
ABSTRACT
Flooded paddy soils are global sources of the greenhouse gas methane. Net methane fluxes from paddy fields reflect the sum of methane generation in the anoxic zone and methane consumption in the oxic zone, which are driven by methanogenic archaea and methanotrophic bacteria, respectively. Furthermore, methanogenic archaea produce methane utilizing hydrogen or acetate generated by hydrogen-producing bacteria or acetogenic bacteria, respectively. Therefore, methane emissions are regulated and orchestrated by hydrogen production, acetogenesis, methanogenesis, and methane oxidation. However, a comprehensive understanding of the microbial consortia that regulate methane emissions has yet to be achieved owing to the lack of simultaneous assessments of microbial drivers involved in these four processes, especially the production of hydrogen and acetate, which are primary substrates for methanogenesis. In this study, the transcriptional profiles of genes encoding the enzymes that catalyze each process were comprehensively investigated by shotgun RNA sequencing analysis (metatranscriptomics). Seasonal and spatial transitions in soil redox potentials and the transcriptional activities of methanogens indicated active methane metabolism in paddy soils 5 weeks after waterlogging, when the soil redox conditions of the surface (0–1 cm depth; S layer) and subsurface (5–7 cm depth; D layer) were the most divergent. Deep metatranscriptomics focusing on such soils suggested that: (1) in the anoxic D layer, rather than in the oxic S layer, Deltaproteobacteria, Acidobacteria, and Planctomycetes actively generate hydrogen; (2) Deltaproteobacteria, Betaproteobacteria, Acidobacteria, and Alphaproteobacteria generate acetate; (3) utilizing these products as substrates for methanogenesis, the archaea Methanocella, Methanoregula, and Methanosaeta actively produce methane; (4) concurrently, in the oxic S layer, methanotrophs related to Methylocystis and Methylogaea oxidize methane. The present study represents the first comprehensive report of the community structure and dynamics of the microbial consortia underpinning the methane emissions from paddy fields.
1. Introduction
Flooded paddy soils are major anthropogenic sources of methane, an important greenhouse gas (Yagi and Minami Citation1990). Global methane emissions from flooded paddy soils are estimated to be 33–40 Tg per year (IPCC Citation2013). Unlike other upland agricultural soils, paddy soils exhibit temporal anaerobic conditions owing to waterlogging and leading to active methanogenesis by strict anaerobic archaea (Itoh et al. Citation2013; Fazli et al. Citation2013). Therefore, the microbial processes involved in methane metabolism in paddy soils are of great interest to researchers, not only for a better understanding of anaerobic microbiology but also to develop techniques for successful, sustainable rice production with a lower environmental burden.
After waterlogging at the beginning of rice-growing season, a vertical oxygen concentration gradient is generated in soils, leading to the development of an anoxic layer at the subsurface, despite the maintenance of an oxic layer at the soil surface (Liesack et al. Citation2000). Such anoxic layer soils are thought to be hotspots of active methanogenesis by strict anaerobic archaea (Liesack et al. Citation2000). Methane is biologically converted from hydrogen/carbon dioxide or acetate by diverse methanogenic archaea (Falz et al. Citation1999; Conrad Citation2007). This means that the active methanogenesis of the anoxic zone in paddy soils is strictly regulated by hydrogen production and acetogenesis under anaerobic conditions. However, some portion of the methane generated by these methanogens is eliminated in the oxic layer at the soil surface under waterlogged conditions. This methane is oxidized to carbon dioxide via C1 metabolism, which is driven by methanotrophic bacteria (Hanson and Hanson Citation1996). Thus, net methane emissions from paddy fields reflect the sum of methane generation and elimination, which rely on three sequential processes: production of substrates (hydrogen and acetate) for methanogenesis, methane generation, and methane oxidation.
However, a comprehensive understanding of the microbial communities driving methane metabolism via these sequential processes has not yet been achieved. There are innumerable studies focusing on methanogenesis and methane oxidation and analyzing the microbial diversity of methanogenic archaea and methanotrophic bacteria in paddy soils in Asia (Lu et al. Citation2000; Hou et al. Citation2000; Kumarswamy et al. Citation2001; Yang and Chang Citation2001; Singh and Dubey Citation2012; Sugano et al. Citation2005; Yan et al. Citation2005; Peng et al. Citation2008; Supparattanapan et al. Citation2009; Watanabe et al. Citation2010) and in Europe (Weber et al. Citation2001; Conrad and Klose Citation2006). In previous studies, the families Methanosarcinaceae and Methanosaetaceae and orders Methanobacteriales, Methanomicrobiales, and Methanocellales have been frequently detected as methanogens, while the families Methylococcaceae, belonging to the Gammaproteobacteria, and Methylocystaceae, belonging to the Alphaproteobacteria, are common methanotrophs (Breidebach and Conrad Citation2014; Eller et al. Citation2001). In addition, there are only a few studies investigating the microbial contributors that produce the primary and essential substrates for methanogenesis, i.e., hydrogen and acetate, in paddy soils (Baba et al. Citation2014; Baba et al. Citation2016). Recently, Jewell et al. used shotgun RNA sequencing analysis (metatranscriptomics) to assess the microbial consortia driving the carbon cycle in an alluvial aquifer environment and revealed a complete picture of the microbial communities involved in methane metabolism, including acetogenesis (Jewell et al. Citation2017). Therefore, simultaneous assessments of the microbial drivers involved in these sequential processes – hydrogen production, acetogenesis, methanogenesis, and methane oxidation – would greatly improve our understanding of methane metabolism in paddy soils.
Shotgun RNA sequencing analysis (metatranscriptomics) using a high-throughput sequencer, which randomly sequences the RNAs present in environmental samples, is an effective method for investigating the community structure and functions of active microorganisms based on the sequence information of transcribed ribosomal and messenger RNAs (Frias-Lopez et al. Citation2008; Urich et al. Citation2008). Recently, we used metatranscriptomics of paddy soil samples to investigate the diversity of active microbial drivers of reductive nitrogen transformation (RNT), including nitrogen fixation, denitrification, and dissimilatory nitrate reduction to ammonia, in paddy soils (Masuda et al. Citation2017). Our results showed that such reactions are mainly driven by Deltaproteobacteria, which are predominant but were previously overlooked as RNT players in paddy soils because of polymerase chain reaction (PCR) biases caused by mismatches in primer sequences and its high GC content (Masuda et al. Citation2017). Therefore, the application of metatranscriptomics should enable us to obtain a comprehensive and less biased profile of the microbial consortia driving methane metabolism.
In order to reveal the community structure of the active microbial consortia driving methane metabolism in paddy soils, we analyzed the microbes in paddy fields with metatranscriptomics. Seasonal and spatial investigations of soil redox potentials and the transcriptional activities of methanogens and methanotrophs identified key moments and hotspots of methane metabolism in the paddy fields. Focusing on such soils, we analyzed the transcriptional profiles of genes involved in hydrogen production, acetogenesis, methanogenesis, and methane oxidation and clarified the key microbial drivers of each process. This is the first study reporting the comprehensive microbial consortia of methane metabolism in paddy soils, including methanogens and methanotrophs, as well as substrate producers for methanogens.
2. Materials and methods
2.1. Study field and soil sampling
The paddy field used in this study is located at Niigata Agricultural Research Institute (Nagaoka, Niigata, Japan: 37°26ˊN, 138°52ˊE). The soil type, cropping history, and soil physicochemical properties of the field were described previously (Itoh et al. Citation2013). The field management in 2012 was basically the same as that performed during the 2009 rice cultivation season (Itoh et al. Citation2013). Fifteen transparent acrylic cores (inner diameter × depth: 10 × 15 cm) were randomly placed in the plough layer (0–10 cm) at the furrow of the paddy field just after puddling on 7 May 2012. Soil samples were collected at five time points: 7 May (W0, just after puddling), 23 May (W2, 2 weeks after waterlogging), 18 June (W6, 6 weeks after waterlogging), 28 August (I, intermittent drainage), and 1 October (CD, after complete drainage). At each sampling event, three cores were collected after removing the surface water, if any, and were immediately frozen in liquid nitrogen, transported to the laboratory with dry ice, and stored at −80°C until use. For the soil cores collected on 23 May, 18 June, 28 August, and 1 October, surface and subsurface layers from depths of 0–1 cm (S layer) and 5–7 cm (D layer), respectively, were cut from each frozen core with a sterilized knife and subjected to the following analyses. For the soil cores collected on 7 May, portions of bulk soil from a depth of 0–10 cm were used for analysis because of its homogeneity just after paddling. Each of the soil samples was represented by three independent replicates.
2.2. Measurement of redox potential and Fe2+ content
In accordance with each sampling event, the redox potential (Eh) at a depth of 5 cm in the plough layer was measured as described previously (Itoh et al. Citation2013). To estimate the redox potentials of the two layers obtained from each soil core as described above, the Fe2+ content of each layer was determined as described previously (Ishii et al. Citation2009) immediately after cutting the layer out of the core.
2.3. Preparation of soil RNA
Nine soil samples were subjected to RNA extraction in triplicate (27 samples in total) and used for metatranscriptomics in this study. Soil RNA was extracted from 2 g (wet weight) of each soil sample using the PowerSoil Total RNA Isolation Kit (Mo Bio Laboratories, Carlsbad, CA, USA). Remaining DNA was removed using the TURBO DNA-free Kit (Applied Biosystems, Foster City, CA, USA) and RNA Clean & Concentrator (Zymo Research, Irvine, CA, USA) as described previously (Masuda et al. Citation2017). The integrity and quantity of the prepared RNAs were determined using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) with Qubit RNA Assay Kits (Invitrogen).
2.4. Shotgun RNA sequencing
Complementary DNA (cDNA) libraries were constructed for metatranscriptomic sequencing using the NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs Inc., Ipswich, MA, USA). Libraries were subjected to paired-end sequencing on an Illumina MiSeq sequencer (Illumina, San Diego, CA, USA) using a MiSeq Reagent Kit v2 (Illumina) for 500 cycles (2 × 250 bp) according to the manufacturer’s instructions. For seasonal and spatial surveys of transcriptional activities of genes involved in methanogenesis and methane oxidation, all 27 cDNA libraries (9 soils × 3 replicates) were sequenced separately in three runs on the sequencer (pilot metatranscriptomics). For deeper survey of transcriptional profiles of the microbial consortia driving methane metabolism, six cDNA libraries (S and D layer soils of W6 with three replicates each) were sequenced separately in two runs (Masuda et al. Citation2017). Original sequence data for this deep metatranscriptomics analysis was derived from our previous work (Masuda et al. Citation2017) and analyzed for the microbial consortia involved in methane metabolism in this study. The total number of reads obtained by pilot and deep metatranscriptomics ranged from 9,578 to 2,468,737 and 5,170,274 to 7,240,814, respectively (Table S1).
2.5. Informatics and statistics
Resulting paired-end sequences were joined together with >30 bp overlapping sequences containing <10% mismatches and then converted from fastq to the fasta format with a Q-score cutoff of >30 using PRINSEQ (Schmieder and Edwards Citation2011). Ribosomal RNA sequences (rRNAseq) in the metatranscriptomics data were identified using a BLAT search against the M5RNA database (E value of <10–5, alignment length of >50 bp) on the MG-RAST server ver. 3.6 (Meyer et al. Citation2008). Messenger RNA sequences (mRNAseq) were readily determined by a BLAT search against the M5nr database (Wilke et al. Citation2012) (E value of <10–5, alignment length of >30 aa) on the MG-RAST server ver. 3.6 (Meyer et al. Citation2008). From all of the assigned mRNAseq, we collected highly expressed gene sequences of key enzymes according to KEGG compound database (Hattori et al. Citation2010) involved in hydrogen production [[FeFe] hydrogenase, [NiFe] hydrogenase, [Fe] hydrogenase], acetogenesis [acetyl-CoA synthase (ACS), acetate kinase (AckA)], methanogenesis [methyl coenzyme M reductase (MCR)], and methane oxidation [particulate methane monooxygenase (PMO), and soluble methane monooxygenase (SMO)]. Obtained sequences were compared to the nr database in December 2015 by BLASTX for more precise assignment (E value of <10–5, alignment length of >30 aa).
2.6. Nucleotide sequence accession number
Sequence data derived from the pilot metatranscriptomics analyses in this study were deposited in the public database of the MG-RAST sever (http://metagenomics.anl.gov/, Meyer et al. Citation2008). Deposit IDs are summarized in Table S1.
3. Results and discussion
3.1. Seasonal and spatial profiles of soil redox properties and the potentials of methane metabolism
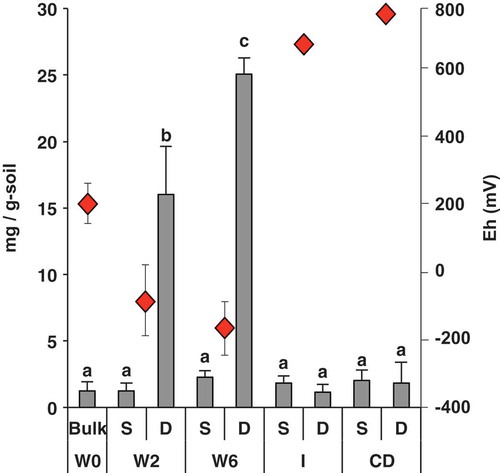
As described in our previous studies (Itoh et al. Citation2013; Masuda et al. Citation2017), soil Eh reached at its lowest value 6 weeks after waterlogging, indicating that an anoxic layer developed and was maintained in the plough layer soils until 6 weeks after waterlogging (W6). Around 6 weeks after waterlogging, abundant methane flux was observed in our previous survey in 2009 (Itoh et al. Citation2013); therefore, it was plausible that methanogenesis occurred on 18 June 2012 (W6) in this survey. Next, we investigated the spatial profiles of the soil redox potentials based on Fe2+ content, which is an indicator of soil reduction levels (Kimura Citation2000; Itoh et al. Citation2013). Although the Fe2+ contents of the S layers of all soil samples were the same, the Fe2+ contents of the D layers of W2 and W6 were significantly higher than those of the corresponding S layers or those of any other soils, including W0, I, and CD (). These significant differences between the S and D layers of W2 and W6 indicated the development of both oxic and anoxic zones in the surface and subsurface soils, respectively, where aerobic and anaerobic reactions could progress.
Figure 1. Seasonal changes in soil redox potential (Eh) and concentrations of Fe2+ in paddy soils from depths of 0–1 cm (S layer) and 5–7 cm (D layer) at different time points. Sampling periods occurred at five stages: waterlogging (W0, just after ploughing; W2, 2 weeks after waterlogging; W6, 6 weeks after waterlogging), intermittent drainage (I), and after complete drainage (CD). Data are shown as means with the standard error of three replicates. Different letters indicate significant differences (ANOVA with Tukey’s test, p < 0.05). Eh data were shown in our previous study (Masuda et al. Citation2017).
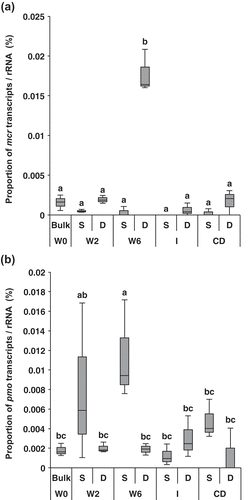
Methanogenesis and methane oxidation are catalyzed by MCR and methane monooxygenase (PMO and SMO), respectively. The transcription of genes encoding these enzymes was investigated through the pilot metatranscriptomics analysis in order to estimate methane production/consumption activities. mcr transcripts were more frequently detected in the D layer soils of W6 than in any other soil samples (p < 0.05, ); in contrast, the most abundant pmo transcripts were detected in the S layer soil of W6 (p < 0.05, ). Transcripts of smo were rarely detected in our metatranscriptomic analysis. These results suggest the existence of a key moment and hotspot of methanogenesis and methane oxidation in the D and S layers of W6, respectively. However, transcripts relevant to the production of substrates for methanogenesis, i.e., hydrogen and acetate, were scarce in this sample compared with those involved in methanogenesis and methane oxidation, resulting in less information regarding hydrogen/acetate producers.
Figure 2. Relative abundances of transcripts relevant to methane generation (mcr) (a) and methane oxidation (pmo) (b) normalized by rRNA abundances. Sampling periods occurred at five stages: waterlogging (W0, just after ploughing; W2, 2 weeks after waterlogging; W6, 6 weeks after waterlogging), intermittent drainage (I), and after complete drainage (CD). Each box plot shows median (black line), first quartile-third quartile percentiles (box range), n = 3. Different letters indicate significant differences (ANOVA with Tukey’s test, p < 0.05).
Based on these seasonal and spatial profiles of soil redox potentials, the relative abundance of transcripts of the genes relevant to methane production/consumption, and the limited detection of transcripts relevant to the production of substrates for methanogenesis, deep metatranscriptomics analysis of the D and S layers of the W6 sample was performed to identify the microbial communities involved in methanogenesis, methane oxidation, and the production of substrates for methanogenesis.
3.2. Diversity of methanogens
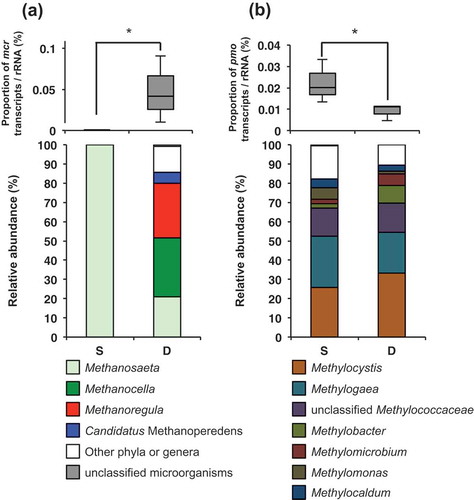
In accordance with the results of the pilot metatranscriptomics analysis as described earlier, deep metatranscriptomics analysis showed that the transcriptional activity of mcr in W6 was significantly higher in the D layer than in the S layer (p < 0.05, , upper panel). Most mcr transcripts in the D layer obtained from deep metatranscriptomics analysis were derived from the genera Methanocella (31.0 ± 0.9%), Methanoregula (28.5 ± 4.0%), or Methanosaeta (21.0 ± 3.7%), belonging to the class Methanomicrobia (, lower panel). The community structure of archaea based on rRNA sequences obtained from metatranscriptomics also showed that Methanosaeta (16.5 ± 1.7%), Methanocella (13.8 ± 1.6%), and Methanoregula (7.0 ± 1.5%) were dominant members of the active archaeal community in this soil (Fig. S1). Previous studies based on polymorphism analyses of the archaeal 16S rRNA gene and mcr revealed that the genera Methanosarcina, Methanosaeta, and Methanocella are general methanogens in paddy soils (Fazli et al. Citation2013; Bao et al. Citation2016). Methanocella, a hydrogenotrophic methanogen, is known to utilize hydrogen and carbon dioxide as substrates in methanogenesis, while Methanosaeta utilize acetate and Methanosarcina utilize both hydrogen/carbon dioxide and acetate (Galagan et al. Citation2002; Sakai et al. Citation2007; Kamagata and Mikami Citation1991). Although the 16S rRNA genes of Methanoregula have also been detected in paddy soils (Nguyen et al. Citation2015; Liu et al. Citation2017), their mcr genes are rarely detected in these soils. Although two strains, Methanoregula boonei and Methanoregula formicica, have been isolated from acidic peat bog and methanogenic sludge, respectively (Bräuer et al. Citation2006; Yashiro et al. Citation2011), the genome sequences of these strains were completed rather recently. Methanoregula is known to utilize hydrogen and carbon dioxide for methane generation (Bräuer et al. Citation2011). Thus, both hydrogenotrophic (Methanocella and Methanoregula) and acetoclastic (Methanosaeta) methanogens may be active in Niigata paddy soils. In addition to these three genera, Methanosarcina is another dominant methanogen based on the rRNA genes sequences belonging to the archaeal community in this soil (Fig. S1). However, mcr transcripts from Methanosarcina were only rarely detected ( lower panel), suggesting that the contribution of Methanosarcina to methane generation might be small in this paddy soil. Methanosarcina is favored over Methanosaeta by high acetate concentrations (Min and Zinder Citation1989; Großkopf et al. Citation1998) and ideal for high hydrogen concentrations compared with Methanocellales methanogens (Lu et al. Citation2005), indicating that the acetate and hydrogen concentration in this paddy soil might be too low for Methanosarcina to generate methane. The differences between the methanogenic communities indicated by rRNA gene sequences and mcr transcripts described above suggest that not all methanogenic archaea perform methanogenesis actively. Some microbes are known to maintain consistent levels of rRNA, even under starvation conditions (Lärdh et al. Citation1992; Fukui et al. Citation1996). Therefore, the methanogenic communities derived from the mcr transcript-based analysis may better reflect the contribution to methanogenesis in the soil.
Figure 3. Relative abundances (upper panels) of transcripts relevant to methane generation (mcr) (a) and methane oxidation (pmo) (b) normalized by rRNA abundances, and the taxonomic composition of the assigned archaeal genera in the W6 period (lower panels). Each box plot shows median (black line), first quartile-third quartile percentiles (box range), n = 3 (upper panels). Data are shown as means of three replicates (lower panels). Asterisks indicate significant differences (Mann–Whitney U-test; * p < 0.05).
3.3. Diversity of methanotrophs
Consistent with the results of the pilot metatranscriptomics analysis described earlier, deep metatranscriptomics analysis revealed that the transcriptional activity of pmo in the W6 sample was significantly higher in the S layer than in the D layer (p < 0.05, , upper panel). Transcripts of smo were also rarely detected in our deep metatranscriptomics. Most of the pmo transcripts obtained from deep metatranscriptomics analysis were derived from the genera Methylogaea (26.8 ± 14.3%), belonging to the class Gammaproteobacteria, and Methylocystis (25.7 ± 0.7%), belonging to the class Alphaproteobacteria (, lower panel). In a previous PCR-based study, pmo transcripts derived from Methylocystis, Methylococcus, and Methylomonas, belonging to the Alpha- and Gammaproteobacteria, were frequently detected in paddy soils (Liu et al. Citation2017), while the detection of Methylogaea-derived pmo transcripts in paddy fields has rarely been reported. This oversight is partially due to the fact that the genome sequence of Methylogaea oryzae, isolated from paddy fields (Geymonat et al. Citation2011), was finished in 2015. Methanotrophs have long been studied, and 14 genera in the class Gammaproteobacteria and five genera in the class Alphaproteobacteria are known (Islam et al. Citation2015). However, in this study, most pmo transcripts were derived from a few genera, in particular Methylocystis and Methylogaea (, lower panel), suggesting that these two taxa are capable of adapting to the environmental conditions in paddy soils.
3.4. Diversity of hydrogen-producing bacteria
Organic matter in paddy soils is converted to the substrates necessary for the production of methane, i.e., hydrogen and acetate, by fermenting bacteria (Takai Citation1970; i.e., acetogenesis, Equation (1)). We thus attempted to identify the microbial communities that produce these substrates for methane generation using metatranscriptomics analysis.
Hydrogenases are categorized based on metal atoms at their active site into three types: [FeFe] hydrogenase, [NiFe] hydrogenase, and [Fe] hydrogenase (Vignais and Billoud Citation2007). Although any transcripts of [Fe] hydrogenase gene were not detected in our metatranscriptomics, those of [FeFe] and [NiFe] hydrogenase genes were detected (). The relative abundances of the transcripts of [FeFe] and [NiFe] hydrogenase genes were significantly higher in D layer soils than in S layer soils (p < 0.05, , upper panel). Such uneven distributions of such transcriptional activities in anoxic and oxic soil layers may be expected because hydrogen generation is an anaerobic reaction.
Figure 4. Relative abundances (upper panels) of gene transcripts relevant to hydrogenase ([FeFe] hydrogenase and [NiFe] hydrogenase) (a) and acetogenesis (acetyl-CoA decarbonylase/synthase (ACD/ACSS) and acetate kinase (AckA)) (b) normalized by rRNA abundances, and the taxonomic composition of the assigned bacterial phyla in the W6 period (lower panels). The most dominant bacterial phylum, Proteobacteria, was further classified and shown at the class level as Alpha-, Beta-, Gamma-, and Deltaproteobacteria. Each box plot shows median (black line), first quartile-third quartile percentiles (box range), n = 3 (upper panels). Data are shown as means of three replicates (lower panels). Asterisks indicate statistically significant differences (Mann–Whitney U-test; *p < 0.05).
![Figure 4. Relative abundances (upper panels) of gene transcripts relevant to hydrogenase ([FeFe] hydrogenase and [NiFe] hydrogenase) (a) and acetogenesis (acetyl-CoA decarbonylase/synthase (ACD/ACSS) and acetate kinase (AckA)) (b) normalized by rRNA abundances, and the taxonomic composition of the assigned bacterial phyla in the W6 period (lower panels). The most dominant bacterial phylum, Proteobacteria, was further classified and shown at the class level as Alpha-, Beta-, Gamma-, and Deltaproteobacteria. Each box plot shows median (black line), first quartile-third quartile percentiles (box range), n = 3 (upper panels). Data are shown as means of three replicates (lower panels). Asterisks indicate statistically significant differences (Mann–Whitney U-test; *p < 0.05).](/cms/asset/5e221524-b729-465f-919b-3c031506efe7/tssp_a_1457409_f0004_oc.jpg)
In the present study, most transcripts of [FeFe] hydrogenase gene were derived from bacteria belonging to the phylum Planctomycetes (23.8 ± 11.4%), Acidobacteria (17.3 ± 2.1%), and the class Deltaproteobacteria (8.8 ± 4.9%) (, lower panel), specifically the genera Thermogutta, Holophaga, and Desulfovibrio (Fig. S2–S6). It is highly possible that these bacteria actively produce hydrogen in paddy soils. This is in contrast to the results of previous PCR-based studies, in which bacteria belonging to the phyla Bacteroidetes, Chloroflexi, Firmicutes, and Proteobacteria, specifically the class Clostridia, and the genera Dehalococcoides, Desulfovibrio, and Pelobacter were detected as hydrogen-generating bacteria in Japanese paddy soils (Baba et al. Citation2014; Citation2016). This difference in diversity of [FeFe] hydrogenase gene derived from hydrogen producers might be due to the difference in soil samples and analytical method used in both studies.
Basically, [NiFe] hydrogenase catalyzes hydrogen oxidation; however, Geobacter sulfurreducens harbors two distinct types of [NiFe] hydrogenases (Hya and Hyb); Hya-type [NiFe] hydrogenases are considered to catalyze hydrogen production, whereas Hyb-type hydrogenases catalyzes hydrogen oxidation (Coppi et al. Citation2004; Coppi Citation2005). In this study, most transcripts of [NiFe] hydrogenase gene were derived from bacteria belonging to the classes Deltaproteobacteria (39.5 ± 1.3%) and Betaproteobacteria (8.3 ± 2.0%) and the phylum Acidobacteria (11.6 ± 1.5%) (, lower panel), specifically the genera Anaeromyxobacter, Geobacter, Sulfuricella, and Candidatus Solibacter (Fig. S2–S5). Transcripts of [NiFe] hydrogenase genes derived from these genera could not be identified as Hya or Hyb type because their sequences showed low similarity against [NiFe] hydrogenase genes of G. sulfurreducens. However, it is possible that Anaeromyxobacter catalyze hydrogen production because Anaeromyxobacter sp. Fw109-5, frequently detected in our study, harbors two pairs of [NiFe] hydrogenase on their genome likewise G. sulfurreducens (NCBI Protein ID: ABS24740, ABS27704, NP_951185, NP_951839). In addition, hydrogen is also by-product of nitrogen fixation catalyzed by nitrogenase (Hoffman et al. Citation2014; Equation 2). Our previous metatranscriptomics study detected many transcripts of nitrogenase gene derived from Anaeromyxobacter and Geobacter in waterlogged paddy soils (Masuda et al. Citation2017). Therefore, Anaeromyxobacter and Geobacter were suggested to be key microbes producing hydrogen via two pathways: (1) organic acid fermentation (Equation 1) involved in Hya [NiFe] hydrogenase and (2) nitrogen fixation (Equation 2) catalyzed by nitrogenase.
Our previous work based on a clone library analysis of prokaryotic 16S rRNA genes in the same paddy fields detected increases in Syntrophobacterales and Syntrophorhabdaceae after waterlogging (Itoh et al. Citation2013), two groups known to produce hydrogen via symbioses with methanogens, as indicated by in vitro experiments (Kato and Watanabe Citation2010; Qiu et al. Citation2008). However, the metatranscriptomics analysis in this study detected few gene transcripts of [FeFe] hydrogenase and [NiFe] hydrogenase derived from these groups in the paddy field, suggesting that they contribute less to the hydrogen production needed for methanogenesis than the other anaerobic bacteria, i.e., Thermogutta, Desulfovibrio, Anaeromyxobacter, and Geobacter, which were frequently detected in this study.
3.5. Diversity of acetogens
The other substrate of methane in paddy soils is acetate generated by microorganisms through various pathways. Genes encoding enzymes relevant to acetate generation (acetogenesis) are as follows: acetyl-CoA decarbonylase/synthase (ACD/ACSS) and AckA (Jewell et al. Citation2017; Schäfer et al. Citation1993; Shieh and Whitman Citation1987). Their relative abundances were significantly higher in D layer soils than in S layer soils (p < 0.05, , upper panel). This appears to be reasonable, as acetogenesis is active in D layer soils, where methanogenesis is also active. Gene transcripts derived from the classes Alpha-, Beta-, and Deltaproteobacteria and the phyla Acidobacteria (, lower panel), specifically the genera Bradyrhizobium, Burkholderia, Anaeromyxobacter, and Candidatus Solibacter, were frequently detected (Fig. S2–S5). It is well known that most acetogens are members of the phylum Firmicutes, though acetogens are also found among the Spirochaetes, Deltaproteobacteria, and Acidobacteria (Drake et al. Citation2006). In a previous study using culture-based methods, a few classifications of anaerobic acetogens, e.g., the phylum Firmicutes and genera Desulfovibrio and Pelobacter, were isolated from paddy soil (Rosencrantz et al. Citation1999; Kato et al. Citation2015). Acetogenesis was previously detected in paddy soils by high-pressure liquid chromatography (Krylova et al. Citation1997), and only a few studies have analyzed acetogenic bacterial communities in paddy soils using metagenomics/metatranscriptomics or PCR-based methods for amplifying genes. However, to the best of our knowledge, this is the first report to determine the complete acetogenic bacterial community in paddy soils.
3.6. Microbial consortia driving methane metabolism
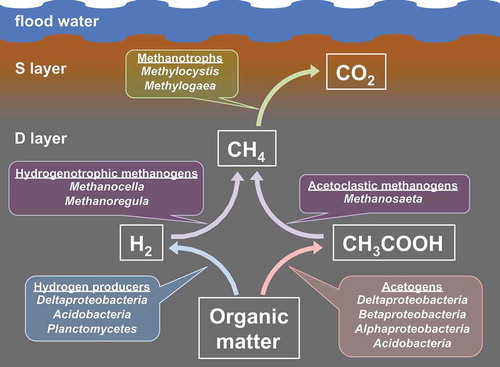
Fe2+ content of W6 samples was significantly higher in D layer than S layer, indicating that anoxic condition developed in D layer (). At the same time, our deep metatranscriptomics analysis determined that the D layer was responsible for active methanogenesis, hydrogen production, and acetogenesis, and the S layer was responsible for methane oxidation. It is reasonable that both methanogenesis and the production of substrates for methane (hydrogen and acetate) were active in the same soil locations.
Moreover, our deep metatranscriptomics analysis is the first to comprehensively clarify the microbial consortia relevant to all of the main processes in the methane cycle (i.e., hydrogen production, acetogenesis, methanogenesis, and methane oxidation) in paddy soils. Specifically, we found that Methanosaeta, Methanocella, and Methanoregula were predominant as methanogens; Methylocystis and Methylogaea were predominant as methanotrophs; Deltaproteobacteria, Acidobacteria, and Planctomycetes were predominant hydrogen producers; and Alpha-, Beta-, Delta-proteobacteria, and Acidobacteria were predominant acetogens in paddy soils ().
Figure 5. Overview of methane metabolism in paddy soils, as suggested by the results of this study.
Hydrogen and acetate are pivotal substrates/nutrients not only for methanogenic archaea but also for other facultative and obligate anaerobes; these microbes drive anaerobic processes actively occurring in paddy soils such as denitrification and metal/sulfate reductions (Hori et al. Citation2010; Itoh et al. Citation2013). Therefore, diverse bacteria converting organic matter to hydrogen and acetate may support the whole biogeochemical processes occurring in paddy soils under anoxic conditions. As a means of competing with other consumers for substrates, methanogens develop symbiotic relations with some bacteria. Hydrogen-mediated syntrophy is widely observed in anoxic environments; for instance, Syntrophobacterales and Syntrophorhabdaceae of Deltaproteobacteria with hydrogenotrophic methanogens (Kato and Watanabe Citation2010; Qiu et al. Citation2008). In addition, electric syntrophy, which involves direct interspecies electron transfer via conductive mineral particles, has also been reported between acetoclastic methanogens and Geobacter of Deltaproteobacteria (Kouzuma et al. Citation2015; Holmes et al. Citation2017). It is possible that methanogens developed symbiotic relations with some genera, such as Geobacter and Anaeromyxobacter, of Deltaproteobacteria detected in this study, for competitive success in the presence of other substrate consumers.
Our metatranscriptomics analysis frequently detected transcripts of Anaeromyxobacter genes involved in hydrogen production and acetogenesis (Fig. S2), suggesting that predominant Anaeromyxobacter microbes may strongly support methanogenesis via substrate production and provision to methanogens. Anaeromyxobacter is an obligate anaerobe and iron reducer common in paddy soils (Sanford et al. Citation2002; Hori et al. Citation2010). Remarkably, recent metagenomics and metatranscriptomics studies have indicated that Anaeromyxobacter plays a pivotal role in the nitrogen cycle in terrestrial environments, especially RNT such as nitrous oxide reduction to nitrogen gas, dissimilatory nitrate reduction to ammonium, and nitrogen fixation (Orellana et al. Citation2014; Masuda et al. Citation2017). The present metatranscriptomics study provides novel and strong evidence that Anaeromyxobacter, predominating in paddy soils, plays a role in not only iron reduction and the nitrogen cycle but also the carbon cycle in the paddy soil environment. Additional metatranscriptomic analyses in different soil environments and co-culture experiments of Anaeromyxobacter with methanogens are needed to verify this previously unknown ecological function of Anaeromyxobacter in terrestrial environments.
Table_Sup2.pdf
Download PDF (47.8 KB)Table_Sup1.pdf
Download PDF (33.2 KB)SupFig5.pdf
Download PDF (83 KB)SupFig5.pdf
Download PDF (82.7 KB)SupFig4.pdf
Download PDF (85.1 KB)SupFig3.pdf
Download PDF (85.4 KB)SupFig2_.pdf
Download PDF (85 KB)SupFig1.pdf
Download PDF (72.1 KB)Acknowledgments
We thank the technical staff of Niigata Agricultural Research Institute for their assistance with field management. We also thank Shigeto Otsuka, Kazuo Isobe and Haoyang Shen of the University of Tokyo for their helpful discussion. This study was supported by JSPS KAKENHI Grant Numbers JP22248038, JP25252013, JP15K14675, and JP17H01464, Japan.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Baba R, Asakawa S, Watanabe T 2016: H2-producing bacterial community during rice straw decomposition in paddy field soil: estimation by an analysis of [FeFe]-hydrogenase gene transcripts. Microbes Environ., 31, 226–233. doi:10.1264/jsme2.ME16036
- Baba R, Kimura M, Asakawa S, Watanabe T 2014: Analysis of [FeFe]-hydrogenase genes for the elucidation of a hydrogen-producing bacterial community in paddy field soil. FEMS Microbiol. Lett., 350, 249–256. doi:10.1111/fml.2014.350.issue-2
- Bao Q, Huang Y, Wang F, Nie S, Nicol GW, Yao H, Ding L 2016: Effect of nitrogen fertilizer and/or rice straw amendment on methanogenic archaeal communities and methane production from a rice paddy soil. Appl. Microbiol. Biotechnol., 100, 5989–5998. doi:10.1007/s00253-016-7377-z
- Bräuer SL, Cadillo-Quiroz H, Ward RJ, Yavitt JB, Zinder SH 2011: Methanoregula boonei gen. nov., sp. nov., an acidiphilic methanogen isolated from an acidic peat bog. Int. J. Syst. Evol. Microbiol., 261, 45–52. doi:10.1099/ijs.0.021782-0
- Bräuer SL, Cadillo-Quiroz H, Yashiro E, Yavitt JB, Zinder SH 2006: Isolation of a novel acidiphilic methanogen from an acidic peat bog. Nature, 442, 192–194. doi:10.1038/nature04810
- Breidebach B, Conrad R 2014: Seasonal dynamics of bacterial and archaeal methanogenic communities in flooded rice fields and effect of drainage. Front. Microbiol., 5, 752.
- Conrad R 2007: Microbial ecology of methanogens and methanotrophs. Adv. Agron., 96, 1–63.
- Conrad R, Klose M 2006: Dynamics of the methanogenic archaeal community in anoxic rice soil upon addition of straw. Eur. J. Soil Sci., 57, 476–484. doi:10.1111/j.1365-2389.2006.00791.x
- Coppi MV 2005: The hydrogenases of Geobacter sulfurreducens: a comparative genomic perspective. Microbiology, 151, 1239–1254. doi:10.1099/mic.0.27535-0
- Coppi MV, O’Neil RA, Lovley DR 2004: Identification of an uptake hydrogenase required for hydrogen-dependent reduction of Fe (III) and other electron acceptors by Geobacter sulfurreducens. J. Bacteriol, 186, 3022–3028. doi:10.1128/JB.186.10.3022-3028.2004
- Drake HL, Küsel K, Matthies C 2006: Acetogenic Prokaryotes. In The Prokaryotes, Eds. Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E, 3rd ed., Vol. 2, pp. 354–420. Ecophysiology and Biochemistry Springer SBM, New York.
- Eller G, Stubner S, Frenzel P 2001: Group-specific 16S rRNA targeted probes for the detection of type I and type II methanotrophs by fluorescence in situ hybridisation. FEMS Microbiol. Lett., 198, 91–97. doi:10.1111/j.1574-6968.2001.tb10624.x
- Falz KZ, Holliger C, Großkopf R, Liesack W, Nozhevnikova AN, Müller B, Wehrli B, Hahn D 1999: Vertical distribution of methanogens in the anoxic sediment of Rotsee (Switzerland). Appl. Environ. Microbiol., 65, 2402–2408.
- Fazli P, Man HC, Shah UK, Idris A 2013: Characteristics of methanogens and methanotrophs in rice fields: a review. As. Pac. J. Mol. Biol. Biotechnol., 1, 3–17.
- Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW, DeLong EF 2008: Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U. S. A., 105, 3805–3810. doi:10.1073/pnas.0708897105
- Fukui M, Suwa Y, Urushigawa Y 1996: High survival efficiency and ribosomal RNA decaying pattern of Desulfobacter latus, a highly specific acetate-utilizing organism, during starvation. FEMS Microbiol. Ecol., 19, 17–25. doi:10.1111/j.1574-6941.1996.tb00194.x
- Galagan JE, Nusbaum C, Roy A et al. 2002: The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res., 12, 532–542. doi:10.1101/gr.223902
- Geymonat E, Ferrando L, Tarlera SE 2011: Methylogaea oryzae gen. nov., sp. nov., a mesophilic methanotroph from a rice paddy field. Int. J. Syst. Evol. Microbiol., 61, 2568–2572. doi:10.1099/ijs.0.028274-0
- Großkopf R, Janssen PH, Liesack W 1998: Diversity and structure of the methanogenic community in anoxic rice paddy soil microcosms as examined by cultivation and direct 16S rRNA gene sequence retrieval. Appl. Environ. Microbiol., 64, 960–969.
- Hanson RS, Hanson TE 1996: Methanotrophic bacteria. Microbiol. Mol. Biol. Rev., 60, 439–471.
- Hattori M, Tanaka N, Kanehisa M, Goto S 2010: SIMCOMP/SUBCOMP: chemical structure search servers for network analyses. Nucleic Acid Res., 38, 652–656. doi:10.1093/nar/gkq367
- Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC 2014: Mechanism of nitrogen fixation by nitrogense: the next stage. Chem. Rev., 114, 4041–4062. doi:10.1021/cr400641x
- Holmes DE, Shrestha PM, Walker DJF, Dnag Y, Nevin KP, Woodard TL, Lovley DR 2017: Metatranscriptomic evidence for direct interspecies electron transfer between Geobacter and Methanothrix species in methanogenic rice paddy soils. Appl. Environ. Microbiol., 83, e00223–17. doi:10.1128/AEM.00223-17
- Hori T, Müller A, Igarashi Y, Conrad R, Friedrich MW 2010: Identification of iron-reducing microorganisms in anoxic rice paddy soil by 13C-acetate probing. ISME J., 4, 267–278. doi:10.1038/ismej.2009.100
- Hou AX, Wang ZP, Chen GX, Patrick WH 2000: Effects of organic and N fertilizers on methane production in a Chinese rice soil and its microbiological aspect. Nutr. Cycl. Agroecosyst., 58, 333–338. doi:10.1023/A:1009875509876
- IPCC Climate Change 2013: the Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. T.F. Stocker, D. Qin, G.K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, P.M. Midgley (eds). Cambridge University Press, Cambridge, UK.
- Ishii S, Yamamoto M, Kikuchi M, Oshima K, Hattori M, Otsuka S, Senoo K 2009: Microbial populations responsive to denitrification-inducing conditions in paddy soil, as revealed by comparative 16S rRNA gene analysis. Appl. Environ. Microbiol., 75, 7070–7078. doi:10.1128/AEM.01481-09
- Islam T, Larsen Ø, Torsvik V, Øvreås L, Panosyan H, Murrell JC, Birkeland NK, Bodrossy L 2015: Novel methanotrophs of the family methylococcaceae from different geographical regions and habitats. Microorganisms, 3, 484–499. doi:10.3390/microorganisms3030484
- Itoh H, Ishii S, Shiratori Y, Oshima K, Osuka S, Hattori M, Senoo K 2013: Seasonal transition of active bacterial and archaeal communities in relation to water management in paddy soils. Micobes Environ., 28, 370–380. doi:10.1264/jsme2.ME13030
- Jewell TN, Karaoz U, Bill M, Chakraborty R, Brodie EL, Williams KH, Beller HR 2017: Metatranscriptomic analysis reveals unexpectedly diverse microbial metabolism in a biogeochemical hot spot in an alluvial aquifer. Front. Microbiol., 8, 40. doi:10.3389/fmicb.2017.00040
- Kamagata Y, Mikami E 1991: Isolation and characterization of a novel thermophilic Methanosaeta strain. Int. J. Syst. Bacteriol., 41, 191–196. doi:10.1099/00207713-41-2-191
- Kato S, Watanabe K 2010: Ecological and evolutionary interactions in syntrophic methanogenic consortia. Microbes Environ., 25, 145–151. doi:10.1264/jsme2.ME10122
- Kato S, Yumoto I, Kamagata Y 2015: Isolation of acetogenic bacteria that induce biocorrosion by utilizing metallic iron as the sole electron donor. Appl. Environ. Microbiol., 81, 67–73. doi:10.1128/AEM.02767-14
- Kimura M 2000: Anaerobic microbiology in waterlogged rice fields. In Soil Biochemistry, Eds. Bollarg JM, Stotzky G, Vol. 10, pp. 35–138. Marcel Dekker Inc., New York.
- Kouzuma A, Kato S, Watanabe K 2015: Microbial interspecies interactions: recentfindings in syntrophic consortia. Front. Microbiol., 6, 477. doi:10.3389/fmicb.2015.00477
- Krylova NI, Janssen PH, Conrad R 1997: Turnover of propionate in methanogenic paddy soil. FEMS Microbiol. Ecol., 23, 107–117. doi:10.1111/j.1574-6941.1997.tb00395.x
- Kumarswamy S, Ramakrishnan B, Sethunathan N 2001: Methane production and oxidation in an anoxic rice soil as influenced by inorganic redox species. J. Environ. Qual., 30, 2195–2201. doi:10.2134/jeq2001.2195
- Lärdh K, Cohen PS, Kjelleberg S 1992: Ribosomes exist in large excess over the apparent demand for protein synthesis during carbon starvation in marine Vibrio sp. strain-CCUG-15956. J. Bacteriol., 174, 6780–6788. doi:10.1128/jb.174.21.6780-6788.1992
- Liesack W, Schnell S, Revsbech NP 2000: Microbiology of flooded rice paddies. FEMS Microbiol. Rev., 24, 625–645. doi:10.1111/j.1574-6976.2000.tb00563.x
- Liu J, Xu H, Jiang Y, Zhang K, Hu Y, Zeng Z 2017: Methane emissions and microbial communities as influenced by dual cropping of Azolla along with early rice. Sci. Rep., 7, 40635. doi:10.1038/srep40635
- Lu Y, Lueders T, Friedrich MW, Conrad R 2005: Detecting active methanogenic populations on rice roots using stable isotope probing. Environ. Microbiol., 7, 326–336. doi:10.1111/emi.2005.7.issue-3
- Lu Y, Wassmann R, Neue HU, Huang C, Bueno CS 2000: Methanogenic responses to exogenous substrates in anaerobic rice soils. Soil Biol. Biochem., 32, 1683–1690. doi:10.1016/S0038-0717(00)00085-7
- Masuda Y, Itoh H, Shiratori Y, Isobe K, Otsuka S, Keishi S 2017: Predominant but previously-overlooked prokaryotic drivers of reductive nitrogen transformation in paddy soils, revealed by metatranscriptomics. Microbes Environ., 32, 180–183. doi:10.1264/jsme2.ME16179
- Meyer F, Paarmann D, D’Souza M et al. 2008: The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 9, 386. doi:10.1186/1471-2105-9-386
- Min H, Zinder SH 1989: Kinetics of acetate utilization by two thermophilic acetotrophic methanogens: methanosarcina sp. strain CALS-1 and Methanothrix sp. strain CALS-1. Appl. Environ. Microbiol., 55, 488–491.
- Nguyen SG, Guevarra RB, Kim J, Ho CT, Trinh MV, Unno T 2015: Impacts of initial fertilizers and irrigation systems on paddy methanogens and methane emission. Water Air. Soil Pollut., 226, 309. doi:10.1007/s11270-015-2501-8
- Orellana LH, Rodriguez-R LM, Higgins S, Chee-Sanford JC, Sanford RA, Ritalahti KM, Löffler FE, Konstantinidis KT 2014: Detecting nitrous oxide reductase (nosZ) genes in soil metagenomes: method development and implications for the nitrogen cycle. MBio, 5, e01193–14. doi:10.1128/mBio.01193-14
- Peng J, Lü Z, Rui J, Lu Y 2008: Dynamics of methanogenic archaeal community during plant residue decomposition in an anoxic rice field soil. Appl. Environ. Microbiol., 74, 2894–2901. doi:10.1128/AEM.00070-08
- Qiu YL, Hanada S, Ohashi A, Harada H, Kamagata Y, Sekiguchi Y 2008: Syntrophorhabdus aromaticivorans gen. nov., sp. nov., the first cultured anaerobe capable of degrading phenol to acetate in obligate syntrophic associations with a hydrogenotrophic methanogen. Appl. Environ. Microbiol., 74, 2051–2058. doi:10.1128/AEM.02378-07
- Rosencrantz D, Rainey FA, Janssen PH 1999: Culturable populations of Sporomusa spp. and Desulfovibrio spp. in the anoxic bulk soil of flooded rice microcosms. Appl. Environ. Microbiol., 65, 3526–3533.
- Sakai S, Imachi H, Sekiguchi Y, Ohashi A, Harada H, Kamagata Y 2007: Isolation of key methanogens for global methane emission from rice paddy fields: a novel isolate affiliated with the clone cluster rice cluster I. Appl. Environ. Microbiol., 73, 4326–4331. doi:10.1128/AEM.03008-06
- Sanford RA, Cole JR, Tiedje JM 2002: Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl. Environ. Microbiol., 68, 893–900. doi:10.1128/AEM.68.2.893-900.2002
- Schäfer T, Selig M, Schönheit P 1993: Acetyl-CoA synthetase (ADP forming) in archaea, a novel enzyme involved in acetate formation and ATP synthesis. Arch. Microbiol., 159, 72–83. doi:10.1007/BF00244267
- Schmieder R, Edwards R 2011: Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. doi:10.1093/bioinformatics/btr026
- Shieh JS, Whitman WB 1987: Pathway of acetate assimilation in autotrophic and heterotrophic methanococci. J. Bacteriol., 169, 5327–5329. doi:10.1128/jb.169.11.5327-5329.1987
- Singh A, Dubey SK 2012: Temporal variations in methanogenic community structure and methane production potential of tropical rice ecosystems. Soil Biol. Biochem., 48, 162–166. doi:10.1016/j.soilbio.2012.01.022
- Sugano A, Tsuchimoto H, Cho TC, Kimura M, Asakawa S 2005: Succession of methanogenic archaea in rice straw incorporated into a Japanese rice field: estimation by PCR-DGGE and sequence analyses. Archaea, 1, 391–397. doi:10.1155/2005/582597
- Supparattanapan S, Saenjan P, Quantin C, Maeght JL, Grunberger O 2009: Salinity and organic amendment effects on methane emission from rain-fed saline paddy field. Soil Sci. Plant Nutr., 55, 142–149. doi:10.1111/j.1747-0765.2008.00330.x
- Takai Y 1970: The mechanism of methane fermentation in flooded paddy soil. Soil Sci. Plan Nut., 16, 238–244. doi:10.1080/00380768.1970.10433371
- Urich T, Lanzén A, Qi J, Huson DH, Schleper C, Schuster SC 2008: Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One, 3, e2527. doi:10.1371/journal.pone.0002527
- Vignais PM, Billoud B 2007: Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev., 107, 4206–5801. doi:10.1021/cr050196r
- Watanabe T, Wang G, Taki K, Ohashi Y, Kimura M, Asakawa S 2010: Vertical changes in bacterial and archaeal communities with soil depth in Japanese paddy fields. Soil Sci. Plant Nutr., 56, 705–715. doi:10.1111/j.1747-0765.2010.00511.x
- Weber S, Lueders T, Friedrich MW, Conrad R 2001: Methanogenic populations involved in the degradation of rice straw in anoxic paddy soil. FEMS Microbiol. Ecol., 38, 11–20. doi:10.1111/j.1574-6941.2001.tb00877.x
- Wilke A, Harrison T, Wilkening J, Field D, Glass EM, Kyrpides N, Mavrommatis K, Meyer F 2012: The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinformatics, 13, 141. doi:10.1186/1471-2105-13-141
- Yagi K, Minami K 1990: Effect of organic matter application on methane emission from some Japanese paddy fields. Soil Sci. Plant Nutr., 36, 599–610. doi:10.1080/00380768.1990.10416797
- Yan X, Yagi K, Akiyama H, Akimoto H 2005: Statistical analysis of the major variables controlling methane emission from rice fields. Glob. Change Biol., 11, 1131–1141. doi:10.1111/j.1365-2486.2005.00976.x
- Yang SS, Chang HL 2001: Effect of green manure amendment and flooding on methane emission from paddy fields. Chemosphere, 3, 41–49.
- Yashiro Y, Ehara M, Miyazaki M, Yamaguchi T, Imachi H 2011: Methanoregula formicica sp. Nov., a methane-producing archaeon isolated from methanogenic sludge. Int. J. Syst. Evol. Microbiol., 61, 53–59. doi:10.1099/ijs.0.014811-0