
ABSTRACT
Damage and susceptibility are key concepts in conservation but are rarely explicitly defined. This paper provides definitions for these terms and applies them to mineral collections. Minerals are often overlooked in the context of heritage conservation due to their assumed stability. While many appear to be stable under ambient conditions, at least 10% of known mineral species are susceptible to temperature, moisture, light, or pollutant levels common in museum stores and displays. These susceptible minerals are represented in museums as natural history specimens (as crystals and inclusions within rocks and fossils), pigments in paintings, and deterioration products of other minerals, metals, and many other object types. A new online resource, the Mineral Susceptibility Database, has been designed to facilitate the preservation of minerals by providing relevant information in a single, accessible location. Data were collated and synthesised from various fields of research, many of which are not easily accessible to museum professionals. As an open repository of interdisciplinary research, the Mineral Susceptibility Database encourages informed decision-making and advocates cross-disciplinary communication, both of which are necessary to improve the care of mineral and geological materials.
Introduction
‘A problem which frequently confronts a museum curator is the proper preservation of certain choice specimens of minerals.’ — A.L. Parsons (Citation1922, 59)
Minerals, like other types of objects in museum collections, may be subject to damage and deterioration. The first publications discussing mineral instability within the museum context appeared in the first half of the twentieth century (Parsons Citation1922, Citation1926; Bannister Citation1937). Yet as Parsons’ (Citation1922) opening line (above) suggests, mineral instability has been a long-standing and bemoaned issue.
Interest in mineral instability grew within the museum sector throughout the twentieth century, culminating in an intensive period of research from the mid-1970s through to 2000 (e.g. Howie Citation1979a, Citation1984, Citation1992a; King Citation1985; Waller, Andrew, and Tétreault Citation2000). While the quantity of information and discussion on the topic has greatly increased throughout the past century, many questions regarding best practice for mineral collections care have remained unanswered to this day.
As a response to numerous calls for further research into this subject (Parsons Citation1922; King Citation1985; Howie Citation1992a; Waller, Andrew, and Tétreault Citation2000; Baars and Horak Citation2018), the collaborative research project ‘Preservation of geological collections’ was established between the University of Oxford, National Museum Cardiff, BSRIA Ltd, and OR3D Ltd. This paper is one early result of this project and aims to answer the following critical questions:
What are ‘damage’ and ‘susceptibility’?
More specifically, what constitutes ‘damage’ to a mineral?
Why and how are minerals ‘susceptible’?
What are the environmental conditions which induce a mineral’s susceptibility and potentially result in damage?
Historic treatments and current guidance for mineral collections
As with any museum collection, there are two potential approaches for managing and mitigating the deterioration of mineral collections: interventive treatment and preventive practices. Interventive treatments for mineral specimens have historically included:
Boiling in water (Fenlon and Petrera Citation2019);
Refrigeration (King Citation1985; Waller Citation1992);
Oven drying (King Citation1985; Fenlon and Petrera Citation2019);
Liquid storage in linseed oil, mineral oil, carbon tetrachloride, paraffin, glycerol, silicone liquid, water, or alcohol (Howie Citation1979a, Citation1979b; Waller Citation1992; Fenlon and Petrera Citation2019);
Suspension over or immersion in ammonia or ethanolamine thioglycolate solutions (Howie Citation1979a, Citation1979b; Larkin Citation2011; Caracanhas Cavallari, Brincalepe Salvador, and Rodrigues da Cunha Citation2014; Fenlon and Petrera Citation2019; Irving and Hadland Citation2019);
Spot treatments or poultices using morpholine or ethanolamine thioglycolate (Howie Citation1979a, Citation1979b; Larkin Citation2011; Caracanhas Cavallari, Brincalepe Salvador, and Rodrigues da Cunha Citation2014; Fenlon and Petrera Citation2019);
Coating and consolidation (Howie Citation1984; King Citation1985; Larkin Citation2011) with linseed oil, vaseline (Tennent and Baird Citation1985), shellac, paraffin wax, (poly)vinyl acetate, or polybutyl methacrylate (Howie Citation1979a, Citation1979b; Caracanhas Cavallari, Brincalepe Salvador, and Rodrigues da Cunha Citation2014; Fenlon and Petrera Citation2019; Irving and Hadland Citation2019); and
Anti-bacterial and anti-insect treatments and fumigants (Fenlon and Petrera Citation2019; Irving and Hadland Citation2019).
Many of these treatments, largely designed to control pyrite (FeS2) deterioration, are no longer used due to chemical toxicity, methodological inefficacy, or treatment failure. Those that are still in use—chiefly ammonia or ethanolamine thioglycolate treatments and the occasional use of coatings and consolidants (Larkin Citation2011)—are limited almost exclusively to larger institutions, as these methods are time and resource intensive and require specialist knowledge and equipment.
Given the lack of accessible, efficacious treatment options, improving preventive practices and collections care is the preferable route for preserving mineral specimens (c.f. Kerbey and Horak Citation2006; Graham Citation2018). However, this requires an understanding of favourable storage conditions for the various mineral species. At present, at least ten percent of the approximately 5,739 known species (International Mineralogical Association Citation2021) require unique, species-specific environmental conditions. Obtaining the relevant data for these minerals is currently difficult and tedious at best because no single resource exists that would allow conservators and curators to access the information they require.
Howie’s 1992 publication, The Care and Conservation of Geological Material, is the most comprehensive text on mineral instability to date, providing information for more than 300 mineral species and devoting a whole chapter to pyrite and marcasite (both FeS2). As such, the book is frequently cited in publications, standards, and guidelines. However, the publication (Howie Citation1992a) is very much of its time. It was written as ‘a “state of the art” text [… . to] encourage others to tackle many areas where problems continue to cause loss and decay’ (Howie Citation1992a: xi), just as Parsons (Citation1922) had urged 70 years earlier. While the data may have been accurate at time of publication, nearly 30 years have elapsed without revision or addition. This is because little progress has been made within the museum sector since.
PAS 198:2012 (BSI Citation2012; withdrawn on publication of BS EN 16892:2018)—the most up-to-date standard on collections care for the museum sector—provides wide-ranging recommendations for the environmental and pollution-related requirements of many different cultural heritage materials. However, PAS 198 provides insufficient advice on the appropriate storage conditions for geological collections (BSI Citation2012 Tables E.1 and G.2). Table E.1, meant to outline the effects of temperature and relative humidity (RH) on materials, only covers pyrite and marcasite deterioration and does so briefly. Other humidity-sensitive minerals are grouped together in one sentence that makes no reference to which minerals are affected by humidity and at what conditions. Whereas in Table G.2, ‘Pollutant-material interaction in enclosures’, only broad mineral groups are mentioned—‘carbonate, borate, phosphate and similar soluble minerals of weak acids’ (BSI Citation2012, 40)—and are stated to only be susceptible to formic and acetic acids. Documents written specifically for geological collections do not offer much additional information. These include those developed by the British Museums and Galleries Commission in 1993—updated in 2004 by the Museums Libraries and Archives Council (MLA) (Stanley Citation2004)—and by the Geological Curators’ Group (GCG) (Brunton, Besterman, and Cooper Citation1984).
Appendix E of the MLA Standards (Stanley Citation2004) provides relative humidity and temperature recommendations for the display and storage of geological specimens. However, only four categories of specimen types are suggested: ‘general’, ‘sensitive’, ‘pyrites and marcasites’, and ‘sub-fossil bone, tusks, teeth, fossils with shale or clay matrix’. This crude delineation inadequately reflects the diversity of mineral collections, as highlighted by the texts cited (Howie Citation1992a; Nassau Citation1992; Waller Citation1992) within the document. Similar oversimplifications also occur in the GCG Guidelines (Brunton, Besterman, and Cooper Citation1984). In this text, the authors mentioned several minerals which will change at a given moisture content or under humidity fluctuations. These changes include the likelihood of hydration state changes of sulfates and some zeolites, the vulnerability of halides to deliquesce, and the hygroscopicity of clay minerals. Yet the authors still recommended a blanket approach of 50 ± 5% RH, the then de facto standard across the museum sector, which arose likely due to titular prestige rather than any technical or scientific evidence (see Henderson Citation2018). A sole, unspecific caveat was provided for vulnerable materials: ‘Specialized collections or specimens may require high or low regimes of humidity’ (Brunton, Besterman, and Cooper Citation1984, C16). This statement is echoed in the MLA Standards: ‘Sensitive minerals and other materials: depends on mineral or material’ (Stanley Citation2004, 64). Yet neither document provides the detail necessary for determining the appropriate storage conditions for ‘sensitive’ minerals.
Whilst these three documents reference some important works, the brevity of the sections handling mineral instability and the length of time that has elapsed since they were last updated highlights the paucity of research on this subject within the museum sector. This is greatly contrasted by the collections care requirements for other heritage materials that have been better researched. For example, Appendix G of the MLA Standards (Stanley Citation2004) lists a wide range of archival materials and their specific environmental requirements. These are supported by national and international standards (BS 4783, 5454, 5687; ISO 5466, 6051) developed after decades of research. Comparable efforts to study geological material have not yet been undertaken, even though such research is fundamental for the improvement of collection care standards for minerals and related materials. It is impossible for current guidelines and standards to become a comprehensive resource without first addressing a catalogue of questions (Waller Citation1992; Baars and Horak Citation2018) surrounding the storage of mineral specimens. The first of which is ‘What is damage? And how is it presented in minerals?’
What is ‘damage’?
The cornerstone of museum work is the mitigation of damage to collections to improve their longevity and continued access and use. The basis of damage mitigation must be a thorough and unambiguous understanding of what damage actually means for a specific material. Disagreement on what is meant by ‘damage’ can result in confusing, even contradictory condition assessments. What one considers ‘damage’ may be seen by another as part of an object’s history. Thus, a shared definition of damage enables a consensus on how to best monitor, detect, and assess collection items, and defines when treatments are considered necessary. Therefore, it is critical to have an established definition of damage to carry out this work as effectively as possible.
But this is easier said than done. Damage is a complex and highly subjective concept because it implies not only a detrimental change in state but also a negative change in values (Strlič et al. Citation2013) and uses. This is reflected in the common definition of damage as: ‘physical harm that impairs the value, usefulness, or normal function of something’ (Oxford University Press Citation2021a).
Changes in state involve changes to the intrinsic, material properties of an object (Ashley-Smith Citation1995), whilst changes in value, and subsequently use, entail alterations in extrinsic, human-assigned concepts (Ashley-Smith Citation1999; Appelbaum Citation2007). Yet not all change in an object’s state results in a change of value. Nor is all change negative (). In some cases, change may even be beneficial to the object’s value (e.g. evidence of wear or use). Hence, whilst damage is usually associated with change, change does not always equate to damage (Ashley-Smith Citation1999; Meul Citation2008) as it may not necessarily affect value.
Figure 1. A schematic representation of how an agent of change produces damage. Damage is not a straightforward process of cause and effect. Rather, it is the result of a perception that an object’s value and or use has been negatively affected due to changes in state caused by exposure to a given agent of change.
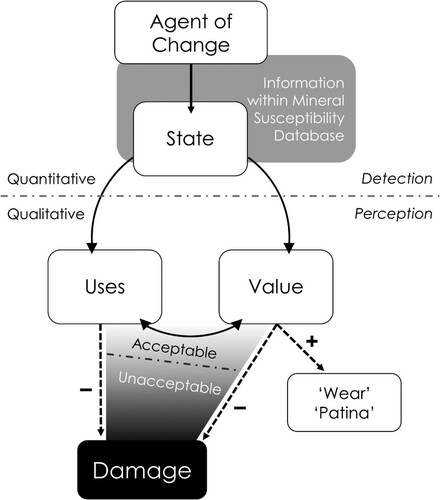
Change may be accumulated rapidly or gradually, and certain types of change may be considered acceptable, or indeed inevitable (Michalski Citation1994; Rose and Hawks Citation1995; Waller Citation1995). Yet at some point, a threshold is reached where the amount of change accrued is perceived as unacceptable and negatively affects the object’s values and uses (Appelbaum Citation2007; Robb et al. Citation2013; Strlič et al. Citation2013). The decision whether a type or amount of change is classified as unacceptable—and hence considered damage—is dependent on the clarity of the object’s values and uses (see Allmon Citation1994; Appelbaum Citation2007; Baars Citation2010; Robb et al. Citation2013), many of which rely heavily on associated contextual information (Robb et al. Citation2013). This emphasises that damage is an observer- and context-dependent concept (Ashley-Smith Citation1999; Appelbaum Citation2007).
We hope that our proposed definition for damage succinctly reflects this complexity: damage is a material change which is perceived to have negatively affected an item’s value-defining aspects.
Determining unacceptable change
It is important to demarcate the threshold between acceptable and unacceptable change (the dashed line within the shaded region of ) in order to define an appropriate conservation response and to suggest when intervention and treatment are necessary (Howie Citation1979a; Strlič et al. Citation2013). But how is this possible when damage and value are subjective and variable? While some now see it as ‘an illusion’ (Muñoz-Viñas Citation2002, 26) that damage can be defined objectively, Strlič et al. (Citation2013) suggested this may be achieved through decoupling ‘value’ from ‘change’.
The decision to focus on material change, rather than change in value, is largely due to the complexity and subjectivity of the latter. Value is a multifaceted and subjective concept which varies with the object being assessed, the assessor, and the context (Ashley-Smith Citation1995; Baars Citation2010; Robb et al. Citation2013; Strlič et al. Citation2013). Thus, the interpretation of values and its terminology will also vary from person to person. Each individual will apply their own frame of reference to determine what the terms mean for a given context (Taylor Citation2013), often introducing interpretational bias (Taylor and Stevenson Citation1999; Taylor Citation2013). This is demonstrated most notably during condition assessments when subjective and imprecise terminology— such as ‘good’, ‘bad’, ‘stable’, or ‘degrading’ (Ashley-Smith 199; Taylor Citation2013; Gioventù Citation2018; Kosek and Barry Citation2019)—are used for ranking condition. The inherent subjectivity of value and value terminology makes it difficult to measure value consistently, let alone to define, and compare across institutions, regions, cultures, and eras.
Material change, on the other hand, is often consistently quantifiable using scientific methods. The use of measurements generated by instrumentation, rather than human observation, is advantageous in minimising subjectivity and error (Ashley-Smith Citation1995; Baars and Horak Citation2018), although neither can be completely removed (e.g. sampling and confirmation biases). The resultant data may be recorded objectively in a standardised manner, allowing for communication and comparison across time and space.
Various analytical techniques have been employed in examining museum collections, including minerals. These techniques include, but are not limited to:
Measuring weight changes,
Photography,
Colorimetry,
Infrared (IR) spectroscopy,
Raman spectroscopy,
Mössbauer spectroscopy,
Micro-computed tomography (micro-CT),
X-ray diffraction (XRD),
Scanning electron microscopy (SEM),
Energy dispersive X-ray spectroscopy (EDS or EDX), and
X-ray absorption near-edge structure (XANES).
While all such analytical methods are effective for identifying physical and chemical changes – generally through the comparison of fresh and degraded samples – they are not all equally applicable for museum use. The ideal technique is one that is accessible, cost effective, quick, straightforward to use, and requires little to no destructive sampling. These criteria are vital when assessing and monitoring large collections but may be compromised for high value or priority collections or when answering specific research questions. Most of the above methods will result in a compromise of some sort; many require destructive sampling, and some require a long time (>1 h per specimen) to acquire data of sufficient quality. In addition, many analytical techniques are still largely inaccessible to the museum sector due to equipment and operating cost, as well as the expertise required to use the equipment.
Of the methods listed above, weight measurements, photography, colorimetry, XRD, IR, and Raman spectroscopy are likely the most practical methods to employ within a museum setting (Royce Citation2019). However, each technique explores different properties and consequently has its own limitations. As such, no one method can be considered a stand-alone technique to answer the gamut of questions relevant to the care and conservation of museum collections. Nevertheless, information produced by analytical methods may be used in the development of exposure limits or doses to various agents of change through dosimetry (e.g. Waller, Andrew, and Tétreault Citation2000; Odlyha et al. Citation2011; del Hoyo-Meléndez, Mecklenburg, and Teresa Doménech-Carbó Citation2011; Grøntoft et al. Citation2016; Hackney Citation2016). This may enable the prediction of change and the prioritisation of subsequent treatment options, if any (Strlič et al. Citation2013; Baars and Horak Citation2018).
Vulnerability and susceptibility
Conservation as a discipline exists because many heritage items are vulnerable to damage and require careful management to preserve them. Unsurprisingly, there is much reference in conservation literature to the vulnerability of heritage items (e.g. Sabbioni et al. Citation2009; Ortiz and Ortiz Citation2016). What is less clear, and rarely defined, is what is meant when an item is said to be vulnerable.
Vulnerability is commonly defined as: ‘the quality or state of being exposed to the possibility of being attacked or harmed, either physically or emotionally’ (Oxford University Press Citation2021b). Like damage, the definition of vulnerability embodies an inherent intrinsic-extrinsic duality. The object’s properties are again the intrinsic factor. Yet this time the extrinsic factor is the external conditions (rather than human-assigned concepts). However, unlike damage, the intrinsic and extrinsic factors of vulnerability are mutually dependent. Without either the propensity or the circumstances for vulnerability, one cannot claim an object to be vulnerable. This is emphasised by the need to state the context when discussing vulnerability (for example, ‘vulnerable to wear and tear’).
These same concepts apply to susceptibility; ‘the state or fact of being likely or liable to be influenced or harmed by a particular thing’ (Oxford University Press Citation2021c). While vulnerability and susceptibility have similar definitions and are often used interchangeably, vulnerability has a more emotional connotation than susceptibility due to societal implications; where those most vulnerable within society are often defined as children, the elderly, and those who are disabled or immunocompromised. The conservation implications of vulnerability and susceptibility, however, are the same and the choice of terminology is a matter of preference. The authors prefer ‘susceptibility’, which will be used henceforth in this paper.
Conservation implications of susceptibility
The susceptibility of heritage objects is governed by their physical and chemical properties in relation to the surrounding environmental conditions. An object’s properties determine which conditions are favourable for stability (or metastability; Waller Citation1991), and the object’s response (i.e. change) to unfavourable conditions (i.e. agents of change). As the object’s physical and chemical properties are inherent and will not spontaneously change without reason, we could say that susceptibility is an inherent, secondary property of an object. Susceptibility may be acquired or altered through the addition of new material or a previous change. For example, a repair undertaken using a chemical consolidant can make an item susceptible to further physical damage if the consolidant subsequently fails (Chiantore and Lazzari Citation2001).
Whether an object’s susceptibility is expressed, however, is dependent on the probability of exposure to unfavourable conditions. For example, a pigment’s photosensitivity is not expressed until the pigment is exposed to visible or ultraviolet light. It is only after some degree of exposure that the pigment will respond through colour and or chemical changes.
As a result of being context-dependent, an object’s susceptibility is relative compared to those of other objects. Not only can the object's responses vary wildly, but so can the sets of conditions required to evoke these responses. Within museums, this is exemplified by the contrasting RH requirements for different material and collection types (Erhardt and Mecklenburg Citation1994). Additionally, some objects may be susceptible to conditions that rarely or never occur in a given environment. The museum environment encompasses a rather narrow range of conditions compared to those possible on, in, and beyond Earth. Some minerals cannot persist within the museum environment, as they require high temperatures and or pressures to exist. One such example is high quartz (β-quartz; SiO2). Above 573°C at 1bar (∼1atm) pressure, the hexagonal crystal structure of β-quartz is thermodynamically preferred. When cooled below 573°C, the β-quartz unit cell structure immediately collapses into the denser, trigonal cell structure of low quartz (α-quartz; SiO2) (Heaney Citation1994) through bond bending and kinking (Heaney and Veblen Citation1991). While this structural adjustment is reversible, it requires heating to the transition temperature. Thus, while α-quartz specimens will not structurally rearrange into β-quartz under typical museum conditions, all β-quartz specimens in collections are actually α-quartz paramorphs displaying a β-quartz habit (Hudson Institute of Mineralogy Citation2021). This example emphasises that not all susceptibilities are relevant within the museum context.
Thus, the degree of susceptibility an object has towards unfavourable conditions can be considered the product of both the probability and the effects of exposure. It is important to understand the material and how it responds to potential environmental parameters to identify susceptibility and determine which pose as deterioration risks within the museum setting. This process includes the accurate identification of an object’s composition, as knowledge of its material properties is imperative for the determination of appropriate storage conditions (Baars, Royce, and Cotterell Citation2021).
Why minerals are susceptible to change
A mineral is a homogeneous, naturally occurring solid with a defined and unique chemical composition and crystalline structure. As such, each mineral requires unique conditions to form. It is at these formation conditions that the mineral is stable and can persist indefinitely until a significant change in conditions occurs.
Conditions at which a mineral is stable are termed its stability field, which may be depicted in a stability (or phase) diagram (). Stability fields are classically defined by temperature, pressure, and the composition of compounds present in the surrounding environment (Hazen and Ausubel Citation2016). Other conditions relevant to phase stability include acidity and alkalinity (pH), and the presence of energy sources such as light or radiation.
Figure 2. Phase diagram of SiO2 polymorphs. While coesite has the largest stability field of the silica polymorphs, it cannot exist under atmospheric pressure at Earth’s surface, as the structure of α-quartz is energetically preferred. Image used with permission courtesy Akhavan (Citation2014).
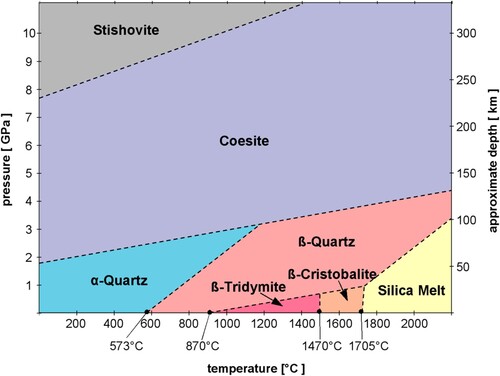
A mineral's stability field is governed by both the chemical composition and crystal structure of a mineral. Minerals with greater stability are able to withstand greater variations in environmental conditions. For example, coesite’s (SiO2) monoclinic structure is stable over a wider range of temperatures and pressures compared to those of the other silica minerals ().
A narrow stability field, on the other hand, means that a mineral can only exist under tight, well-defined conditions. Even slight variations in conditions—be it the presence of other elements, variation in cation ratios, or the pH of the formation solution—can mean the formation of, or alteration to, a different mineral species (Hazen and Ausubel Citation2016).
When conditions are beyond a mineral's stability field, the mineral is either metastable or unstable, depending upon the amount of energy in the system, the ease of transition, and the presence of potentially facilitative components. When metastable—such as aragonite (CaCO3) or the allotropes of tin (Sn)—minerals may persist beyond their stability field but have insufficient energy to change into the phase that is energetically stable at those conditions (Nesse Citation2012). This means that the reaction will not occur until the ambient conditions change so that there is a sufficiently great difference in free energy between the current metastable phase and the energetically preferred phase (e.g. significant undercooling is required for tin’s structural rearrangement to occur; Ojima, Taneda, and Takasaki Citation1993; Zeng et al. Citation2014). Or the reaction does occur but at such a slow rate that it may take a very long time (in some cases, thousands of years) to completely alter to a different phase (Putnis Citation1992), for example aragonite's structural transformation to calcite (CaCO3) (Crawford and Hoersch Citation1972). This differs from instability, which occurs when there is sufficient energy available to initiate a reaction that causes the mineral to alter into an energetically more stable phase (Putnis Citation1992; Nesse Citation2012) at a rate detectable within a human time frame. For many mineral reactions, this results in the formation of a new mineral species, yet other chemical compounds may also be produced, including gases, liquids, and non- crystalline solids.
How minerals change in a museum environment
Approximately 10% of mineral species are susceptible to change under the conditions present in indoor environments (Royce Citation2021). This paper defines the indoor museum setting to be approximately 1 bar pressure, −20–50°C, 1–99% RH, and exposure to light and common indoor pollutants (see Grzywacz Citation2006, Appendix 1). While this range does include some values that may be considered ‘extremes’, it covers conditions which may occur in buildings without insulation or HVAC, or during rare events, such as equipment failure, unusual weather conditions, flooding, and localised heating by spotlights. This paper, however, does not cover any minerals or changes which occur beyond the stated parameters, as they will rarely be present in museum collections.
Means of mitigating damage from physical forces – such as shock, vibration, and abrasion – have been discussed in other texts (Brunton, Besterman, and Cooper Citation1984; Howie Citation1984; King Citation1986; Stanley Citation2004) and are thus not covered below. Therefore, we focus on four agents of change: adverse temperatures, adverse moisture levels, light, and atmospheric pollutants. At present, more than 550 minerals have been identified (Royce Citation2021) as being susceptible to these agents under museum conditions (). Furthermore, many of these species are widely represented in collections, with some considered to be rare and difficult to acquire (Howie Citation1979a, Citation1984; Hazen and Ausubel Citation2016).
Table 1. A list of reactions that may occur to minerals in a museum environment, their causes, and definitions. Most of these reactions are irreversible, and even those that are reversible can produce permanent change.
Adverse temperature
Temperature and humidity are intimately related, however there are a handful of minerals that are affected by temperature only, regardless of a change in or maintenance of RH. One such example is lansfordite (MgCO3·5H2O), which will dehydrate into nesquehonite (MgCO3·3H2O) if temperatures are above approximately 10°C (Waller Citation1992), despite maintaining 100% RH. Nesquehonite can subsequently lose additional water and CO2 when kept under ambient conditions (Robie and Hemingway Citation1972).
While lansfordite’s transition to nesquehonite is from one solid state to another, dissolution into a liquid phase is possible for some species. Nitrocalcite (Ca(NO3)2·4H2O) will enter solution as a result of dissociation (i.e. the splitting of a compound into smaller units) into the highly soluble trihydrate (Ca(NO3)2·3H2O) and liquid water when exposed to ∼30°C at a fairly low (20–30%) RH (Waller Citation1992). Similarly, hydrated sodium metasilicates (Na2SiO3·nH2O) can melt into a liquid phase at temperatures greater than 47.85°C (Baker, Woodward, and Pabst Citation1933).
Rapid changes in temperature can also lead to fracture. Differential temperatures between a specimen’s interior and exterior result in increasing stresses, which are consequently released through cracking or spalling (Waller Citation1992; Horak Citation1994). Native sulfur (S8) is a well-known example (Howie Citation1984; King Citation1985; Waller Citation1992) that shatters with handling and localised heating from lighting. Waller (Citation1992) lists several characteristics that increase the likelihood of a mineral to fracture, including easy cleavage and high brittleness.
Temperature variation can also produce less dramatic, but equally significant changes in some minerals. Trögerite ((H3O)(UO2)(AsO4)·3H2O) undergoes a reversible structural rearrangement over 18–28°C (de Benyacar and de Abeledo Citation1974). Above this range, trögerite exhibits a tetragonal structure, but below 18°C the mineral displays a low symmetry, pseudo-tetragonal form. Similar structural transformations are believed to occur to the other minerals in the metatorbernite group (de Benyacar and de Abeledo Citation1974).
Adverse moisture and relative humidity
The effects of inappropriate levels or fluctuations in RH are varied and complex, as they are dependent on material type (c.f. Erhardt and Mecklenburg Citation1994). RH is involved in numerous reactions () including phase changes (i.e. hydration, deliquescence, efflorescence) and oxidation. As a result, more than 400 minerals are susceptible to RH, most of which have different stability ranges that often do not overlap (Royce Citation2021).
Hygroscopic materials respond to RH fluctuations by absorbing or desorbing moisture (Howie Citation1979a) to maintain equilibrium with the environment. Yet this process is not instantaneous and may induce stresses throughout the specimen with large RH fluctuations (Erhardt and Mecklenburg Citation1994). These stresses may result in dimensional changes, cracking, and delamination (Howie Citation1984; Erhardt and Mecklenburg Citation1994). Changes in hydration states are also frequent, leading to an altered composition. The loss of structural water can lead to shrinkage, fractures, and even disintegration (Howie Citation1984; Waller Citation1992).
Water-soluble minerals will also absorb moisture from the atmosphere and deliquesce. Halite (NaCl) is a prime example, entering solution when the RH is above 75.3% at 20°C (Feldman Citation2000). If deliquescent conditions are temporary, the specimen can slump, round, or flatten when in a semi-liquid phase (Waller Citation1992). When conditions persist for prolonged lengths of time, the transparency and lustre of a specimen can be affected (), resulting in a translucent or opaque, matte surface finish. Deliquescent minerals can also be affected by hydrolysis or oxidation when in solution (Waller Citation1992).
Figure 3. A specimen of halite (NaCl; OUNHM MIN.26782), featuring the characteristic rounding of corners and edges, and an opaque matte surface due to deliquescence. Image used with permission of Oxford University Natural History Museum.

Similarly, water-soluble minerals can migrate through a porous medium, given a sufficiently high RH, the exact value of which is species-specific. This process occurs with cycles of fluctuating RH, where the salts enter solution when the RH is above the deliquescence point and recrystalise when the RH drops below it (Howie Citation1979b; Erhardt and Mecklenburg Citation1994). Crystallisation at the surface (efflorescence) often results in surface disfiguration (Howie Citation1979b). Recrystallisation can also occur within the material matrix, causing mechanical stress and damage. The presence of soluble salts can facilitate oxidation reactions and increase susceptibility to pollutants (Erhardt and Mecklenburg Citation1994).
Example: pyrite (FeS2)
Pyrite has long been a bane to all who have encountered it (c.f., Torrens Citation1977; Lowson Citation1982). Its decay is the most frequently documented form of mineral deterioration in geological collections (Howie Citation1992b; Blount Citation1993; Rouchon et al. Citation2012; Odin et al. Citation2014, Citation2018; Miles Citation2019) due to the mineral’s ubiquitous nature. Pyrite is often quoted as the most abundant metal sulfide on Earth’s surface (Kullerud and Yoder Citation1959; Lowson Citation1982; Eggleston, Ehrhardt, and Stumm Citation1996; Guevremont et al. Citation1998a; Rimstidt and Vaughan Citation2003; Dos Santos, de Mendonça Silva, and Duarte Citation2016; Miles Citation2019). This is because pyrite is formed in a variety of environments (Kullerud and Yoder Citation1959; Guevremont et al. Citation1998a; Rickard and Luther Citation2006) and occurs in all major rock types (Kullerud and Yoder Citation1959; Howie Citation1992b; Larkin Citation2011).
The deterioration mechanisms and pathways of pyrite are complex and variable, and are heavily dependent on the conditions present, including water and oxygen concentration, pH, and the presence of catalytic ions, compounds, and biological agents (Lowson Citation1982). Yet for both chemical and electrochemical oxidation pathways, the presence of oxygen or water, or both, is required for oxidation to occur (Guevremont et al. Citation1998b; Rosso, Becker, and Hochella Citation1999).
When only molecular oxygen (O2) is available, oxidation is slow, forming discrete oxide patches around defects (Rosso, Becker, and Hochella Citation1999). These defects are non-stoichiometric, sulfur-deficient sites (Guevremont et al. Citation1998a, Citation1998b; Rosso, Becker, and Hochella Citation1999; Dos Santos, de Mendonça Silva, and Duarte Citation2016) which expose iron to the atmosphere. Iron has a higher affinity for molecular oxygen than sulfur (Guevremont et al. Citation1998b; Rosso, Becker, and Hochella Citation1999), resulting in the preferential adsorption of oxygen at iron sites and the formation of Fe-O groups (Rosso, Becker, and Hochella Citation1999).
Pyrite oxidises similarly when exposed to only water, albeit at a faster rate than when exposed to only molecular oxygen (Guevremont et al. Citation1998b). Water is also preferentially adsorbed onto defects (Guevremont et al. Citation1998b; Rosso, Becker, and Hochella Citation1999; cf. Yalcin et al. Citation2020), producing Fe-O and Fe-OH bonds. The formation of these bonds causes nearby sulfur sites to become slightly more electropositive, increasing their susceptibility to water. Water can then react with these sulfur sites to produce S-O bonds, the precursors to sulfate formation (Rosso, Becker, and Hochella Citation1999).
Yet when pyrite is exposed to both water and molecular oxygen, the rate of oxidation is significantly greater than the sum of O2-only and H2O-only oxidation, indicating a synergy between O2 and H2O (Guevremont et al. Citation1998b). Oxidation is most aggressive when equal quantities of O2 and H2O are present (Rosso, Becker, and Hochella Citation1999). Additionally, Usher et al. (Citation2004) established that the oxygen from the water, rather than molecular oxygen, is the primary source of oxygen in the resulting sulfate products.
The products of both the chemical and electrochemical oxidation of pyrite are ferrous sulfate and sulfuric acid. Both sulfuric acid and ferrous sulfate are hygroscopic and often may react further with atmospheric moisture (Howie Citation1992b; Jerz and Rimstidt Citation2004). Sulfuric acid may scorch labels () and specimen housing materials (Howie Citation1992b; Waller, Andrew, and Tétreault Citation2000; Larkin Citation2011; Miles Citation2019) and is capable of dissolving or altering other mineral species (Howie Citation1992b; Waller, Andrew, and Tétreault Citation2000; Jerz and Rimstidt Citation2004).
Figure 4. A veinstone specimen affected by pyrite decay. A section has spalled off the body; both feature characteristic yellow and white sulfate efflorescence. Also note the ‘scorching’ of the label, caused by sulfuric acid, which has defaced the accession number. Image courtesy National Museum Cardiff.

The composition and hydration state of the sulfates varies with the ambient RH and the composition of the specimen undergoing oxidation (Jerz and Rimstidt Citation2004; Odin et al. Citation2014). Ferrous sulfate also frequently undergoes an additional series of reactions. It may hydrate, dehydrate, hydrolyse, or oxidise depending on the ambient conditions. These reactions produce ferroso-ferric sulfates, basic sulfate-hydrates, and ferric sulfates (Howie Citation1979a, Citation1992b; Blount Citation1993; Jambor, Nordstrom, and Alpers Citation2000; Jerz and Rimstidt Citation2004), which may react even further to form a complex series of products.
The molar volumes of product sulfates are much larger than those of the initial pyrite (), resulting in volumetric expansion (Wiese, Powell, and Fyfe Citation1987; Howie Citation1992b; Jerz and Rimstidt Citation2004). If formed within a specimen, this expansion produces stresses that frequently result in the specimen cracking, spalling (), and eventually disintegrating (Wiese, Powell, and Fyfe Citation1987; Howie Citation1992b; Blount Citation1993; Jerz and Rimstidt Citation2004; Larkin Citation2011; Miles Citation2019).
Table 2. Molar volumes of pyrite and some common iron sulfate reaction products, illustrating the increases in molar volume during pyrite oxidation reactions.
Light
Light absorbed by specimens may cause phase transitions as well as colour change or loss. Light can also activate or accelerate reactions with other decay agents (Nassau Citation1992; Child Citation1994a). Many photosensitive minerals only undergo colour change (Nassau Citation1992). A limited number of these display reversible colour change upon removal of the light source or through irradiation (Howie Citation1984; Nassau Citation1992; Horak Citation1994), as only the electronic states within these minerals have been altered. This is demonstrated in many coloured α-quartz (SiO2) varieties, including amethyst. Amethyst's purple colouration is attributed to ferric iron (Fe3+) which can either be a substitution for silicon or interstitially included in the quartz structure (Rossman Citation1994). When exposed to ionising radiation, Fe3+ is oxidised to Fe4+ and produces the violet colour (Rossman Citation1994). Amethyst’s colour may be lost upon heating (Nassau Citation1992; Rossman Citation1994) or exposure to ultraviolet radiation (Currier Citation1985; Kane Citation1985), as non-ionising radiation reduces the iron back to Fe3+. However, colour ‘may be restored by ionizing radiation, if the heating is not excessive’ (Rossman Citation1994, 442).
Light may induce chemical reactions—such as oxidation (Howie Citation1984) and elemental liberation (Howie Citation1992c; Nassau Citation1992)—that may result in the formation of a different mineral species and irreversible colour change. Photosensitivity can be inherent to the mineral (due to its structure and or chemical composition) or produced by impurities which often act as colourants. The degree of photosensitivity may also vary with the specimen’s origin (Nassau Citation1992), formation conditions, and associated mineralogy.
It is likely that many minerals are susceptible to only specific wavelengths within the visible, infrared, and ultraviolet regions of the electromagnetic spectrum, and that the reaction rates are intensity- and wavelength-dependent (Villmann and Weickhardt Citation2018). Research of realgar’s (As4S4) photo-induced alteration to pararealgar (As4S4) has evidenced this (Douglass, Shing, and Wang Citation1992; Kyono Citation2007; Jovanovski and Makreski Citation2020), but comparable research for other photosensitive minerals is lacking. Relatedly, there has been no investigation of how a photosensitive mineral’s reactions differ across types of lighting (e.g. sunlight, LED, halogen) and under varying illuminance.
Example: cinnabar (α-HgS)
Cinnabar is a notoriously photosensitive mineral, irreversibly blackening upon light exposure. Several localities have been identified that produce photosensitive specimens (Dreyer Citation1939; McCormack Citation2000). At these locations, cinnabar is associated with various mercury halide minerals (McCormack Citation2000; Neiman, Balonis, and Kakoulli Citation2015) and often contains trace amounts (∼1 wt%) of alkali halogens, usually chlorides (Dreyer Citation1939; McCormack Citation2000; Keune and Boon Citation2005). At these concentrations, halogens are integral catalysts for the photochemical redox reaction which produces the blackening (Keune and Boon Citation2005; Anaf, Janssens, and De Wael Citation2013; Neiman, Balonis, and Kakoulli Citation2015). Moisture also plays a key role in the reaction (Saunders and Kirby Citation2004; Neiman, Balonis, and Kakoulli Citation2015); increasing RH results in a greater colour change from red to black.
This surficial blackening of cinnabar () is attributed to the formation of colloidal metallic mercury nanoparticles (Dreyer Citation1939; Keune and Boon Citation2005; Anaf, Janssens, and De Wael Citation2013; Da Pieve et al. Citation2013; Neiman, Balonis, and Kakoulli Citation2015), rather than the long assumed metacinnabar (βHgS) (Gettens, Feller, and Chase Citation1972), calomel ([Hg2]2 + Cl2), or corderoite (Hg2+3S2Cl2) (Da Pieve et al. Citation2013; Neiman, Balonis, and Kakoulli Citation2015), although the latter two minerals are produced during the reaction process.
Figure 5. A specimen of cinnabar (HgS; OUNHM MIN.15474), displaying a dark, silvery appearance, attributable to the surficial formation of metallic mercury. Image used with permission of Oxford University Natural History Museum.

While the exact reaction pathway is still uncertain, the general pathway for mineral specimen blackening is believed to be as follows. Cinnabar first transforms into corderoite and or kenhsuite (both Hg2+3S2Cl2) upon exposure to light, moisture, and chloride ions (Keune and Boon Citation2005; Radepont et al. Citation2011). However, corderoite is structurally unstable when exposed to light and oxygen (Da Pieve et al. Citation2013), and will degrade into calomel, metallic mercury, and sulfur (Keune and Boon Citation2005). Kenhsuite, an even less stable polymorph of corderoite (Radepont et al. Citation2011), is also photosensitive and will likewise decompose; first structurally into corderoite and then into the subsequent products of calomel, metallic mercury, and sulfur. Calomel may be subsequently reduced (Neiman, Balonis, and Kakoulli Citation2015) to mercuric chloride and metallic mercury. Additional reactions may occur and produce several other chlorine- and sulfur-containing species (Keune and Boon Citation2005; Anaf, Janssens, and De Wael Citation2013), which may or may not contain mercury (Radepont et al. Citation2011).
Pollutants
Museum pollutants are unwanted gases or particulates that cause or accelerate deterioration. Indoor sources of pollutants include human activity, display and storage materials, and sometimes the collection objects themselves (Waller, Andrew, and Tétreault Citation2000; Eggert et al. Citation2004; Stanley Citation2004; Grzywacz Citation2006). Gaseous pollutants (such as carboxylic acids, sulfur dioxide, mercury vapour and hydrogen sulfide) may be more concentrated inside tightly sealed storage or display cabinets (Tétreault, Sirois, and Stamatopoulou Citation1998; Waller, Andrew, and Tétreault Citation2000; Schieweck et al. Citation2005).
Indoor pollutants are known to cause damage to limestone, metals, glass, and ceramics, but few studies have investigated the effects of indoor pollutants on geological collections. Significant sulfur gases include hydrogen sulfide (Howie Citation1979b), carbonyl sulfide, elemental sulfur, and sulfur dioxide (Waller, Andrew, and Tétreault Citation2000; Eggert et al. Citation2004). These reduced sulfur gases may be emitted from geological specimens or housing materials (Waller, Andrew, and Tétreault Citation2000; Eggert et al. Citation2004; Lussier and Smith Citation2007), and damage adjacent minerals, paper labels, trays, and even wooden drawers. For example, these gases produce copper sulfide and sulfate efflorescence on copper-containing materials () and cause pitting to the substrate (Eggert et al. Citation2004). The formation and composition of the efflorescence is dependent on the ambient temperature, RH, and pollutant type and concentration. Reduced sulfur gases also tarnish metallic minerals, such as those containing silver (Waller, Andrew, and Tétreault Citation2000).
Figure 6. A specimen labelled as domeykite (Cu3As; OUNHM MIN.27550), displaying patches of a dark navy-blue efflorescence and lines of a cocoa brown efflorescence, which have likely formed by the off-gassing of storage material or the deterioration of neighbouring specimens. Image used with permission of Oxford University Natural History Museum.

Carboxylic acids (formic and acetic acids, formaldehyde, and acetaldehyde) generally originate from wooden housing materials (e.g. oak, chestnut, cherry, MDF), adhesives, sealants, varnishes, plastics (Child Citation1994b; Grzywacz Citation2018; Gibson and Watt Citation2010), paper, and cardboard. These acids corrode limestone, egg and other calcareous shells, and metals including lead, copper, zinc, tin, and iron (Tétreault, Sirois, and Stamatopoulou Citation1998, Citation2003; Raychaudhuri and Brimblecombe Citation2000; Waller, Andrew, and Tétreault Citation2000). Reaction rates accelerate with increasing acid concentration and RH (Tétreault, Sirois, and Stamatopoulou Citation1998, Citation2003).
Example: calcite (CaCO3)
Calcareous material—including shell (Tennent and Baird Citation1985; Caracanhas Cavallari, Brincalepe Salvador, and Rodrigues da Cunha Citation2014), limestone, and marble (Stipp, Gutmannsbauer, and Lehmann Citation1996)—is particularly susceptible to acids. Frequently discussed in museum literature is Byne’s Disease, a disfiguring white or grey efflorescence occurring on shells and other calcareous material exposed to carboxylic acids (Tennent and Baird Citation1985; Caracanhas Cavallari, Brincalepe Salvador, and Rodrigues da Cunha Citation2014). Yet less discussed are the effects of sulfur dioxide and nitric acid on calcareous materials, even though they are equally damaging.
When calcite is exposed to ambient conditions, its surfaces react with atmospheric water to produce a hydrated layer of strongly chemisorbed hydroxyl (-OH) groups, altering the surface composition to Ca(OH)(CO3H) (Stipp, Gutmannsbauer, and Lehmann Citation1996; Al-Hosney and Grassian Citation2004, Citation2005; Usher, Baltrusaitis, and Grassian Citation2007). This hydrolysis species can even form at low (∼10%) RH (Al-Hosney et al. Citation2005), as the formation satisfies any dangling bonds that may be on the mineral surface (Stipp, Gutmannsbauer, and Lehmann Citation1996; Usher, Baltrusaitis, and Grassian Citation2007).
The Ca(OH)(CO3H) layer readily adsorbs water (Al-Hosney and Grassian Citation2005) and forms a water film as the RH increases. A monolayer is formed at 20% RH (Baltrusaitis and Grassian Citation2009). The multilayer gains a liquid-like structure above 50% RH (Al-Hosney and Grassian Citation2005) and bulk-like properties above 90% RH (Usher, Baltrusaitis, and Grassian Citation2007).
This adsorbed water plays a major role in the uptake of atmospheric pollutants (Al-Hosney and Grassian Citation2005). Surface reactivity and reaction rates are significantly enhanced by the presence of a water film (Krueger Citation2003; Al-Hosney and Grassian Citation2005; Al-Hosney et al. Citation2005; Prince et al. Citation2008; Baltrusaitis and Grassian Citation2009) and increase with increasing RH. Additionally, surface adsorbed water enhances further pollutant uptake on the mineral surface and aids in mobilising surface ions (Stipp, Gutmannsbauer, and Lehmann Citation1996; Tétreault, Sirois, and Stamatopoulou Citation1998; Al-Hosney and Grassian Citation2005; Prince et al. Citation2008). This increases the solution’s acidity and exposes fresh CaCO3 for reaction (Al-Hosney and Grassian Citation2005; Al-Hosney et al. Citation2005; Usher, Baltrusaitis, and Grassian Citation2007), allowing for greater CaCO3 dissolution. The reaction can then extend into the mineral bulk and produce pitting and etching (Chiarello, Wogelius, and Sturchio Citation1993; Usher, Baltrusaitis, and Grassian Citation2007).
Under ambient conditions, a pollutant species—such as sulfur dioxide, nitric acid, acetic acid, or formic acid—reacts with hydrated calcite surfaces to form the respective product through an ephemeral intermediate, carbonic acid (H2CO3) (Al-Hosney and Grassian Citation2004). Surface material is incorporated in the formation of the reaction product, resulting in significant erosion and disfigurement of surface features (Usher, Baltrusaitis, and Grassian Citation2007). Products may be amorphous or crystalline (Al-Hosney et al. Citation2005; Usher, Baltrusaitis, and Grassian Citation2007) and appear as patches or a contiguous layer. However, when exposed to nitric acid, an aqueous electrolyte ultimately forms on calcite surfaces, as calcium nitrate is unstable at ambient RH; its deliquescence point is between 7 and 18% RH (Al-Abadleh et al. Citation2003; Krueger Citation2003).
Challenges at large
The previous section illustrates that most information on mineral stability is generated by disciplines such as earth sciences, chemistry, and material science. However, much of this research in unavailable to museum professionals, both physically (due to the lack of open access publications) and verbally. For many museum professionals, literature from other fields may appear laden with specific and technical jargon, strange graphs, and terrifying equations. It is undeniable that these can overwhelm and confuse even the most scientifically inclined curators if they are unfamiliar with the subject being presented (Hoyles Citation2020). Thus, the lack of easily accessible and digestible information tailored for a layman’s understanding significantly hampers knowledge transfer into the museum sector.
Yet the largest obstruction of knowledge exchange comes from a lack of awareness and effective communication (Tennent Citation1994; Viñas Citation2002; Henderson Citation2018). Many museum professionals are unaware that relevant knowledge is available from other sectors. And many researchers from other sectors are equally unaware of their findings’ implications for museums and heritage (c.f. Emmons Citation1945, 88). Those who do acknowledge such uses fail to effectively communicate across disciplines, as their work is rarely advertised or published within museum literature.
The Mineral Susceptibility Database
An openly accessible database was created as part of this research project to address these challenges. The Mineral Susceptibility Database (MSD) aims to be a comprehensive reference for museum professionals – and a starting point for further research – when assessing the conditions required by their mineral collections and objects. It consolidates current relevant research from various fields (including museums, earth science, chemistry, and material science) into one freely accessible location. By being a repository of interdisciplinary research, the database:
encourages informed decision-making,
increases awareness of which disciplines and institutions are performing relevant research,
exposes additional research applications and opportunities, and
advocates cross-disciplinary research and communication.
The Database began as a project to collate the data from The Care and Conservation of Geological Material (Howie Citation1992a) into a single spreadsheet. The spreadsheet soon grew to include data from other publications which contained similar tables and data (Parsons Citation1922, Citation1926; Bannister Citation1937; King Citation1982, Citation1983, Citation1985; O’Donoghue Citation1983; Howie Citation1984; Hazen and Ausubel Citation2016). It was at this point that the authors decided that the database would be a useful resource to share with others.
It is anticipated that the Database will continue to grow by the addition of new scientific information from relevant journals and publications. Articles pertinent to mineral stability are reviewed for applicable data, which are then inserted into the database. While this may sound straightforward, the data review process is rather complex, laborious, and time consuming. Papers are progressively weeded out upon examining the abstract and experimental design. If the experiment was performed under atmospheric conditions, the rest of the paper is scrutinised, with key data, quotes, and reaction processes highlighted. More often than not, however, the reported data are not in a format compatible with the MSD. Sometimes relevant data are not stated in text, but in a table or a figure. Or the units require conversion. Thus, while the data presented in the MSD are replicated in good faith from trusted sources, we recommend returning to the original document and confirming parameters with the corresponding author.
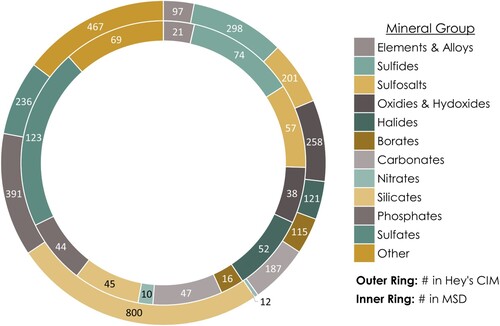
At the time of publication, the MSD contained 987 entries for 596 mineral species, which represented 17% of total species listed in the 3rd edition of Hey’s Chemical Index of Minerals (CIM) (), and 10% of total identified species. Additionally, the MSD included four ‘precious’ non-minerals (amber, pearl, obsidian, and coral), 18 minerals which are not Hey indexed (these are species which have been identified since the publication of the third edition of Hey's Chemical Index of Minerals in 1993 (Hey's CIM, see Clark (Citation1993)) and five discredited mineral species, as examples of the latter may still be present and catalogued in museum collections under their old, now discredited name.
Figure 7. A graphical representation of the distribution of minerals across major mineral groups. The outer ring displays the number of those included in the 3rd edition of Hey’s Chemical Index of Minerals (CIM, see Clark (Citation1993)), while the inner ring shows the number of those included in the Mineral Susceptibility Database (MSD). By comparing the two rings, one can see that certain mineral groups are better represented in the MSD than others.
A total of 67% of the database entries are water-related (), with the next largest susceptibility group being light at 14%. Water-related entries include minerals which are hygroscopic, water soluble, and susceptible to RH changes (e.g. hydration, deliquescence). The three predominant responses within the MSD entries are dehydration (17%), water-solubility (14%), and oxidation (exclusive of photo-oxidation: 14%; inclusive of photo-oxidation: 21%) (). These data are generally experimental, quantitative, fairly robust, and come from the geoscience and material science literature. Data for other reaction types are far fewer and generally more qualitative.
Figure 8. Distribution of susceptibility data entries within the MSD grouped by agent of change.
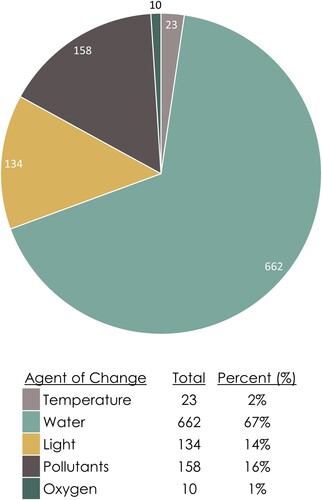
Figure 9. Distribution of susceptibility data entries within the MSD grouped by response to an agent of change.
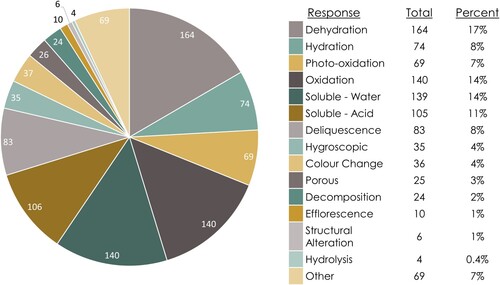
The overwhelming presence of hydration and oxidation reaction data shows that hydration and oxidation state changes are both common and important reactions that occur under atmospheric conditions.
The prevalence of hydration and oxidation reactions may be explained by:
Reaction types other than hydration and oxidation state changes are not as common or less important at atmospheric conditions.
It is assumed that these reactions are less common or important at atmospheric conditions.
It is assumed that the research is being performed but it is actually not.
The research is being performed but has not yet produced results or publications.
The research is being or has been performed but is inaccessible (for any number of reasons).
Researchers are just unaware of all the possible reactions any one mineral can undergo.
How to use and access the MSD
The MSD is comprised of three tables: Mineral Identification (ID), Susceptibility, and Solubility. These tables are organised using the Hey CIM groupings, rather than by structural groups (e.g. Dana or Strunz) or alphabetically, to emphasise how groups of chemically related minerals respond similarly to a given agent of change. Likewise, chemical formulas are included to relate chemistry to susceptibility, reaction types, and products. It is hoped that these choices will lead to a better understanding of mineral susceptibility and facilitate decision making in a museum collections context.
All three tables contain the International Mineralogical Association (IMA) approved name and formula, and the Hey Index number. The Mineral ID table contains relevant properties and information for each mineral in the MSD. This includes: Moh’s hardness, tenacity, health and safety data, other names (e.g. common names such as vermillion and fool’s gold), and notes. The notes column contains a variety of information such as: hygroscopicity, porosity, known polymorphs, magnetism, sensitivity to physical forces, and IMA changes in status and nomenclature.
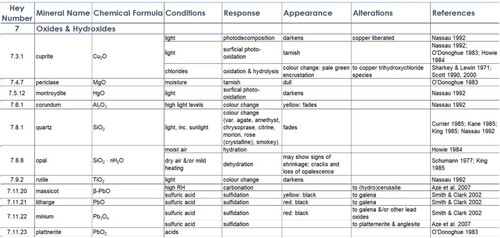
Susceptibility data are presented in the Susceptibility table (). Each entry is referenced by at least one source, and is organised into the following categories:
Environment: the conditions at which the mineral will change or react,
Response: the reaction process occurring at the stated conditions,
Appearance: how the mineral will visibly change, and
Alteration: how the mineral will chemically change.
Figure 10. An example of MSD Susceptibility entries for some oxide minerals.
Solubility data are similarly referenced and structured:
Chemical: the substance in which the mineral is soluble,
Degree of solubility
Alteration: the resultant products of dissolution.
The database is freely available online at ORA-Data (the University of Oxford’s data repository) and Reference for Mineral Care (this research project’s website; http://mineralcare.web.ox.ac.uk) as a pdf. The MSD is additionally supplemented by Mineral Spotlights at Reference for Mineral Care. These Spotlights are short, referenced articles written with the non-expert in mind, and expand upon some of the more complex and interesting mineral reactions listed in the MSD (e.g. tin, vivianite, and realgar).
Future work
Existing entries and references evidence current research hotspots (e.g. sulfates due to Martian research), and the institutions and individuals performing these studies. Empty and unquantified fields in entries, on the other hand, expose current knowledge gaps and provide research opportunities. Some additional data required includes:
fleshing out the reaction types, parameters, and products for current entries,
further references for pre-existing entries,
new entries, and
other organisational system numbers (e.g. (Nickel-) Strunz and Dana Classifications).
The latter will improve searchability for institutions using other organisational systems. We also hope to migrate the database to a more permanent, purpose-based platform to enhance usability, interactivity, and maintenance. Additional features could then be added, such as further general information available per mineral as well as pictures exemplifying deterioration.
Discussion
In this paper, we defined damage to facilitate decisions for extending collection lifetimes (c.f. Henderson Citation2018). In this case, we applied our definition to mineral collections, yet it may be equally applicable to other collection types.
Damage is dependent on both an object’s inherent susceptibility to its environment and its extrinsic, stakeholder-ascribed values and uses. Since environments, stakeholders, values, and uses are guaranteed to change over an object’s lifetime, we must accept that ‘damage’ – or rather the perception of a negative change – is unavoidable. We can, however, attempt to mitigate damage and extend an object’s useful life through conservation strategies aimed at slowing material change. What a useful life is considered to be – be it extended use, retained value, or material stability (Henderson Citation2018) – for any material inevitably means object preservation to some degree (be it physically or digitally), as an object begets the values and uses ascribed to it.
In order to improve preservation, one must first know what exactly is in the collection, the environment(s) they require, and current storage conditions. This may seem like an obvious and easy task to complete. However, ‘many professionals are locked in to monitoring and responding to RH change rather than any change in an object’s state’ (Henderson Citation2018, 34), and it is very possible that the state of the collection store is different than the numbers suggest. Things may be fine under fluctuating conditions (Henderson Citation2018), or perhaps the specimens have turned to powder in their boxes (), but the only way to know is to open the drawers and look at the objects themselves rather than the environmental conditions which are mere proxies for potential damage.
Figure 11. (a) A specimen of melanterite (FeSO4 · 7H2O; OUNHM MIN.26443) and (b) a specimen of epsomite (MgSO4 · 7H2O; NMW.26.151.GR–). Since accession, the specimens have fallen apart and produced powder due to dehydrating into lesser-hydrated sulfates (rozenite (FeSO4 · 4H2O) and hexahydrate (MgSO4 · 6H2O), respectively). Images used with permission of Oxford University Natural History Museum and National Museum Cardiff, respectively.

But are the objects indeed what we think they are? There are instances where an object is misidentified, sometimes to its detriment (Baars, Royce, and Cotterell Citation2021). As exemplified above in the section on how minerals change in a museum environment, storage requirements differ according to a material’s composition, and that ideal conditions may be at odds with those prevalent within an institution. Thus, it is crucial to correctly identify an object’s composition – ideally pre-accession – in order to determine appropriate conditions in which to store and display the object (Baars, Royce, and Cotterell Citation2021).
This paper does not suggest a correct answer nor the best choices to make when addressing mineral collections care—such as which specimens to prioritise—for we cannot know every possible context. However, what we do provide is the material information necessary to facilitate informed, evidence-based decisions. Additionally, we advocate for a stepwise evaluation of change before characterising it as damage (); beginning with an investigation of possible agents of change, followed by an objective assessment of an object’s changes in state, and then an appraisal of these changes in light of stakeholder values and uses. If the overall result is a sufficiently negative change, it is only then that the change to the object should be attributed as damage.
Figure 12. A schematic representation of our proposed, stepwise evaluation of damage. These four steps can be viewed as a simplification of the diagram presented at the beginning of this paper ().

Conclusions
We have defined damage to heritage items as ‘a material change which is perceived to have negatively affected an item’s value-defining aspects’. We also examined the terms ‘vulnerability’ and ‘susceptibility’ within a heritage context and applied them to mineral collections in order to characterise mineral instability. About 10% of known mineral species are susceptible to temperature, moisture, light, and pollutant levels common within museum environments, of which we have only listed a handful of examples. The full list of currently identified susceptible mineral species is provided in the Mineral Susceptibility Database (MSD). The MSD has been designed to facilitate decision-making during the development of preservation strategies through collating relevant information from various fields in a single accessible location. Through both this paper and the Database, we encourage an awareness of mineral susceptibility, informed decision making, and cross-disciplinary research and communication to improve the care of minerals and geological collections.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Akhavan, A.C., 2014. Overview of Silica Polymorphs. The Quartz Page. Accessed 25 October 2021. http://www.quartzpage.de/gen_mod.html.
- Al-Abadleh, H. A., B. J. Krueger, J. L. Ross, and V. H. Grassian. 2003. “Phase Transitions in Calcium Nitrate Thin Films.” Chemical Communications 3 (22): 2796–2797.
- Al-Hosney, H. A., S. Carlos-Cuellar, J. Baltrusaitis, and V. H. Grassian. 2005. “Heterogeneous Uptake and Reactivity of Formic Acid on Calcium Carbonate Particles: A Knudsen Cell Reactor, FTIR and SEM Study.” Physical Chemistry Chemical Physics 7 (20): 3587–3595.
- Al-Hosney, H. A., and V. H. Grassian. 2004. “Carbonic Acid: An Important Intermediate in the Surface Chemistry of Calcium Carbonate.” Journal of the American Chemical Society 126 (26): 8068–8069.
- Al-Hosney, H. A., and V. H. Grassian. 2005. “Water, Sulfur Dioxide and Nitric Acid Adsorption on Calcium Carbonate: A Transmission and ATR-FTIR Study.” Physical Chemistry Chemical Physics 7 (6): 1266–1276.
- Allmon, W. D. 1994. “The Value of Natural History Collections.” Curator: The Museum Journal 37 (2): 83–89.
- Alpers, C. N., D. K. Nordstrom, and J. W. Ball. 1989. “Solubility of Jarosite Solid Solutions Precipitated from Acid Mine Waters, Iron Mountain, California.” USA Sciences Geologiques - Bulletin 42: 281–298.
- Anaf, W., K. Janssens, and K. De Wael. 2013. “Formation of Metallic Mercury during Photodegradation/Photodarkening of α-HgS: Electrochemical Evidence.” Angewandte Chemie International Edition 52 (48): 12568–12571.
- Appelbaum, B. 2007. Conservation Treatment Methodology. Amsterdam: Butterworth-Heinemann.
- Ashley-Smith, J. 1995. Definitions of Damage. Annual Meeting of the Association of Art Historians. Accessed 25 October 2021. https://cool.conservation-us.org/byauth/ashley-smith/damage.html.
- Ashley-Smith, J. 1999. Risk Assessment for Object Conservation. Oxford: Butterworth-Heinemann.
- Baars, C. 2010. “Dare To Prepare? The Value of Preparing and Sampling Historically Important Museum Collections.” The Geological Curator 9 (4): 237–242.
- Baars, C., and J. Horak. 2018. “Storage and Conservation of Geological Collections—a Research Agenda.” Journal of the Institute of Conservation 41 (2): 154–168.
- Baars, C., Royce, K., and Cotterell, T. 2021. The Importance of Correct Mineral Identification for the Determination of Appropriate Specimen Storage Conditions in Geological Museum Collections. The Geological Curator, in the press.
- Baker, C. L., H. T. Woodward, and A. Pabst. 1933. “Four Crystalline Hydrates of Sodium Metasilicate.” American Mineralogist 18: 206–215.
- Baltrusaitis, J., and V. H. Grassian. 2009. “Calcite Surface in Humid Environments.” Surface Science 603 (17): L99–L104.
- Bannister, F. A. 1937. “The Preservation of Minerals and Meteorites.” The Museums Journal 36 (11): 465–476.
- Blount, A. M. 1993. “Nature of the Alterations Which Form on Pyrite and Marcasite During Collection Storage.” Collection Forum 9 (1): 1–16.
- Brunton, C. H. C., T. P. Besterman, and J. A. Cooper. 1984. Guidelines for the Curation of Geological Materials: Geological Society Miscellaneous Paper No. 17.
- BSI. 2012. PAS 198:2012 - Specification for managing environmental conditions for cultural collections. London: British Standards Institution.
- Caracanhas Cavallari, D., R. Brincalepe Salvador, and B. Rodrigues da Cunha. 2014. “Dangers to Malacological Collections: Bynesian Decay and Pyrite Decay.” Collection Forum 28 (1–2): 35–46.
- Chiantore, O., and M. Lazzari. 2001. “Photo-oxidative Stability of Paraloid Acrylic Protective Polymers.” Polymer 42: 17–27.
- Chiarello, R. P., R. A. Wogelius, and N. C. Sturchio. 1993. “In-situ Synchrotron X-ray Reflectivity Measurements at the Calcite-Water Interface.” Geochimica et Cosmochimica Acta 57 (16): 4103–4110.
- Child, R. E. 1994a. “The Effect of the Museum Environment on Geological Collections.” In Conservation of Geological Collections, edited by R. E. Child, 1–3. London: Archetype Publications.
- Child, R. E. 1994b. “Environmental Effects on Geological Material: Salt Efflorescence and Damage.” In Conservation of Geological Collections, edited by R. E. Child, 18–22. London: Archetype Publications.
- Clark, A. (ed.) 1993. Hey's Chemical Index of Minerals, 3rd edn. London: Chapman and Hall.
- Crawford, W. A., and A. L. Hoersch. 1972. “Calcite-aragonite Equilibrium from 50°C to 150°C.” American Mineralogist 57 (5–6): 995–998.
- Currier, R. H. 1985. “Natural Fading of Amethyst.” Gems & Gemology 21 (2): 115–116.
- Da Pieve, F., C. Hogan, D. Lamoen, J. Verbeeck, F. Vanmeert, M. Radepont, M. Cotte, K. Janssens, X. Gonze, and G. Van Tendeloo. 2013. “Casting Light on the Darkening of Colors in Historical Paintings.” Physical Review Letters 111: 208302. doi:10.1103/PhysRevLett.111.208302.
- de Benyacar, M. A. R., and M. E. J. de Abeledo. 1974. “Phase Transition in Synthetic Troegerite at Room Temperature.” American Mineralogist 59 (7–8): 763–767.
- del Hoyo-Meléndez, J. M., M. F. Mecklenburg, and M. Teresa Doménech-Carbó. 2011. “Determination of the Annual Light Exposure Received by Two-Dimensional Museum Objects Displayed on Vertical Surfaces Using Photometric Measurements.” Studies in Conservation 56 (1): 31–40.
- Dos Santos, E. C., J. C. de Mendonça Silva, and H. A. Duarte. 2016. “Pyrite Oxidation Mechanism by Oxygen in Aqueous Medium.” The Journal of Physical Chemistry C 120 (5): 2760–2768.
- Douglass, D. L., Chichang Shing, and Ge Wang. 1992. “The Light-Induced Alteration of Realgar to Pararealgar.” American Mineralogist 77 (11–12): 1266–1274.
- Dreyer, R. M. 1939. “Darkening of Cinnabar in Sunlight.” American Mineralogist 24: 457–460.
- Eggert, G., M. Weichert, H. Euler, and B. Barbier. 2004. Some news about ‘black spots’. In: Metal 04: proceedings of the International Conference on Metals Conservation, Canberra, Australia, 4–8 October 2004, 142–148.
- Eggleston, C. M., J. J. Ehrhardt, and W. Stumm. 1996. “Surface Structural Controls on Pyrite Oxidation Kinetics; an XPS-UPS, STM, and Modeling Study.” American Mineralogist 81 (9–10): 1036–1056.
- Emmons, R. C. 1945. “A Mineralogist’s Obligation.” American Mineralogist 30 (3–4): 87–96.
- Erhardt, D., and M. Mecklenburg. 1994. “Relative Humidity re-Examined.” Studies in Conservation 39 (sup. 2): 32–38.
- Feldman, S. R. 2000. Sodium Chloride. In Kirk-Othmer Encyclopedia of Chemical Technology. doi:10.1002/0471238961.1915040902051820.a01.pub3.
- Fenlon, A., and L. Petrera. 2019. “Pyrite Oxidation: A History of Treatments at the Natural History Museum, London.” The Geological Curator 11 (1): 9–18.
- Gettens, R. J., R. L. Feller, and W. T. Chase. 1972. “Vermilion and Cinnabar.” Studies in Conservation 17: 45–69.
- Gibson, L. T., and C. M. Watt. 2010. “Acetic and Formic Acids Emitted from Wood Samples and Their Effect on Selected Materials in Museum Environments.” Corrosion Science 52 (1): 172–178. doi:10.1016/j.corsci.2009.08.054.
- Gioventù, E. 2018. “Comparison of Three Methods for Evaluating the Condition of the Collection in the Royal Apartments of the Pitti Palace, Florence.” Studies in Conservation 63 (sup. 1): 355–358.
- Graham, F. 2018. Caring for natural history collections - Preventive conservation guidelines for collections. Accessed 25 October 2021. https://www.canada.ca/en/conservation-institute/services/preventive-conservation/guidelines-collections/natural-history.html.
- Grøntoft, T., D. Thickett, P. Lankester, S. Hackney, J. H. Townsend, K. Ramsholt, and M. Garrido. 2016. “Assessment of Indoor air Quality and the Risk of Damage to Cultural Heritage Objects Using MEMORI®Dosimetry.” Studies in Conservation 61 (sup. 1): 70–82.
- Grzywacz, C. M. 2006. Monitoring for Gaseous Pollutants in Museum Environments. Los Angles: Getty Publications.
- Grzywacz, C. M. 2018. Detection and mitigation of museum pollutants: An update. Accessed 25 October 2021. https://resources.culturalheritage.org/osg-postprints/1992-2/gryzwacz/.
- Guevremont, J. M., D. R. Strongin, and M. A. A. Schoonen. 1998a. “Thermal Chemistry of H2S and H2O on the (100) Plane of Pyrite; Unique Reactivity of Defect Sites.” American Mineralogist 83 (11–12): 1246–1255.
- Guevremont, J. M., A. R. Elsetinow, D. R. Strongin, J. Bebie, and M. A. A. Schoonen. 1998b. “Structure Sensitivity of Pyrite Oxidation; Comparison of the (100) and (111) Planes.” American Mineralogist 83 (11–12): 1353–1356.
- Hackney, S. 2016. “Colour Measurement of Acid-Detector Strips for the Quantification of Volatile Organic Acids in Storage Conditions.” Studies in Conservation 61: 55–69.
- Hazen, R. M., and J. H. Ausubel. 2016. “On the Nature and Significance of Rarity in Mineralogy.” American Mineralogist 101 (6): 1245–1251.
- Heaney, P. J. 1994. “Structure and Chemistry of the Low-Pressure Silica Polymorphs.” In Silica: Physical Behavior, Geochemistry and Materials Applications, edited by P. J. Heaney, C. T. Prewitt, and G. V. Gibbs, 1–40. Washington D.C.: Mineralogical Society of America.
- Heaney, P. J., and D. R. Veblen. 1991. “Observations of the α-β Phase Transition in Quartz: A Review of Imaging and Diffraction Studies and Some new Results.” American Mineralogist 76 (5–6): 1018–1032.
- Henderson, J. 2018. “Reflections on the Psychological Basis for Suboptimal Environmental Practices in Conservation.” Journal of the Institute of Conservation 41 (1): 32–45.
- Horak, J. M. 1994. “Environmental Effects on Geological Material: Light Induced Changes of Minerals.” In Conservation of Geological Collections, edited by R. E. Child, 23–30. London: Archetype Publications.
- Howie, F. M. P. 1979a. “Museum Climatology and the Conservation of Palaeontological Material.” Special Papers in Palaeontology 22: 193–125.
- Howie, F. M. P. 1979b. “Physical Conservation of Fossils in Existing Collections.” The Geological Curator 2 (5): 269–280.
- Howie, F. M. P. 1984. “Conservation and Storage: Geological Material.” In Manual of Curatorship: A Guide to Museum Practice, edited by J. M. A. Thompson, 308–317. London: Butterworths: Museums Association.
- Howie, F. M. P. 1992a. The Care and Conservation of Geological Material: Minerals, Rocks, Meteorites, and Lunar Finds. Oxford: Butterworth-Heinemann.
- Howie, F. M. P. 1992b. “Pyrite and Marcasite.” In The Care and Conservation of Geological Materials: Minerals, Rocks, Meteorites and Lunar Finds, edited by F. M. Howie, 70–84. Oxford: Butterworth- Heinemann.
- Howie, F. M. P. 1992c. “Sulphides and Allied Minerals in Collections.” In The Care and Conservation of Geological Materials: Minerals, Rocks, Meteorites and Lunar Finds, edited by F. M. Howie, 56–69. Oxford: Butterworth-Heinemann.
- Hoyles, C. 2020. “I Can’t do Maths.” UCL Portico Magazine 7: 48–49.
- Hudson Institute of Mineralogy. 2021. Quartz-beta In: mindat.org. Accessed 25 October 2021. https://www.mindat.org/min-7395.html.
- International Mineralogical Association. 2021. The New IMA List of Minerals – A Work in Progress. Accessed 25 October 2021. http://cnmnc.main.jp.
- Irving, J., and P. Hadland. 2019. “Anoxic Storage for ‘Pyrite Decay’ at Oxford University Museum of Natural History - An Exercise in Cost-Efficiency as Well as Long-Term Preservation.” The Geological Curator 11 (1): 39–54.
- Jambor, J. L., D. K. Nordstrom, and C. N. Alpers. 2000. “Metal-sulfate Salts from Sulfide Mineral Oxidation.” In Sulfate Minerals: Crystallography, Geochemistry, and Environmental Significance, edited by C. N. Alpers, J. L. Jambor, and D. K. Nordstrom, 303–350. Washington, D.C.: Mineralogical Society of America.
- Jerz, J. K., and J. D. Rimstidt. 2004. “Pyrite Oxidation in Moist air.” Geochimica et Cosmochimica Acta 68 (4): 701–714.
- Jovanovski, G., and P. Makreski. 2020. “Intriguing Minerals: Photoinduced Solid-State Transition of Realgar to Pararealgar—Direct Atomic Scale Observation and Visualization.” ChemTexts 6 (1): 1–14.
- Kane, R. E. 1985. “Gem Trade Lab Notes.” Gems & Gemology 21 (1): 43–48.
- Kerbey, H. C., and J. M. Horak. 2006. “Maintaining Standards in the Care of Petrology and Mineralogy Collections at the National Museum Wales.” Collection Forum 21: 42–57.
- Keune, K., and J. J. Boon. 2005. “Analytical Imaging Studies Clarifying the Process of the Darkening of Vermilion in Paintings.” Analytical Chemistry 77 (15): 4742–4750.
- King, R. J. 1982. “The Care of Minerals, Section 1: The Cleaning of Minerals.” Journal of the Russell Society 1 (1): 42–53.
- King, R. J. 1983. “The Care of Minerals, Section 2: The Development of Minerals.” Journal of the Russell Society 1 (2): 54–77.
- King, R. J. 1985. “The Care of Minerals, Section 3A: The Curation of Minerals.” Journal of the Russell Society 1 (3): 94–114.
- King, R. J. 1986. “The Care of Minerals, Section 3B: The Curation of Minerals.” Journal of the Russell Society 1 (4): 129–151.
- Kosek, J., and C. Barry. 2019. “Investigating the Condition of Iron Gall ink Drawings: Developing an Assessment Survey.” Journal of the Institute of Conservation 42 (3): 191–209.
- Krueger, B. J. 2003. “The Transformation of Solid Atmospheric Particles Into Liquid Droplets Through Heterogeneous Chemistry: Laboratory Insights Into the Processing of Calcium Containing Mineral Dust Aerosol in the Troposphere.” Geophysical Research Letters 30 (3): 3–6.
- Kullerud, G., and H. S. Yoder. 1959. “Pyrite Stability Relations in the Fe-S System.” Economic Geology 54 (5): 533–572.
- Kyono, A. 2007. “Experimental Study of the Effect of Light Intensity on Arsenic Sulfide (As4S4) Alteration.” Journal of Photochemistry and Photobiology A: Chemistry 189 (1): 15–22.
- Larkin, N. R. 2011. “Pyrite Decay: Cause and Effect, Prevention and Cure. NatSCA News, 21, 35–43. Lowenthal, D., 1994. The Value of Age and Decay.” In Durability and Change: The Science, Responsibility, and Cost of Sustaining Cultural Heritage, edited by W. E. Krumbein, P. Brimblecombe, D. E. Cosgrove, and S. Staniforth, 39–50. Chichester: Wiley and Sons.
- Lowson, R. T. 1982. “Aqueous Oxidation of Pyrite by Molecular Oxygen.” Chemical Reviews 82 (5): 461–497.
- Lussier, S. M., and G. D. Smith. 2007. “A Review of the Phenomenon of Lead White Darkening and its Conversion Treatment.” Studies in Conservation 52 (sup. 1): 41–53.
- McCormack, J. K. 2000. “The Darkening of Cinnabar in Sunlight.” Mineralium Deposita 35 (8): 796–798.
- Meul, V. 2008. “Safeguarding the Significance of Ensembles: Value Assessments in Risk Management for Cultural Heritage.” In ICOM Committee for Conservation 15th Triennial Conference New Delhi, Preprints, edited by J. Bridgland, 1048–1055. Allied Publishers Pvt. Ltd.
- Michalski, S. 1994. “A Systematic Approach to Preservation: Description and Integration with Other Museum Activities.” Studies in Conservation 39 (sup. 2): 8–11.
- Miles, K. 2019. “The Role of Pyrite in Fossilisation and its Potential for Instability.” The Geological Curator 11 (1): 3–8.
- Muñoz-Viñas, S. 2002. “Contemporary Theory of Conservation.” Studies in Conservation 47 (Sup. 1): 25–34.
- Nassau, K. 1992. “Conserving Light Sensitive Minerals and Gems.” In The Care and Conservation of Geological Materials: Minerals, Rocks, Meteorites and Lunar Finds, edited by F. M. Howie, 11–24. Oxford: Butterworth-Heinemann.
- Neiman, M. K., M. Balonis, and I. Kakoulli. 2015. “Cinnabar Alteration in Archaeological Wall Paintings: An Experimental and Theoretical Approach.” Applied Physics A: Materials Science and Processing 121 (3): 915–938.
- Nesse, W. D. 2012. Introduction to Mineralogy. 2nd ed. Oxford: Oxford University Press.
- Odin, G. P., V. Rouchon, O. Béthoux, and D. Ren. 2018. “Gypsum Growth Induced by Pyrite Oxidation Jeopardises the Conservation of Fossil Specimens: An Example from the Xiaheyan entomofauna (Late Carboniferous, China).” Palaeogeography, Palaeoclimatology, Palaeoecology 507: 15–29.
- Odin, G. P., F. Vanmeert, K. Janssens, H. Lelièvre, J. D. Mertz, and V. Rouchon. 2014. “Accelerated Ageing of Shales of palaeontological Interest: Impact of Temperature Conditions.” Annales de Paleontologie 100 (2): 137–149.
- Odlyha, M., S. Jakiela, C. J. Bergsten, J. M. Slater, A. Niklasson, J.-E. Svensson, A. Cavicchioli, et al. 2011. Dosimetry for monitoring in organ pipes and in microclimate frames for paintings. In: Metal 2010: proceedings of the interim meeting of the ICOM-CC Metal Working Group, October 11–15, 2010, Charleston, South Carolina, USA, 430–436.
- O’Donoghue, M. 1983. The Encyclopedia of Minerals and Gemstones. 2nd ed. New York: Crescent Books.
- Ojima, K., Y. Taneda, and A. Takasaki. 1993. “Direct Observation of α → β Transformation in tin by Transmission Electron Microscopy.” Physica Status Solidi (A) 139 (1): 139–144.
- Ortiz, R., and P. Ortiz. 2016. “Vulnerability Index: A New Approach for Preventive Conservation of Monuments.” International Journal of Architectural Heritage 10 (8): 1078–1100.
- Oxford University Press. 2021a. damage. In: Lexico.com. Accessed 25 October 2021. https://www.lexico.com/definition/damage.
- Oxford University Press. 2021b. vulnerability. In: Lexico.com. Accessed 25 October 2021. https://www.lexico.com/definition/vulnerability.
- Oxford University Press. 2021c. susceptibility. In: Lexico.com. Accessed 25 October 2021. https://www.lexico.com/definition/susceptibility.
- Parsons, A. L. 1922. “The Preservation of Mineral Specimens.” American Mineralogist 7 (4): 59–63.
- Parsons, A. L. 1926. “Additional Data Concerning the Preservation of Minerals.” American Mineralogist 11 (4): 79–82.
- Prince, A. P., P. D. Kleiber, V. H. Grassian, and M. A. Young. 2008. “Reactive Uptake of Acetic Acid on Calcite and Nitric Acid Reacted Calcite Aerosol in an Environmental Reaction Chamber.” Physical Chemistry Chemical Physics 10 (1): 142–152.
- Putnis, A. 1992. Introduction to Mineral Sciences. 5th ed. Cambridge: Cambridge University Press.
- Radepont, M., W. De Nolf, K. Janssens, G. Van Der Snickt, Y. Coquinot, L. Klaassen, and M. Cotte. 2011. “The use of Microscopic X-ray Diffraction for the Study of HgS and its Degradation Products Corderoite (α-Hg3S2Cl2), Kenhsuite (γ-Hg3S2Cl2) and Calomel (Hg2Cl2) in Historical Paintings.” Journal of Analytical Atomic Spectrometry 26 (5): 959–968.
- Raychaudhuri, M. R., and P. Brimblecombe. 2000. “Formaldehyde Oxidation and Lead Corrosion.” Studies in Conservation 45 (4): 226–232.
- Rickard, D., and G. W. Luther. 2006. “Metal Sulfide Complexes and Clusters.” In Sulfide Mineralogy and Geochemistry, edited by D. J. Vaughan, 421–504. Washington D.C.: Mineralogical Society of America.
- Rimstidt, J. D., and D. J. Vaughan. 2003. “Pyrite Oxidation: A State-of-the-art Assessment of the Reaction Mechanism.” Geochimica et Cosmochimica Acta 67 (5): 873–880.
- Robb, J., C. Dillon, M. Rumsey, and M. Strlič. 2013. “Quantitative Assessment of Perceived Value of Geological Collections by ‘Experts’ for Improved Collections Management.” The Geological Curator 9 (10): 529–543.
- Robie, R. A., and B. S. Hemingway. 1972. “The Heat Capacities at low-Temperatures and Entropies at 298.15 K of Nesquehonite, MgCO3·3H2O, and Hydromagnesite.” American Mineralogist 57: 1768–1781.
- Robie, R. A., and B. S. Hemingway. 1995. “Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 bar (10^5 Pascals) Pressure and at Higher Temperatures.” Geological Survey Bulletin. doi:10.3133/b2131.
- Rose, C. L., and C. A. Hawks. 1995. “A Preventive Conservation Approach to the Storage of Collections.” In Storage of Natural History Collections: A Preventive Conservation Approach, edited by C. L. Rose, C. A. Hawks, and H. H. Genoways, 1–20. Society for the Preservation of Natural History Collections.
- Rossman, G. R. 1994. “Colored Varieties of the Silica Minerals.” In Silica: Physical Behavior, Geochemistry, and Materials Applications, edited by P. J. Heaney, C. T. Prewitt, and G. V. Gibbs, 433–468. Washington D.C.: Mineralogical Society of America.
- Rosso, K. M., U. Becker, and M. F. Hochella. 1999. “The Interaction of Pyrite {100} Surfaces with O2and H2O; Fundamental Oxidation Mechanisms.” American Mineralogist 84 (10): 1549–1561.
- Rouchon, V., H. Badet, O. Belhadj, O. Bonnerot, B. Lavódrine, J. G. Michard, and S. Miska. 2012. “Raman and FTIR Spectroscopy Applied to the Conservation Report of Paleontological Collections: Identification of Raman and FTIR Signatures of Several Iron Sulfate Species Such as Ferrinatrite and Sideronatrite.” Journal of Raman Spectroscopy 43 (9): 1265–1274.
- Royce, K. 2019. An investigation of analytical methods applied to mineralogical collection assessment. MRes. Thesis. University College London. [unpublished].
- Royce, K. 2021. The Mineral Susceptibility Database. Reference for Mineral Care. Accessed 25 October 2021. https://mineralcare.web.ox.ac.uk/database.
- Sabbioni, C., M. Cassar, P. Brimblecombe, and R. A. Lefevre. 2009. “Vulnerability of Cultural Heritage to Climate Change.” Pollution Atmospherique 202: 157–169.
- Saunders, D., and J. Kirby. 2004. “The Effect of Relative Humidity on Artists’ Pigments.” National Gallery Technical Bulletin 25: 62–72.
- Schieweck, A., B. Lohrengel, N. Siwinski, C. Genning, and Tunga Salthammer. 2005. “Organic and Inorganic Pollutants in Storage Rooms of the Lower Saxony State Museum Hanover, Germany.” Atmospheric Environment 39: 6098–6108. doi:10.1016/j.atmosenv.2005.06.047.
- Stanley, M. 2004. Standards in the Museum Care of Geological Collections 2004. Museums, Libraries and Archives Council (MLA).
- Stipp, S. L. S., W. Gutmannsbauer, and T. Lehmann. 1996. “The Dynamic Nature of Calcite Surfaces in air.” American Mineralogist 81: 1–8.
- Strlič, M., D. Thickett, J. Taylor, and M. Cassar. 2013. “Damage Functions in Heritage Science.” Studies in Conservation 58 (2): 80–87.
- Taylor, J. 2013. “Causes and Extent of Variation in Collection Condition Survey Data.” Studies in Conservation 58 (2): 95–106.
- Taylor, J., and S. Stevenson. 1999. “Investigating Subjectivity Within Collection Condition Surveys.” Museum Management and Curatorship 18 (1): 19–42.
- Tennent, N. H. 1994. “The Role of the Conservation Scientist in Enhancing the Practice of Preventive Conservation and the Conservation Treatment of Artifacts.” In Durability and Change: The Science, Responsibility, and Cost of Sustaining Cultural Heritage, edited by W. E. Krumbein, P. Brimblecombe, D. E. Cosgrove, and S. Staniforth, 165–172. Chichester: John Wiley & Sons, Inc.
- Tennent, N. H., and T. Baird. 1985. “The Deterioration of Mollusca Collections: Identification of Shell Efflorescence.” Studies in Conservation 30 (2): 73–85.
- Tétreault, J., E. Cano, M. van Bommel, D. A. Scott, M. Dennis, M.-G. Barthés-Labrousse, L. Minel, and L. Robbiola. 2003. “Corrosion of Copper and Lead by Formaldehyde, Formic and Acetic Acid Vapours.” Studies in Conservation 48 (4): 237–250.
- Tétreault, J., J. Sirois, and E. Stamatopoulou. 1998. “Studies of Lead Corrosion in Acetic Acid Environments.” Studies in Conservation 43: 17–32.
- Torrens, H. S. 1977. “Note on Edward Jacob.” Newsletter of the Geological Curators Group (‘The Geological Curator’) 1 (9), inside front cover.
- Usher, C. R., J. Baltrusaitis, and V. H. Grassian. 2007. “Spatially Resolved Product Formation in the Reaction of Formic Acid with Calcium Carbonate (101̄4): The Role of Step Density and Adsorbed Water-Assisted Ion Mobility.” Langmuir 23 (13): 7039–7045.
- Usher, C. R., C. A. Cleveland, D. R. Strongin, and M. A. Schoonen. 2004. “Origin of Oxygen in Sulfate during Pyrite Oxidation with Water and Dissolved Oxygen: An in Situ Horizontal Attenuated Total Reflectance Infrared Spectroscopy Isotope Study.” Environmental Science & Technology 38 (21): 5604–5606. doi:10.1021/es0494003.
- Villmann, B., and C. Weickhardt. 2018. “Wavelength Dependence of Light Induced Changes in Reflectance Spectra of Selected Dyes and Pigments.” Studies in Conservation 63 (2): 104–112. doi:10.1080/00393630.2017.1345088.
- Waller, R. 1991. “Correct use of Terms Stable, Metastable, and Unstable.” The Geological Curator 5 (6): 245–246.
- Waller, R. 1992. “Temperature- and Humidity-Sensitive Mineralogical and Petrological Specimens.” In The Care and Conservation of Geological Materials: Minerals, Rocks, Meteorites and Lunar Finds, edited by F. M. Howie, 25–50. Oxford: Butterworth-Heinemann.
- Waller, R. 1995. “Risk Management Applied to Preventive Conservation.” In Storage of Natural History Collections: A Preventive Conservation Approach, edited by C. L. Rose, C. A. Hawks, and H. H. Genoways, 21–28. Society for the Preservation of Natural History Collections.
- Waller, R., K. J. Andrew, and J. Tétreault. 2000. “Survey of Gaseous Pollutant Concentration Distributions in Mineral Collections.” Collection Forum 14 (1–2): 1–32.
- Wiese, R. G., M. A. Powell, and W. S. Fyfe. 1987. “Spontaneous Formation of Hydrated Iron Sulfates on Laboratory Samples of Pyrite- and Marcasite-Bearing Coals.” Chemical Geology 63 (1–2): 29–38.
- Yalcin, S. E., B. A. Legg, M. Yeşilbaş, N. S. Malvankar, and J. F. Boily. 2020. “Direct Observation of Anisotropic Growth of Water Films on Minerals Driven by Defects and Surface Tension.” Science Advances 6 (30): 1–10.
- Zeng, G., McDonald, S.D., Gu, Q.F., Sweatman, K., and Nogita, K., 2014. Effects of Element Addition on the β→α Transformation in tin. Philosophical Magazine Letters, 94 (2), 53–62.