Abstract
Al2O3/CH3SO3H (AMA) is an efficient catalyst for the three-component condensation reaction of aldehyde, 1,3-dicarbonyl compound, and urea or thiourea to afford the corresponding 3,4-dihydropyrimidin-2-(1H)-ones in high isolated yield via this procedure, which works very effectively regardless of the electronic nature of the substituent on the ring, although electron-donating groups precipitate the rate of reaction. The catalyst is recyclable and stable at room temperature, and the reaction protocol is simple, is cost-effective, and gives good isolated yield with high purity.
INTRODUCTION
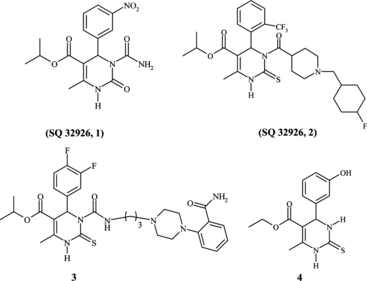
Multicomponent reactions (MCRs) occupy an outstanding position in organic and medicinal chemistry for their high degree of atom economy, applications in combinatorial chemistry, and diversity-oriented synthesis.[ Citation 1 ] The Biginelli reaction,[ Citation 2 ] one of the most useful multicomponent reactions, offers an efficient way to access multifunctionalized 3,4-dihydropyrimidin-2-(1H)-ones (DHPMs) and related heterocyclic compounds.[ Citation 3 ] Such heterocycles (Scheme ) show a wide scope of pharmacological properties including antiviral, antitumor, antibacterial, and anti-inflammatory activities.[ Citation 4 ] Recently, appropriately functionalized DHPM analogs have emerged as orally active antihypertensive agents (1,2)[ Citation 5 ] and α 1a adrenoceptor-selective antagonists (3).[ Citation 6 ] Another highlight in this context has been the identification of the structurally rather simple DHPM monastrol (4) as a novel cell-permeable molecule that blocks normal bipolar spindle assembly in mammalian cells, causing cell cycle arrest.[ Citation 7 ]
Scheme 1 Four of these related heterocyclic compounds 3,4-dihydropyrimidin-2-(1H)-ones (DHPMs) that have pharmacological properties are illustrated above.
Thus the synthesis of these heterocyclic compounds is of much current importance. The search for more suitable preparation of dihydropyrimidinones continues today. The first protocol to prepare the compounds of this type was presented by Biginelli in 1893 and involved a three-component, one-pot condensation.[ Citation 2 ] A major drawback to Biginelli's original reactions, however, was poor to moderate yields.[ Citation 8 ] Recently, many improved procedures have been reported using InBr3,[ Citation 9 ] InCl3,[ Citation 10 ] LiClO4,[ Citation 11 ] FeCl3 · 6H2O or NiCl2 · 6H2O,[ Citation 12 ] p-TsOH,[ Citation 13 ] LaCl3 · 7H2O,[ Citation 14 ]Bi(OTf)3,[ Citation 15 ] La(OTf)3,[ Citation 16 ] BF3 · OEt2,[ Citation 17 ] ionic liquids (BMIm · PF6 andBMIm · BF4),[ Citation 18 ] natural HEU type zeolite, [ Citation 19 ] I2,[ Citation 20 ] N-bromosuccinimide (NBS),[ Citation 21 ] polyaniline–bismoclite complex [ Citation 22 ] and other Lewis acids,[ Citation 23 ] heteropoly acid,[ Citation 24 ] sulfated zirconia,[ Citation 25 ] Sr(NO3)2,[ Citation 26 ] and covalently anchored sulfonic acid onto silica.[ Citation 27 ] However, some of the newer reported methods also suffer from drawbacks such as unsatisfactory yields, cumbersome product isolation procedures, and environmental pollution.[ Citation 9 , Citation 10 , Citation 14 , Citation 16 , Citation 17 ] Moreover, the main disadvantage of almost all existing methods is that the catalysts are destroyed in the workup procedure and cannot be recovered or reused. Therefore, still there is need for versatile, simple, and environmentally friendly processes whereby DHPMs may be formed under milder and practical conditions.
Solid-supported reagents are unique acid catalysts that have become popular over the past two decades. The activity and selectivity of a reagent dispersed on the surface of a support are improved as the effective surface area of the reagent is increased significantly, and hence they are expected to perform more effectively than the individual reagents.[ Citation 28 ]
Low toxicity, air tolerance, low prices, and moisture resistance are other common features that cause the use of solid-supported reagents to be more attractive than alternatives of conventional Lewis acids or metal triflates.
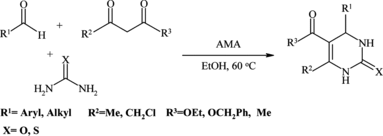
In our last work, it was reported that a mixture of Al2O3/CH3SO3H (AMA) is an inexpensive and effective reagent for Fries rearrangement,[ Citation 29 ] Beckmann rearrangements,[ Citation 30 ] direct conversions of aromatic aldehydes to the corresponding glycol monoesters,[ Citation 31 ] hydration of nitriles into amides,[ Citation 32 ] syntheses of macrocyclic polyether-diesters,[ Citation 33 ] syntheses of new hydroxythioxanthone derivatives,[ Citation 34 ] direct sulfonyltion of phenloes with p-toluensulfonic acid,[ Citation 35 ] and synthesis of coumarin derivatives,[ Citation 36 ] in excellent yields and with high selectivity. Here we report the ability of this reagent as reusable catalyst for the one-pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones and thiones in high yields (Scheme ).
Scheme 2 One-pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones and thiones using Al2O3/CH3SO3H (AMA).
RESULTS AND DISCUSSION
To exploit simple and suitable conditions for synthesis of 3,4-dihydropyrimidin-2-(1H)-ones, the reaction of benzaldehyde 5a, urea 6a, and ethyl acetoacetate 7a was chosen as a model to afford the DHPM 8a (R1 = Ph, R2 = Me, R3 = OEt, X = O), and its behavior was studied under a variety of conditions via thin-layer chromatography (TLC) and 1H NMR and 13C NMR spectroscopy (Table ).
Table 1. Reaction of benzaldehyde (1 mmol), ethyl acetoacetate (1 mmol), and urea (1.2 mmol) under various reaction conditions
A summary of obtained results is provided in Table . At room temperature, the reaction rate was found to be slow and was increased with increase in temperature. At 60 °C, the reaction rate was found to be maximal, and further increase in temperature did not show any enhancement (Table , entries 6 and 7). Entries 1–6 show the effect of various solvents on the yield of reaction. Although toluene, acetonitrile, and dimethyl formamide afforded the product in high yields, we chose ethanol for its low cost and environmental acceptability. Our results show that the reaction does not proceed if no catalyst is employed (Table , entry 8), whereas the yield of Biginelli product is increased to 98% with the addition of AMA (0.1 g, equal to 0.5 mmol H+). Entry 6 described the yields of five consecutive condensations leading to 8a. In these experiments, the product was isolated by filtration and washing the solid residues with ethyl acetate. Thus the remaining catalyst, which always works the same, begins reloading with fresh reagents for further runs. We did not observe any large decrease in the yield, demonstrating the efficiency of alumina methansulfonic acid (AMA) as a catalyst in Biginelli condensations. Therefore, the solid insoluble MeSO3H/Al2O3 is a truly heterogeneous efficient catalyst for this transformation.
The previously mentioned results show the advantages of this method as a new and more suitable way to DHPM synthesis.
Results of the Biginelli reaction catalyzed by AMA are presented in Table . Alumina methansulfonic acid (AMA) works for the condensation of a series of aldehydes, 1,3-dicarbonyl compounds, and urea or thiourea. It is an environmentally, friendly catalyst that works at 60 °deg;C in short reaction times (20 min-3 h). This catalyst works regardless of structural variations in the aldehydes or β-ketoesters.
Table 2. Synthesis of dihydropyrimidin-ones and -thiones (DHPMs) by the condensation of aldehydes, β-dicarbonyls, and urea or thiourea catalyzed by Al2O3/CH3SO3H (AMA) as a recyclable catalyst in ethanol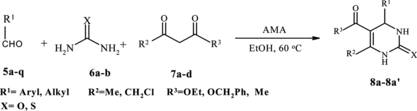
Besides, the β-ketoester, β-diketone (Table , entries 14, 15, 16, and 21) can also be employed without any decrease in yields. Under these conditions, the yields were significantly better in comparison with classical Biginelli procedure. Thus, several pharmacologically relevant substituent patterns could be introduced with high efficiency under the present conditions.
A variety of heterocyclic, aliphatic, and aromatic aldehydes were reacted with urea and β-dicarbonyl compound to afford 3,4-dihydropyrimidin-2-(1H)-ones in high yields using catalytic amounts of AMA (0.1 g, equal to 0.5 mmol H+) under similar reaction conditions.
Various types of substituted benzaldehydes containing either electron-withdrawing or electron-donating substitutions successfully afforded the Biginelli products in high yields (Table , entries 1–28). An important feature of this procedure is the survival of a variety of functional groups such as ethers, nitro, hydroxyl, halides, cyanide, etc. under the reaction conditions. Acid-sensitive substrates such as 4-cyano benzaldehyde are also reacted in high yields without the formation of any side products (Table , entry 11).
Although aromatic aldehydes having either electron-donating or electron-withdrawing substituents reacted efficiently to afford excellent yields of 3,4-dihydropyrimidin-2-(1H)-ones, the aliphatic aldehydes, which are known to be less reactive under conventional Biginelli reaction conditions, also reacted smoothly to afford very high yields (Table , entries 26 and 27).
Thiourea was used as one of the substrates to provide the corresponding DHPMs in reasonable yields (Table , entries 21–24, 27, and 28). Most important, many of the pharmacologically relevant substitution patterns on the aromatic ring could be introduced without any interruption in efficiency.
To access the feasibility of applying this method in a preparative scale, we carried out the one-pot, three-component Biginelli condensation of benzaldehyde with ethyl acetoacetate and urea on a 100-mmol scale (Table , entry 29). As expected, the reaction proceeded similarly to the case in a smallerly scale (Table , entry 1), and the desired 3,4-dihydropyrimidinone was obtained in 98% isolated yield in 25 min.
The merits of the present method are reflected from the fact that it provides better yields of various substituted 3,4-dihydropyrimidinones in shorter reaction times as compared to the recently known methods using heterogeneous catalysts.
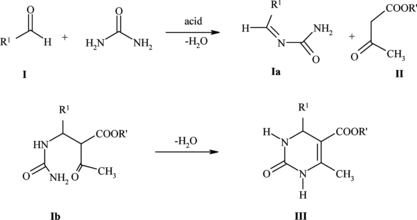
The generally accepted Biginelli reaction mechanism[17,44,45a] (Scheme ) involves the formation of C=N bond from the parent aldehyde (I) and urea followed by (protic or Lewis) acid-catalyzed addition of acetoacetate ester (II) to the aryl (or alkyl)idene–urea (Ia) and cyclodehydration (via Ib), yielding dihydropyrimidinones (III). AMA might promote the reaction by accelerating the formation C=N bond in the rate-determining step.[ Citation 45b ]
Scheme 3 Suggested Biginelli reaction mechanism.
In conclusion, the present procedure provides an efficient and improved modification of Biginelli reactions. Mild reaction condition, ease of workup, high yields, stability and recyclability of the catalyst, large-scale synthesis, and simple procedure are features of this new procedure. Moreover, this method has the ability to tolerate a wide variety of substituents in all three components. When we compare our results (time, yield, reaction conditions) with some results obtained by other groups, as can be seen, our method is simpler, is more efficient, and uses no toxic solvents, and the catalyst could be readily recovered and reused for the one-pot formation of DHPMs. Hence, we believe that this method will find wide application in organic synthesis as well as industry.
EXPERIMENTAL
General Information
NMR spectra were recorded on a Bruker Avance DPX-250 (1H NMR 250 MHz and 13C NMR 62.9 MHz) spectrometer in pure deuterated solvents with tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are in hertz (Hz). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. IR spectra were obtained using a Shimadzu Fourier transform infrared (FT-IR) 8300 spectrophotometer. Mass spectra were determined on a Shimadzu GCMS-QP 1000 EX instrument at 70 or 20 ev. Melting points were determined in open capillary tubes in a Büchi-535 circulating-oil melting-point apparatus. The purity determination of the substrates and reaction monitoring were accomplished by thin-layer chromatography (TLC) on silica-gel PolyGram SILG/UV 254 plates. Chemical materials were purchased from Fluka, Aldrich and Merck companies. Acidic alumina (Al2O3) type 540 C was purchased from Fluka.
Preparation of AMA
Methansulfonic acid (16.52 mL, 255 mmol) was added dropwise over a period of 90 min at 40 °C to a mixture of alumina (51 g, 510 mmol) in dichloromethane (30 mL). After the addition was complete, the mixture was stirred for 2 h, and then the solvent was evaporated under reduced pressure. After removal of CH2Cl2 in a rotary evaporator, the solid powder was kept at 120 °C for 72 h. A white solid of 68.0 g was obtained.
General Procedure for Synthesis of DHPMs
A mixture of the aldehyde (1 mmol), β-dicarbonyl compound (1 mmol), urea or thiourea (1.2 mmol), and alumina-methansulfonic acid (0.1 g, equal to 0.5 mmol H+) in ethanol (5 mL) was heated at 60 °C for the appropriate time (Table ). After completion of the reaction as indicated by TLC, the reaction mixture was cooled to room temperature and was filtered through a sinter funnel. The solid residue was washed with 20 mL hot ethanol (50 °C). The remaining catalyst was reloaded with fresh reagents for further runs. The filtrate was concentrated, and the solid product was recrystallized from ethyl acetate/n-hexane (1/3) or ethanol.
All produced DHPMs were characterized in detailed structural data by IR, 1H NMR, 13C NMR, and elemental analysis as given next.
Data
Unknown compounds or compounds for which incomplete physical data were reported in the literature were characterized by FTIR, NMR (1H, 13C), and elemental analysis.
Ethyl-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8a)
Compound 8a was obtained in 98% yield. Mp 204–206 °C (lit. 202–204°C)[ Citation 37 ]; 1H NMR (250 MHz, CDCl3): 1.19 (t, J = 7.1 Hz, 3H), 2.33 (s, 3H), 4.01 (q, J = 7.2 Hz, 2H), 5.31 (s, 1H), 5.90 (s, 1H), 7.10–7.35 (m, 5H), 8.30 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.1, 18.6, 55.7, 60.0, 101.3, 126.5, 127.0, 128.0, 143.0, 146.2.
Ethyl-6-methyl-4-(4-nitrophenyl)-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8b)
Compound 8b was obtained in 91% yield. Mp 209–211 °C, (lit. 209–211°C) [ Citation 37 ]; 1H NMR (250 MHz, DMSO): 1.04 (t, J = 7.0 Hz, 3H), 2.24 (s, 3H), 3.86 (q, J = 7.0 Hz, 2H), 5.25 (s, 1H), 7.50 (d, J = 7.3 Hz, 2H), 7.85 (s, 1H), 8.20 (d, J = 7.2 Hz, 2H), 8.54 (s, 1H); 13C NMR (62.9 MHz, DMSO): 13.9, 17.8, 53.6, 59.3, 98.1, 123.7, 127.6, 146.6, 149.3, 151.7, 151.9, 165.0.
Ethyl-4-(4-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8c)
Compound 8c was obtained in 95% yield. Mp 212–214 °C, (lit. 213–215°C)[37]; 1H NMR (250 MHz, CDCl3): 1.18 (t, J = 7.2 Hz, 3H), 2.31 (s, 3H), 4.06 (q, J = 7.2 Hz, 2H), 5.35 (s, 1H), 6.80 (s, 1H), 7.22–7.28 (m, 4H), 8.37 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.1, 18.6, 55.0, 60.1, 101.1, 127.9, 128.8, 133.7, 142.2, 146.5, 153.6, 165.4.
Ethyl-4-(4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8d)
Compound 8d was obtained in 96% yield. Mp 203–205 °C, (lit. 201–203°C)[ Citation 37 ]; 1H NMR (250 MHz, DMSO): 1.10 (t, J = 7.0 Hz, 3H), 2.23 (s, 3H), 3.70 (s, 3H), 3.96 (q, J = 7.0 Hz, 2H), 5.10 (s, 1H), 6.88 (d, J = 8.8 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 7.65 (s, 1H), 9.14 (s, 1H); 13C NMR (62.9 MHz, DMSO): 14.5, 18.1, 53.7, 55.4, 59.5, 100.1, 114.1, 127.8, 137.4, 148.4, 152.6, 158.8, 165.8.
Ethyl-4-(3-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8e)
Compound 8e was obtained in 90% yield. Mp 190–192 °C, (lit. 190–193°C)[ Citation 38 ]; 1H NMR (250 MHz, CDCl3): 1.20 (t, J = 7.2 Hz, 3H), 2.34 (s, 3H), 4.06 (q, J = 7.2 Hz, 2H), 5.37 (s, 1H), 6.15 (s, 1H), 7.16–7.40 (m, 4H), 8.37 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.11, 18.61, 55.25, 60.19, 100.0, 124.0, 126.0, 128.0, 130.0, 134.0, 145.0, 146.0, 153.0, 165.0.
Ethyl-6-methyl-4-(3-nitrophenyl)-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8f)
Compound 8f was obtained in 92% yield. Mp 227–229 °C, (lit. 226–227°C)[ Citation 37 ]; 1H NMR (250 MHz, DMSO): 1.07 (t, J = 7.2 Hz, 3H), 2.25 (s, 3H), 3.96 (q, J = 7.2 Hz, 2H), 7.61–7.70 (m, 2H), 7.87 (s, 1H), 8.07–8.36 (m, 2H), 9.34 (s, 1H); 13C NMR (62.9 MHz, DMSO): 13.9, 17.8, 53.6, 59.3, 98.3, 120.9, 122.3, 130.1, 132.9, 146.9, 147.7, 149.4, 151.7, 164.0.
Ethyl-4-(2-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8g)
Compound 8g was obtained in 88% yield. Mp = 216–217 °C, (lit. 215–218°C)[ Citation 9 ]; 1H NMR (250 MHz, CDCl3): 1.13 (t, J = 7.2 Hz, 3H), 2.41 (s, 3H), 3.90 (q, J = 7.0 Hz, 2H), 5.77 (s, 1H), 5.87 (s, 1H), 7.05–7.26 (m, 3H), 7.34–7.51 (m, 1H), 8.69 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 13.9, 18.3, 55.1, 59.9, 98.8, 127.5, 128.0, 129.3, 129.8, 132.6, 139.5, 148.4, 153.1, 165.3.
Ethyl-6-methyl-2-oxo-4-(2-thienyl)-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8h)
Compound 8h was obtained in 95% yield. Mp = 209–210 °C, (lit. 209–210°C)[ Citation 39 ]; 1H NMR (250 MHz, CDCl3): 1.29 (t, J = 7.0 Hz, 3H), 2.27 (s, 3H), 4.13 (q, J = 7.2 Hz, 2H), 5.68 (s, 1H), 6.35 (s, 1H), 6.87–6.95 (m, 2H), 7.17 (d, J = 5.0 Hz, 1H), 8.51 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.2, 18.5, 50.6, 60.2, 101.6, 123.9, 124.8, 126.7, 146.8, 147.3, 153.9, 165.4.
Ethyl-6-methyl-4-(4-methylphenyl)-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8i)
Compound 8i was obtained in 96% yield. Mp 169–171 °C, (lit. 169–171 °C)[ Citation 11 ]; 1H NMR (250 MHz, CDCl3): 1.10 (t, J = 7.2 Hz, 3H), 2.31 (s, 6H), 4.06 (q, J = 7.2 Hz, 2H), 5.34 (s, 1H), 5.86 (s, 1H), 7.02–7.42 (4H, m), 8.25 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.1, 18.6, 21.0, 55.3, 59.9, 101.5, 126.5, 129.3, 137.6, 140.8, 146.2, 153.5, 165.7.
Ethyl-4-(2,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8j)
Compound 8j was obtained in 95% yield. Mp = 157–159 °C, (lit. 158–160°C)40; 1H NMR (250 MHz, CDCl3): 1.10 (t, J = 7.0 Hz, 3H), 2.26 (s, 3H), 3.71 (s, 3H), 3.76 (s, 3H), 4.04 (q, J = 7.0 Hz, 2H), 5.67 (s, 1H), 5.84 (s, 1H), 6.75 (d, J = 2.7 Hz, 1H), 6.70–6.88 (m, 2H), 8.59 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.2, 18.5, 49.9, 55.6, 55.7, 59.9, 98.1, 111.2, 112.1, 113.8, 130.9, 148.5, 150.9, 153.5, 153.7, 165.7.
Ethyl-4-(4-cyanophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8k)
Compound 8k was obtained in 85% yield. Mp 219–221 °C, (lit. 219–222°C)[ Citation 38 ]; 1H NMR (250 MHz, CDCl3): 1.14 (t, J = 7.0 Hz, 3H), 2.33 (s, 3H), 4.01 (q, J = 7.2 Hz, 2H), 5.45 (s, 1H), 6.11 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.62 (d, J = 8.1 Hz, 2H), 8.52 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.2, 18.8, 55.3, 60.3, 100.4, 111.9, 118.5, 127.4, 132.6, 147.1, 148.5, 153.2, 165.2.
Ethyl-4-(2,6-dichlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8l)
Compound 8l was obtained in 88% yield. Mp = 280–283 °C, (lit. 226°C)[23e]; 1H NMR (250 MHz, DMSO): 0.87 (t, J = 7.1 Hz, 3H), 2.15 (s, 3H), 3.80 (q, J = 7.2 Hz, 2H), 6.1 (s, 1H), 7.25 (t, J = 8.67 Hz, 1H), 7.35 (d, J = 7.7 Hz, 2H), 7.69 (s, 1H), 9.25 (s, 1H); 13C NMR (62.9 MHz, DMSO): 13.6, 17.8, 52.1, 58.7, 94.0, 129.3, 135.1, 137.5, 149.7, 150.5, 164.8.
Ethyl-4-(4-isopropylphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8m)
Compound 8m was obtained in 90% yield. Mp 140–142 °C; IR (KBr), υ(cm−1): 3247, 3120, 2962, 2931, 1705, 1651, 1288, 1096, 775, 663; 1H NMR (250 MHz, CDCl3) 1.08–1.21 (m, 9H), 2.29 (s, 3H), 2.73 (m, 1H), 3.98 (q, J = 7.2 Hz, 2H), 5.28 (s, 1H), 6.00 (s, 1H), 7.06 (d, J = 8.2 Hz, 2H), 7.15 (d, J = 8.2 Hz, 2H), 8.64 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.1, 18.5, 23.9, 33.7, 55.2, 59.9, 101.4, 126.5, 126.7, 129.8, 141.2, 146.4, 148.4, 153.9, 165.8. C17H22N2O3 (302.371): calc. C, 67.53%; H, 7.33%; N, 9.26%, found C, 67.59%; H, 7.26%, N, 9.20%.
5-Acetyl-6-methyl-4-(4-methylphenyl)-3,4-dihydro-2(1H)-pyrimidinone (8n)
Compound 8n was obtained in 93% yield. Mp 204–206 °C; IR (KBr), υ(cm−1): 3288, 3120, 2920, 1699, 1616, 1236, 1139, 765, 561; 1H NMR (250 MHz, CDCl3) 2.07 (s, 3H), 2.30 (s, 6H), 5.42 (s, 1H), 6.29 (s, 1H), 7.01–7.22 (m, 4H), 8.59 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 19.5, 21.1, 30.8, 55.9, 110.5, 126.5, 129.7, 137.9, 139.9, 146.1, 153.5, 195.4. C14H16N2O2 (244.292): calc. C, 68.83%; H, 6.60%; N 1.47%, found C, 68.89%; H, 6.50%, N 1.51%.
5-Acetyl-4-(3-chlorophenyl)-6-methyl-3,4-dihydro-2(1H)-pyrimidinone (8o)
Compound 8o was obtained in 86% yield. Mp 280–281 °C; IR (KBr), υ(cm−1): 3290, 3105, 3915, 1706, 1676, 1614, 1362, 1234, 1190, 917, 763, 628; 1H NMR (250 MHz, DMSO): 2.1 (s, 3H), 2.28 (s, 1H), 5.25 (s, 1H), 7.16–7.45 (m, 4H), 7.87 (s, 1H), 9.24 (s,1H); 13 C NMR (62.9 MHz, DMSO): 18.9, 30.4, 53.1, 109.3, 124.9, 126.8, 127.2, 130.4, 133.0, 146.6, 148.7, 152.0, 194.0 C13H13ClN2O2 (264.710): calc. C, 58.99%; H, 4.95%; N, 10.56%, found C, 58.89%, H, 5.00%; N, 10. 61%.
5-Acetyl-6-methyl-4-phenyl-3,4-dihydro-2(1H)-pyrimidinone (8p)
Compound 8p was obtained in 89% yield. Mp = 229–230 °C, (lit. 233–236°C)[16]; 1H NMR (250 MHz, DMSO): 2.16 (s, 1H), 2.27 (s, 1H), 5.24 (s, 1H), 7.25–7.35 (m, 5H), 7.80 (s, 1H), 9.16 (s,1H); 13C NMR (62.9 MHz, DMSO): 18.8, 30.3, 53.7, 109.5, 126.4, 127.5, 128.17, 144.2, 148.1, 152.1, 194.2.
Ethyl-6-(chloromethyl)-4-(4-methylphenyl)-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8q)
Compound 8q was obtained in 90% yield. Mp 164–166 °C; IR (KBr), υ(cm−1): 3363, 3232, 3124, 2927, 2866, 1704, 1654, 1434, 1099, 1018, 771. 1H NMR (250 MHz, CDCl3): 1.20 (t, J = 7.0 Hz, 3H), 2.29 (s. 3H), 4.08 (q, J = 7.0 Hz, 2H), 4.75 (d, J = 13.0 Hz, 1H), 4.83 (d, J = 13.0 Hz, 2H), 5.39 (s, 1H), 6.56 (s, 1H), 7.09–7.026 (m, 4H), 8.01 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.0, 21.1, 55.1, 60.6, 103.5, 126.5, 129.1, 129.4, 129.9, 137.8, 140.1, 143.8, 153.9, 164.6. C16H17ClN2O3 (308.736): calc. C, 58.35%; H, 5.55%, 9.07%, found C, 58.40%; H 5.45%, N, 9.00%.
Ethyl-6-(chloromethyl)-4-(3-nitrophenyl)-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8r)
Compound 8r was obtained in 87% yield. Mp 162–164 °C; IR (KBr), υ(cm−1): 3355, 3232, 3132, 2869, 1704, 1635, 1099, 1022, 914, 732, 455; 1H NMR (250 MHz, DMSO): 1.06 (t, J = 7.0 Hz, 3H), 4.01 (q, J = 7.2 Hz,2H), 4.57 (dd, J 1 = 10.7 Hz J 2 = 3.2 Hz, 1H), 4.61 (dd, J 1 = 10.2 Hz, J 2 = 3.5 Hz, 1H), 5.35 (s, 1H), 7.60–7.71 (m, 2H), 8.0–8.12 (m, 3H), 9.68 (s, 1H); 13C NMR (62.9 MHz, DMSO): 13.6, 53.3, 60.1, 100.8, 121.1, 122.5, 130.2, 132.9, 145.9, 146.5, 146.9, 147.7, 151.9, 163.8. C14H14ClN3O5 (339.733): calc. C, 49.50%, H, 4.15%; N, 12.37%, found C, 49.60%; H 4.10%, N 12.0%.
Ethyl-6-(chloromethyl)-2-oxo-4-phenyl-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8s)
Compound 8s was obtained in 87% yield. Mp 174–176 °C; IR (KBr), υ(cm−1): 3355, 3228, 3124, 2974, 1693, 1647, 1307, 1230, 1099, 1022, 756, 694; 1H NMR (250 MHz, CDCl3): 1.13 (t, J = 7.0 Hz, 3H), 4.03 (q, J = 7.0 Hz, 2H), 4.71 (d, J = 13.2 Hz, 1H), 4.84 (d, J = 13.1 Hz, 1H), 5.35 (s, 1H), 5.78 (s, 1H), 7.14–7.81 (m, 5H), 7.98 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.0, 39.7, 55.8, 60.7, 67.9, 126.6, 127.8, 128.3, 128.9, 142.7, 143.1, 147.0, 148.9, 164. C14H15ClN2O3 (294.736): calc. C, 57.05%: H, 5.13%; N, 9.50%, found C, 57.15%; H, 5.10%; N, 9.45%.
Ethyl-6-methyl-4-(2-naphthyl)-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8t)
Compound 8t was obtained in 91% yield. Mp 196–198 °C; IR (KBr), υ (cm−1): 3222, 3095, 2929, 1703, 1651, 1429, 1284, 1085, 1020, 779, 682, 478; 1H NMR (250 MHz, CDCl3): 1.14 (t, J = 7.2 Hz, 3H), 2.33 (s, 3H), 4.05 (q, J = 7.2 Hz, 2H), 5.52 (s, 1H), 6.24 (s, 1H), 7.35–7.48 (m, 3H), 7.62 (s, 1H), 7.70–7.80 (m, 3H), 8.64 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.0, 18.3, 52.1, 60.0, 98.8, 127.1, 127.9, 129.3, 129.8, 132.6, 139.5, 148.4, 153.1, 165.3. C18H18N2O3 (310.351): calc. C, 69.66%; H, 5.85%; N, 9.03%; found C, 69.78%; H, 5.80%; N, 9.10%.
1-(6-Methyl-4-phenyl-2-thioxo-1,2,3,4-tetrahydro-5-pyrimidinyl)ethanone (8u)
Compound 8u was obtained in 89% yield. Mp 183°C (decomposed) (lit. 185°C decomposed)[42]; 1H NMR (250 MHz, DMSO): 2.16 (s, 3H), 2.37 (s, 3H), 5.28 (s, 1H), 7.26–7.59 (m, 5H), 9.89 (s, 1H), 10.37 (s, 1H); 13C NMR (62.9 MHz, DMSO): 18.2, 30.4, 53.7, 110.4, 126.5, 127.46, 128.6, 142.8, 144.5, 168.0, 174.0, 194.7.
Ethyl-6-methyl-4-phenyl-2-thioxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8v)
Compound 8v was obtained in 85% yield. Mp 209–211 °C (lit. 208–211°C)[9]; 1H NMR (250 MHz, CDCl3): 1.19 (t, J = 7.1 Hz, 3H), 2.35 (s, 3H), 4.10 (q, J = 7.1 Hz, 2H), 5.38 (s, 1H), 7.20–7.35 (m, 5H), 7.48 (s, 1H), 8.12 (s, 1H); 13C NMR (62.9 MHz, CDCl3): 14.1, 18.3, 56.2, 60.4, 102.9, 126.8, 128.4, 128.9, 142.3, 142.7, 165.2, 174.5.
Ethyl-4-(3-nitrophenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8w)
Compound 8w was obtained in 88% yield. Mp 206–208 °C (lit. 206–207°C)[23a]; 1H NMR (250 MHz, CDCl3): 1.08 (t, J = 7.2 Hz, 3H), 2.29 (s, 3H), 4.05 (q, J = 7.2 Hz, 2H), 5.30 (s, 1H), 7.63–7.67 (m, 2H), 8.04 (s, 1H), 8.14 (dd, J 1 = 6.2 Hz, J 2 = 2.2 Hz, 1H), 9.74 (s, 1H), 10.48 (s, 1H); 13C NMR (62.9 MHz, DMSO): 13.9, 17.8, 53.4, 59.7, 99.8, 121.1, 122.7, 130.4, 133.0, 145.4, 145.9, 147.7, 164.8, 174.4.
Ethyl-4-(3-hydroxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8x)
Compound 8x was obtained in 87% yield. Mp 183–184 °C (lit. 184–186°C)[43]; 1H NMR (250 MHz, DMSO): 1.06 (t, J = 7.2 Hz, 3H), 2.26 (s, 3H), 4.01 (q, J = 7.0 Hz, 2H), 5.08 (s, 1H), 7.63 (m, 3H), 7.20 (t, J = 8.7 Hz, 1H), 9.40 (s, 1H), 9.56 (s, 1H), 10.41 (s, 1H); 13C NMR (62.9 MHz, DMSO): 13.9, 17.1, 53.9, 59.5, 100.7, 113.2, 114.5, 116.9, 129.4, 144.7, 144.8, 157.4, 165.1, 174.1.
Ethyl-4-(3-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8y)
Compound 8y was obtained in 94% yield. Mp 163–164°C (lit. 163–165 °C)9; 1H NMR (250 MHz, DMSO):1.10 (t, J = 7.0 Hz, 3H), 2.22 (s,3H), 4.05 (q, J = 7.0 Hz, 2H), 5.04 (s, 1H), 6.58–6.66 (m, 3H), 7.07 (t, J = 8.1 Hz, 1H), 7.66 (s, 1H), 9.32 (s, 1H), 9.34 (s, 1H); 13C NMR (62.9 MHz, DMSO): 14.0, 17.7, 53.7, 59.1, 99.3, 113.0, 114.1, 116.8, 129.2, 146.1, 148.0, 152.0, 157.3, 165.3.
Ethyl-6-methyl-2-oxo-4-pentyl-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8z)
Compound 8z was obtained in 85% yield. Mp 151–154 °C; IR (KBr), υ (cm−1): 3263, 3101, 2939, 3862, 1728, 1651, 1465, 1226, 1087, 1026, 779; 1H NMR (250 MHz, DMSO): 0.80 (t, J = 6.5 Hz, 3H), 1.08–120 (m, 11H), 1.92 (s, 3H), 4.80 (q, J = 6.7 Hz, 2H), 8.09 (s, 1H), 8.19 (s, 1H); 13C NMR (62.9 MHz, DMSO): 13.7, 14.1, 18.0, 21.9, 23.3, 30.9, 36.6, 49.9, 58.9, 99.8, 148.2, 152.4, 165.4; C13H22N2O3 (254.325): calc. C, 61.39%, H, 8.72%, N, 11.01%, found C, 61.45%, H, 8.67%, N, 11.10%.
Ethyl-4-butyl-6-methyl-2-thioxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8a′)
Compound 8a′ was obtained in 83% yield. Mp 138–140 °C; IR (KBr), υ (cm−1): 3186, 2927, 2856, 1710, 1651, 1596, 1434, 1184, 1095, 752, 644, 528; 1H NMR (250 MHz, DMSO): 0.81 (t, J = 6.5 Hz, 3H), 1.1–1.2 (m, 9H), 2.18 (s, 3H), 3.98–4.18 (m, 3H), 9.34 (s, 1H), 10.24 (s, 1H); 13C NMR (62.9 MHz, DMSO): 13.6, 14.1, 17.0, 21.8, 25.5, 35.8, 50.3, 59.4, 100.6, 145.1, 165.1, 174.9; C12H20N2O2S (256.365): calc. C 56.22%; H, 7.86%; N, 10.93%; S, 12.51%, found C, 56.31%; H, 7.81%; N, 10.87%; S, 12.59%.
Benzyl-6-methyl-4-phenyl-2-thioxo-1,2,3,4-tetrahydro-5-pyrimidinecarboxylate (8b′)
Compound 8b′ was obtained in 83% yield. Mp 157–159 °C (lit.156–157°C)[46]; 1H NMR (250 MHz, DMSO): 2.30 (s, 3H), 4.99 (d, J = 12.7 Hz, 1H), 5.10 (d, J = 12.7 Hz, 1H), 5.19 (s, 1H), 7.10–7.31 (m, 10H), 9.66 (s, 1H), 10.38 (s, 1H); 13C NMR (62.9 MHz, DMSO): 17.2, 54.0, 65.1, 100.1, 126.4, 127.5, 127.7, 127.7, 128.2, 128.5, 136.2, 143.2, 145.8, 164.8, 174.1.
ACKNOWLEDGMENT
We gratefully acknowledge the support of this work by the Shiraz University Research Council and Shiraz Faculty of Pharmacy. We are also grateful to Mr. H. Sajedian Fard for helpful cooperation.
Notes
a 0.1 g, equal to 0.5 mmol H+.
b Isolated yield.
c The same catalyst was used for each of five runs.
a Products were characterized by comparison of their spectroscopic data (1H NMR, 13C NMR, and IR) and melting points with those reported in the literature.
b Isolated yield.
c The reaction was carried out on a 100-mmol scale.
Related Research Data
REFERENCES
- For reviews, see (a) Ramón , D. J. ; Yus , M. Asymmetric multicomponent reactions (AMCRs): The new frontie . Angew. Chem. Int. Ed. 2005 , 44 , 1602 – 1634 ; (b) Ramachary , D. B. ; Barbas , C. F. Towards organo-click chemistry: Development of organocatalytic multicomponent reactions through combinations of aldol, Wittig, Knoevenagel, Michael, Diels-Alder, and Huisgen cycloaddition reactions . Chem. Eur. J. 2004 , 10 , 5323 – 5331 ; (c) Denmark , S. E. ; Fan , Y. The first catalytic, asymmetric α-additions of isocyanides: Lewis-base-catalyzed, enantioselective Passerini-type reactions . J. Am. Chem. Soc. 2003 , 125 , 7825 – 7827 ; (d) Andreana , P. R. ; Liu , C. C. ; Schreiber , S. L. Stereochemical control of the Passerini reaction . Org. Lett. 2004 , 6 , 4231 – 4233 ; (e) Cozzi , P. G. ; Rivalta , E. Highly enantioselective one-pot, three-component imino-Reformatsky reaction . Angew. Chem. Int. Ed. 2005 , 44 , 3600 – 3603 ; (f) Armstrong , R. W. ; Combs , A. P. ; Tempest , P. A. ; Brown , S. D. ; Keating , T. A. Multiple-component condensation strategies for combinatorial library synthesis . Acc. Chem. Res. 1996 , 29 , 123 – 131 ; (g) Burke , M. D. ; Schreiber , S. L. A planning strategy for diversity-oriented synthesis . Angew. Chem. Int. Ed. 2004 , 43 , 46 – 58 ; (h) Khodaei , M. M. ; Khosroupor , A. R. ; Jowkar , M. Bi(NO3)3·5H2O-TBAF as an efficient reagent for in situ oxidation: Dihydropyrimidinone formation from benzyl halides . Synthesis 2005 , 1301 – 1304 .
- Biginelli , P. The first synthesis of dihydropyrimidinone by refluxing a mixture of an aldehyde, a β-ketoester, and urea under strongly acidic condition. Gazz. Chim. Ital. 1893, 23, 360–416.
- Kappe , C. O. Biologically active dihydropyrimidones of the Biginelli-type—a literature survey . Eur. J. Med. Chem. 2000 , 35 , 1043 – 1052 .
- (a) Russowsky , D. ; Canto , R. F. S. ; Sanches , S. A. A. ; D'oca , M. G. M. ; Fatima , A. D. ; Carvalho , J. E. D. Synthesis and differential antiproliferative activity of Biginelli compounds against cancer cell lines: Monastrol, oxo-monastrol, and oxygenated analogues . Bioorg Chem. 2006 , 34 , 173 – 178 ; (b) Kappe , C. O. One hundred years of the Biginelli dihydropyrimidine synthesis . Tetrahedron 1993 , 49 , 6937 – 6963 .
- (a) Rovnyak , G. C. ; Atwal , K. S. ; Hedberg , A. ; Kimball , S. D. ; Moreland , S. ; Gougoutas , J. Z. ; O'Reilly , B. C. ; Schwartz , J. ; Malley , M. F. Dihydropyrimidine calcium channel blockers, 4: Basic 3-substituted-4-aryl-1,4-dihydropyrimidine-5-carboxylic acid esters: Potent antihypertensive agents . J. Med. Chem. 1992 , 35 , 3254 – 3263 ; (b) Grover , G. J. ; Dzwonczyk , S. ; McMullen , D. M. ; Normandin , D. E. ; Parham , C. S. ; Sleph , P. G. ; Moreland , S. Pharmacologic profile of the dihydropyrimidine calcium channel blockers SQ 32,547 and SQ 32,926 . J. Cardiovasc. Pharmacol. 1995 , 26 , 289 – 294 .
- (a) Nagarathnam , D. ; Miao , S. W. ; Lagu , B. ; Harrell , M. C. ; Vyas , K. P. ; Gluchowski , C. Design and synthesis of novel α1a-adrenoceptor-selective antagonists, 1: Structure–activity relationship in dihydropyrimidinones . J. Med. Chem. 1999 , 42 , 4764 – 4777 ; (b) Barrow , J. C. ; Nantermet , P. G. ; Nagarathnam , D. ; Forray , C. In vitro and in vivo evaluation of dihydropyrimidinone C-5 amides as potent and selective α1A receptor antagonists for the treatment of benign prostatic hyperplasia . J. Med. Chem. 2000 , 43 , 2703 – 2718 .
- (a) Gartner , M. ; Sunder-Plassmann , N. ; Seiler , J. ; Utz , M. ; Vernos , I. ; Surrey , T. ; Giannis , A. Development and biological evaluation of potent and specific inhibitors of mitotic Kinesin Eg5 . Chem. Bio. Chem. 2005 , 6 , 1173 – 1177 ; (b) Mayer , T. U. ; Kapoor , T. M. ; Mitchison , T. J. ; Schreiber , S. Dissecting cellular processes using small molecules: Identification of colchicine-like, taxol-like, and other small molecules that perturb mitosis . Chem. Biol. 2000 , 7 , 275 – 276 .
- Barluenga , J. ; Thomas , M. ; Ballesterus , A. ; Lopez , A. 1,4-Cycloaddition of 1,3-diazabutadienes with enamines: An efficient route to the pyrimidine ring . Tetrahedron Lett. 1989 , 30 , 4573 – 4576 .
- Fu , N. Y. ; Yuan , Y. F. ; Cao , Z. ; Wang , S. W. ; Wang , J. T. ; Peppe , C. Indium(III) bromide-catalyzed preparation of dihydropyrimidinones: Improved protocol conditions for the Biginelli reaction. Tetrahedron 2002, 58, 4801–4807.
- Ranu , B. ; Hajra , A. ; Jana , U. Indium(III) chloride-catalyzed one-pot synthesis of dihydropyrimidinones by a three-component coupling of 1,3-dicarbonyl compounds, aldehydes, and urea: An improved procedure for the Biginelli reaction . J. Org. Chem . 2000 , 65 , 6270 – 6272 .
- Yadav , J. S. ; Reddy , B. V. S. ; Srinivas , R. ; Venugopal , C. ; Ramalingam , T. LiClO4-catalyzed one-pot synthesis of dihydropyrimidinones: An improved protocol for Biginelli reaction . Synthesis 2001 , 1341 – 1345 .
- Lu , J. ; Ma , H. Iron (III)-catalyzed synthesis of dihydropyrimidinones: Improved conditions for the Biginelli reaction . Synlett 2000 , 63 – 64 ; (b) Lu , J. ; Bai , Y. Catalysis of the Biginelli reaction by ferric and nickel chloride hexahydrates: One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones . Synthesis 2002 , 466 – 470 .
- Jin , T. ; Zhang , S. ; Li , T. p-Toluensulfonic acid-catalyzed efficient synthesis ofdihydropyrimidines improved high yielding protocol for the Biginelli reaction . Synth. Commun. 2002 , 32 , 1847 – 1851 .
- Lu , J. ; Bai , Y. ; Wang , Z. ; Yang , B. ; Ma , H. One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones using lanthanum chloride as a catalyst . Tetrahedron Lett. 2000 , 41 , 9075 – 9078 .
- Varala , R. ; Alam , M. M. ; Adapa , S. R. Bismuth triflate–catalyzed one-pot synthesis of 3,4-dihydropyrimidin-2 (1H)-ones: An improved protocol for the Biginelli reaction . Synlett 2003 , 67 – 70 .
- Ma , Y. ; Qian , C. ; Wang , L. ; Yang , M. Lanthanide triflate–catalyzed Biginelli reaction: One-pot synthesis of dihydropyrimidinones under solvent-free conditions . J. Org. Chem. 2000 , 65 , 3864 – 3868 .
- Hu , E. H. ; Slider , D. R. ; Dolling , U. H. Unprecedented catalytic three-component one-pot condensation reaction: An efficient synthesis of5-alkoxycarbonyl-4-aryl-3,4-dihydropyrimidin-2(1H)-ones . J. Org. Chem. 1998 , 63 , 3454 – 3457 .
- Eng , J. ; Deng , Y. Ionic liquids–catalyzed Biginelli reaction under solvent-free conditions . Tetrahedron Lett. 2001 , 42 , 5917 – 5919 .
- Ajbakhsh , M. ; Mohajerani , B. ; Heravi , M. M. ; Ahmadi , A. N. Natural HEU type zeolite-catalyzed Biginelli reaction for the synthesis of 3,4-dihydropyrimidin-2(1H)-one derivatives . J. Mol. Catal. A 2005 , 236 , 216 – 219 .
- Srinvas , K. V. N. ; Dash , B. Iodine-catalyzed one-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones and thiones: A simple and efficient procedure for the Biginelli reaction . Synthesis 2004 , 2091 – 2093 .
- Hazarkhani , H. ; Karimi , B. N-Bromosuccinimide as an almost neutral catalyst for efficient synthesis of dihydropyrimidinones under microwave irradiation . Synthesis 2004 , 1239 – 1242 .
- Gangadasu , B. ; Palaniappan , S. ; Rao , V. J. One-pot synthesis of dihydropyrimidinones using polyaniline-bismoclite complex: A facile and reusable catalyst for the Biginelli reaction . Synlett 2004 , 1285 – 1287 .
- (a) Kappe , C. O. ; Kumar , D. ; Varma , R. S. Microwave-assisted high-speed parallel synthesis of 4-aryl-3,4-dihydropyrimidin-2 (1H)-ones using a solventless Biginelli condensation protocol. Synthesis 1999, 1799–1803; (b) Bigi , F. ; Carloni , S. ; Frullanti , B. ; Maggi , R. ; Sartori , G. A revision of the Biginelli reaction under solid acid catalysis: Solvent-free synthesis of dihydropyrimidines over montmorillonite KSF. Tetrahedron Lett. 1999, 40, 3465–3468; (c) Radha Rani , V. ; Srinivas , N. ; Radhakishan , M. ; Kulkarni , S. J. ; Raghavan , K. V. Zeolite-catalyzed cyclocondensation reaction for the selective synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Green Chem. 2001, 3, 305–306; (d) Gohain , M. ; Prajapati , D. ; Sandhu , J. S. A novel cu-catalysed three-component one-pot synthesis of dihydropyrimidin-2(1H)-ones using microwaves under solvent-free conditions. Synlett 2004, 235–238; (e) Joseph , J. K. ; Jain , S. L. ; Sain , B. Ion exchange resins as recyclable and heterogeneous solid acid catalysts for the Biginelli condensation: An improved protocol for the synthesis of 3,4-dihydropyrimidin-2-ones. Journal of Molecular Catalysis A: Chemical 2006, 247, 99–102.
- (a) Rafiee , E. ; Jafari , H. A practical and green approach towards synthesis of dihydropyrimidinones: Using heteropoly acids as efficient catalysts . Bioorg. Med. Chem. Lett. 2006 , 16 , 2463 – 2466 ; (b) Maradur , S. P. ; Gokavi , G. S. Heteropoly acid catalyzed synthesis of 3,4-dihydropyrimidin-2(1H)-ones . Catal. Commun. 2007 , 8 , 279 – 284 ; (c) Heravi , M. M. ; Bakhtiari , K. ; Bamoharram , F. F. 12-Molybdophosphoric acid: A recyclable catalyst for the synthesis of Biginelli-type 3,4-dihydropyrimidine-2(1H)-ones . Catal. Commun. 2006 , 7 , 373 – 376 ; (d) Mishra , B. G. ; Kumar , D. ; Rao , V. S. H3PW12O40 catalyzed expeditious synthesis of 3,4-dihydropyrimidin-2(1H)-ones under solvent-free conditions . Catal. Commun. 2006 , 7 , 457 – 459 ; (e) Amini , M. M. ; Shaabani , A. ; Bazgir , A. Tangstophosphoric acid (H3PW12O40): An efficient and eco-friendly catalyst for the one-pot synthesis of dihydropyrimidin-2(1H)-ones . Catal. Commun. 2006 , 7 , 843 – 847 ; (f) Rafiee , E. ; Shahbazi , F. One-pot synthesis of dihydropyrimidones using silica-supported heteropoly acid as an efficient and reusable catalyst: Improved protocol conditions for the Biginelli reaction . J. Mol. Catal. A 2006 , 250 , 57 – 61 .
- Kumar , D. ; Sundaree , M. S. ; Mishra , B. G. Sulfated zirconia–catalyzed one-pot benign synthesis of 3,4-dihydropyrimidin-2(1H)-ones under microwave irradiation . Chem. Lett. 2006 , 35 , 1074 – 1075 .
- Liu , C. ; Wang , J. ; Li , Y. One-pot synthesis of 3,4-dihydropyrimidin-2(1H)(thio)ones using strontium(II) nitrate as a catalyst . J. Mol. Catal. A 2006 , 258 , 367 – 370 .
- Gupta , R. ; Paul , S. ; Gupta , R. Covalently anchored sulfonic acid onto silica as an efficient and recoverable interphase catalyst for the synthesis of 3,4-dihydropyrimidinones/thiones . J. Mol. Catal. A 2007 , 266 , 50 – 54 .
- (a) Corma , A. ; Garcia , H. Silica-bound homogenous catalysts as recoverable and reusable catalysts in organic synthesis . Adv. Synth. Catal. 2006 , 348 , 1391 – 1412 ; (b) Niknam , Kh. ; Karimi , B. ; Zolfigol , M. A. Silica sulfuric acid promoted aromatization of 1,2-dihydroquinolines by using NaNO2 as oxidizing agent under mild and heterogeneous conditions . Catal. Commun. 2007 , 8 , 1427 – 1430 ; (c) Niknam , K. ; Zolfigol , M. A. ; Khorramabadi-Zad , A. ; Zare , R. ; Sheygan , M. Silica sulfuric acid as an efficient and recyclable catalyst for the methoxymethylation of alcohols under solvent-free conditions . Catal. Commun. 2006 , 7 , 494 – 498 .
- Sharghi , H. ; Kaboudin , B. Alumina in methansulfonic acid (AMA) as a new efficent reagent for direct acylation of phenol derivaties and Fries rearrengement: A convenient synthesis of o-hydroxyalkylketones. J. Chem. Res. Synop. 1998, 628–629.
- Sharghi , H. ; Hosseini Sarvari , M. One-step Beckmann rearrangement from carbonyl compounds and hydroxylamine hydrochloride in Al2O3/CH3SO3H (AMA) as a new reagent . J. Chem. Res, Synop. 2001 , 446 – 449 .
- Sharghi , H. ; Hosseini Sarvari , M. Highly selective methodology for the directconversion of aromatic aldehydes to glycol . J. Org. Chem. 2003 , 68 , 4096 – 4099 .
- Sharghi , H. ; Hosseini Sarvari , M. A facile hydration of nitriles into amides by Al2O3/MeSO3H (AMA) . Synth. Commun. 2003 , 33 , 205 – 210 .
- Sharghi , H. ; Hosseini Sarvari , M. Al2O3/MeSO3H (AMA) as a new reagent with high selective ability for monoesterification of diols . Tetrahedron 2003 , 59 , 3627 – 3633 .
- Sharghi , H. ; Salimi Beni , A. R. A novel and efficient method for the synthesis of new hydroxythioxanthone derivatives . Synthesis 2004 , 17 , 2900 – 2904 .
- Sharghi , H. ; Shahsavari-Fard , Z. Al2O3/MeSO3H (AMA): A useful system for direct sulfonylation of phenols with p-toluenesulfonic acid . J. Iranian Chem. Soc. 2005 , 2 , 48 – 53 .
- Sharghi , H. ; Jokar , M. Al2O3/MeSO3H (AMA) as a novel heterogeneous system for synthesis of coumarins under mild conditions . Heterocycles 2007 , 71 , 2721 – 2733 .
- Jin , T. ; Zhang , S. ; Guo , J. ; Li , T. A simple and efficient synthesis of 3,4-dihydropyrimidin-2-ones catalysed by amidosulfonic acid . J. Chem. Res. Synop. 2002 , 37 – 39 .
- Paraskar , A. S. ; Dewkar , G. K. ; Sudalai , A. Cu(OTf)2: A reusable catalyst for high-yield synthesis of 3,4-dihydropyrimidin-2(1H)-ones . Tetrahedron Lett. 2003 , 44 , 3305 – 3308 .
- Sabitha , G. ; Reddy , G. S. K. K. ; Reddy , Ch. S. ; Yadav , J. S. One-pot synthesis of dihydropyrimidinones using iodotrimethylsilane: Facile and new improved protocol for the Biginelli reaction at room temperature . Synlett 2003 , 858 – 860 .
- Ananda Kumar , K. ; Suresh Reddy , C. Mn(OAc)3 · 2H2O-mediated three-component, one-pot, condensation reaction: An efficient synthesis of 4-aryl-substituted 3,4-dihydropyrimidin-2-ones . Tetrahedron Lett. 2001 , 42 , 7873 – 7875 .
- Yarim , M. ; Sarac , S. ; Ertan , M. ; Batu , O. ; Erol , K. Synthesis, structural elucidation, and pharmacological properties of some 5-acetyl-3,4-dihydro-6-methyl-4-(substituted phenyl)-2(1H)-pyrimidinones . Farmaco. 1999 , 54 , 359 – 363 .
- Sharma , S. D. ; Kaur , V. ; Bhutani , P. ; Khurana , J. P. S. Studies on fused β-lactams: Synthesis of 1-aza analogs of cephem. J. Bull. Chem. Soc. Jpn. 1992, 8, 2246–2250.
- Kappe , C. O. ; Shishkin , O. V. ; Uray , G. ; Vernido , P. X-ray structure, conformational analysis, enantioseparation, and determination of absolute configuration of the mitotic kinesin Eg5 inhibitor monastrol . Tetrahedron 2000 , 56 , 1859 – 1862 .
- Folkers , K. ; Johnson , T. B. Researches on pyrimidines, CXXXVI: The mechanism of formation of tetrahydropyrimidines by the Biginelli reaction . J. Am. Chem. Soc. 1933 , 55 , 3784 – 3791 .
- Kappe , O. C. A reexamination of the mechanism of the Biginelli dihydropyrimidine synthesis: Support for an N-acyliminium ion . J. Org. Chem. 1997 , 62 , 7201 – 7204 ; (b) Cepanec , I. ; Litvic , M. ; Filipan-Litivic , M. ; Grungold , I. Antimony(III) chloride-catalysed Biginelli reaction: A versatile method for the synthesis of dihydropyrimidinones through a different reaction mechanism . Tetrahedron 2007 , 63 , 11822 – 11827 .
- Ahmed , N. ; Lier , J. E. V. TaBr5-catalyzed Biginelli reaction: One-pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones/thiones under solvent-free conditions . Tetrahedron Lett. 2007 , 48 , 5407 – 5409 .