doi:10.1016/j.molp.2015.03.011
- Mora-Macías J, Ojeda-Rivera JO, Gutiérrez-Alanís D, Yong-Villalobos L, Oropeza-Aburto A, Raya-González J, Jiménez-Domínguez G, Chávez-Calvillo G, Rellán-Álvarez R, Herrera-Estrella L 2017: Malate-dependent Fe accumulation is a critical checkpoint in the root developmental response to low phosphate. Proc. Natl. Acad. Sci., 114, E3563–E3572. doi:10.1073/pnas.1701952114
- Provart NJ, Gil P, Chen W, Han B, Chang H-S, Wang X, Zhu T 2003: Gene expression phenotypes of Arabidopsis associated with sensitivity to low temperatures. Plant Physiol., 132, 893–906. doi:10.1104/pp.103.021261
- Qian P, Sun R, Ali B, Gill RA, Xu L, Zhou W 2014: Effects of hydrogen sulfide on growth, antioxidative capacity, and ultrastructural changes in oilseed rape seedlings under aluminum toxicity. J. Plant Growth Regul., 33, 526–538.
- Rodriguez L, Gonzalez-Guzman M, Diaz M, et al. 2014: C2-domain abscisic acid-related proteins mediate the interaction of PYR/PYL/RCAR abscisic acid receptors with the plasma membrane and regulate abscisic acid sensitivity in arabidopsis, Plant Cell Online, 26. 4802–4820. doi:10.1105/tpc.114.129973
- Sakano K 1998: Revision of biochemical pH-Stat: involvement of alternative pathway metabolisms. Plant Cell Physiol., 39, 467–473. doi:10.1093/oxfordjournals.pcp.a029393
- Sarry JE, Kuhn L, Ducruix C, et al. 2006: The early responses of Arabidopsis thaliana cells to cadmium exposure explored by protein and metabolite profiling analyses. Proteomics, 6. 2180–2198. doi:10.1002/pmic.200500543
- Sawaki Y, Iuchi S, Kobayashi Y, et al. 2009: STOP1 regulates multiple genes that protect arabidopsis from proton and aluminum toxicities. Plant Physiol., 150. 281–294. doi:10.1104/pp.108.134700
- Shaff JE, Schultz BA, Craft EJ, Clark RT, Kochian LV 2010: GEOCHEM-EZ : a chemical speciation program with greater power and flexibility. Plant Soil, 330, 207–214.
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003: Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res., 2498–2504. doi:10.1101/gr.1239303
- Smeets K, Opdenakker K, Remans T, Van Sanden S, Van Belleghem F, Semane B, Horemans N, Guisez Y, Vangronsveld J, Cuypers A 2009: Oxidative stress-related responses at transcriptional and enzymatic levels after exposure to Cd or Cu in a multipollution context. J. Plant Physiol., 166, 1982–1992. doi:10.1016/j.jplph.2009.06.014
- Suzuki Y, Kawazu T, Koyama H 2004: RNA isolation from siliques, dry seeds, and other tissues of Arabidopsis thaliana. Biotechniques, 37, 542–544.
- Tattini M, Loreto F, Fini A, Guidi L, Brunetti C, Velikova V, Gori A, Ferrini F 2015: Isoprenoids and phenylpropanoids are part of the antioxidant defense orchestrated daily by drought-stressed Platanus × acerifolia plants during Mediterranean summers. New Phytol., 207, 613–626. doi:10.1111/nph.13380
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M 2004: MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J., 37, 914–939. doi:10.1111/j.1365-313X.2004.02016.x
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A 2003: PANTHER: a library of protein families and subfamilies indexed by function. Genome Res., 13, 2129–2141. doi:10.1101/gr.772403
- Tokizawa M, Kobayashi Y, Saito T, Kobayashi M, Iuchi S, Nomoto M, Tada Y, Yamamoto YY, Koyama H 2015: Sensitive to proton rhizotoxicity1, calmodulin binding transcription activator2, and other transcription factors are involved in aluminum-activated malate transporter1 expression. Plant Physiol., 167, 991–1003. doi:10.1104/pp.114.256552
- Wu SJ, Ding L, Zhu JK 1996: SOS1, a genetic locus essential for salt tolerance and potassium acquisition. Plant Cell, 8, 617–627. doi:10.1105/tpc.8.4.617
- Xu JM, Fan W, Jin JF, Lou HQ, Chen WW, Yang JL, Zheng SJ 2017: Transcriptome analysis of al-induced genes in buckwheat (Fagopyrum esculentum Moench) root apex: new insight into al toxicity and resistance mechanisms in an al accumulating species. Front. Plant Sci., 8, 1–14. doi:10.3389/fpls.2017.01141
- Yamamoto YY, Yoshioka Y, Hyakumachi M, Maruyama K, Yamaguchi-Shinozaki K, Tokizawa M, Koyama H 2011: Prediction of transcriptional regulatory elements for plant hormone responses based on microarray data. BMC Plant Biol., 11, 39. doi:10.1186/1471-2229-11-39
- Yokosho K, Yamaji N, Ma JF 2014: Global transcriptome analysis of al-induced genes in an al-accumulating species, common buckwheat (Fagopyrum esculentum Moench). Plant Cell Physiol., 55, 2077–2091. doi:10.1093/pcp/pcu135
- You J, Liu X, Zhang B, Xie Z, Hou Z, Yang Z 2015: Seasonal changes in soil acidity and related properties in ginseng artificial bed soils under a plastic shade. J. Ginseng Res., 39, 81–88. doi:10.1016/j.jgr.2014.08.002
- Zhao C-R, Ikka T, Sawaki Y, Kobayashi Y, Suzuki Y, Hibino T, Sato S, Sakurai N, Shibata D, Koyama H 2009: Comparative transcriptomic characterization of aluminum, sodium chloride, cadmium and copper rhizotoxicities in Arabidopsis thaliana. BMC Plant Biol., 9, 32. doi:10.1186/1471-2229-9-32