Abstract
Conditions for improved preparation of the crystalline iodine(III) reagents [1,2-C6H4(C(CF3)2OI)]2O and 1,2-C6H4(CH2OMe)(I=NTs) are presented together with their x-ray structures. The former is prepared by hydrolysis of 1,2-C6H4(C(CF3)2OICl (x-ray) and allows the preparation of cyclic sulfates from alkenes; the latter fashions sulfamidate under related conditions.
INTRODUCTION
Hypervalent iodine species have become cornerstone oxidants in organic synthesis.[ Citation 1 ] In routine transformations, Dess–Martin periodane 1,[ Citation 2 ] iodosylbenzene 2,[ Citation 3 ] and nitrene precursor 3 [ Citation 4 ] (Scheme ) normally deliver the expected alcohol dehydrogenation or catalytic epoxidation/aziridination products in good yields. Highly reliable experimental conditions have been described for the preparation of 1–3, and the former is also commercially available. However, when more esoteric IIII or IVreagents are called upon to optimize a given synthetic transformation, then optimal preparative procedures for these are not always available in the primary literature. For example, only scant elements of the synthetic sequences leading to 4 and 5 have made fleeting appearances, and no detailed robust experimental procedures are available.[ Citation 5 , Citation 6 ] In this note, we give optimized preparations of these compounds together with some applications that we needed in a separate project.
Scheme 1 Common (1–3) and poorly described (4 and 5) hypervalent iodine reagents.
RESULTS AND DISCUSSION
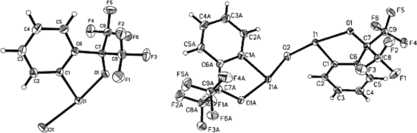
Dimeric 4 is prepared by the synthetic sequence shown in Scheme . Chloro species 6 is an excellent precursor, but it has not been fully described in the literature.[ Citation 5 ] Crystallization of as colorless tablets allows its facile purification and x-ray analysis, which clearly shows the T geometry of the hypervalent iodine centre (Fig. a). The oxophilicity of the I(III) center is evident from the comparison of the lengths of the I–Cl (2.448 Å) and I–O (2.109 Å) bonds (Fig. a). The bromo analog of 6 has been recently been used by Braddock in the bromination of alkenes and adopts a similar geometry.[ Citation 7 ] Hydrolysis of the I–Cl bond in 5 was possible in a biphasic system using potassium hydride (KOH) and a phase-transfer catalyst (PTC) (Scheme ). Monitoring of the reaction by thin-layer chromotography (TLC) showed the formation of two new compounds of differing Rf value. Isolation of both compounds by silica-gel column chromatography gave acceptable yields of the desired dimer 4, along with known 7.[ Citation 8 ] The former could be crystallized as colorless blocks (Fig. b), again greatly facilitating its purification.
Scheme 2 Preparation of 1,1′-oxy-bis(3,3-bis(trifluoromethyl)-3(1H)-1,2-benziodoxole) (4).
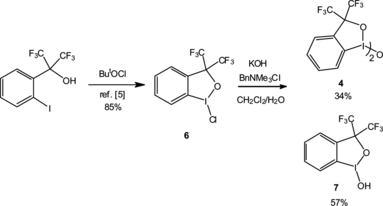
Figure 1 (a) Molecular structure of 6, left-hand side. Selected bond distances and angles: I–C(1) 2.108 (2), I–O 2.109 (6), I–Cl 2.448 (14) Å; Cl–I–O 171.63 (4), C(1)–I–Cl 92.60 (6), C(1)–I–O 79.24 (7)°. (b) Molecular structure of 4, right-hand side. Selected bond distances and angles: I(1)–O(2) 2.0296 (15), I(1)–C(1) 2.097 (2), I(1)–O(1) 2.1548 (15), O(2)–I (1A) 2.0300 (15), I(1A)–C(1A) 2.105 (2), I(1A)-O(1A), 2.1407 (16) Å; O(2)–I(1)–O(1) 167.72 (6), O(2)–I(1A)–O(1A) 168.00 (6)°.
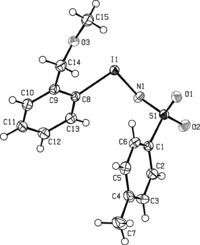
Although it is very popular as a nitrene source, compound 3 shows a couple of negative features: first, its polymeric structure renders it highly insoluble, and second, it is rather susceptible to hydrolysis reactions leading to its degradation to iodosylbenzene 2. In our use of 3, this proved a major problem, and we speculated that an ortho-chelated version of 3 would be more resistant to hydrolysis events. We prepared 5 by the chemistry of Scheme . Diacetate 9 has been mentioned in passing in the literature, but no experimental details or physical data were given.[ Citation 6 ] In our experience, formation of 9 is solvent sensitive—use of neat 37% peracetic acid gave the best results, but the presence of a CH2Cl2 cosolvent led to lower yields (41–56%), mainly due to overoxidation at the benzylic CH2 position, affording 2-iodobenzoic acid. When carrying out the neat AcOOH oxidation on a larger scale, longer times were required to ensure efficient contact mixing of all the reaction components. For example, reaction at a 4-mmol scale took 45 min, whereas that at 6 mmol took 60 min using the same 6:AcOOH ratio (1:2 equivalents). However, in all cases, a high yield of 9 was realized on completion of the reaction. Rewardingly, the new nitrene source 5 was appreciably soluble and crystallised on slow evaporation of methanol solutions (Fig. ). Additionally, compound 5 was found to be thermally stable and relatively nonhygroscopic. Exposure of 5 to atmopheric moisture for 2 days left the compound unchanged, while stirring it in a biphasic CH2Cl2/H2O system for 24 h resulted in the recovery of 5 in 75% yield.
Scheme 3 Preparation of 12-(N-(p-toluenesulfonyl)imino)iodobenzylmethyl ether (5).
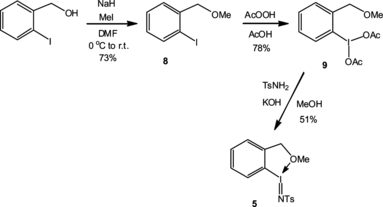
Figure 2 Selected bond distances and angles for (5): C(8)–I(1) 2.13, O(3)–I(1) 2.65 Å; N(1)–I(1)–C(8) 98.48°. Data averaged from two independent molecules.
Reagents 2–5 have been compared in the direct oxidative functionalization of alkenes in the presence of SO3 · DMF.[ Citation 9 ] Two alkenes were used in this brief comparison (Table ). Use of the bis compound 4 allowed high yields of cyclic sulfate to be attained. Additionally, the greater hydrolytic resistance of 5 allowed a significant increase in the sulfamidate yield. Unfortunately, the highly Lewis acidic conditions of these reactions still allowed N/O-exchange to a significant degree, leading to a significant contamination by 12. Interestingly, the sulfamidates are isolated as single regio isomers, which are assigned on the basis of nuclear Overhauser effect (NOE) analysis of compound 11 as summarized in Scheme .
Scheme 4 NOE studies confirming structure of 11.
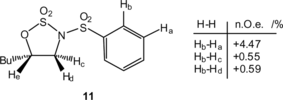
Table 1. Formation of cyclic sulfates and sulamidates using reagents –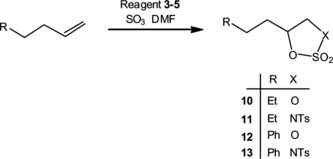
EXPERIMENTAL
General
Chromatography was performed using Fluorochem (35–70 micron) silica gel and TLC analysis using Merck Kieselgel 60 F245+366. Spectroscopic instrumentation used has been described before.[ Citation 10 ] Dimethylformamide (DMF) was dried over activated 4A molecular sieves. Methanol was distilled from Mg(OMe)2. Hygroscopic SO3 · DMF was stored under argon. The purity of AcOOH and hypervalent iodine reagents was confirmed by iodometric titration.[ Citation 11 ] Compound 6 was prepared by a literature procedure.[ Citation 5 ] Only limited data have appeared for compound 6; in our case, it was purified by recrystallization from CH2Cl2 to give the title compound as colorless tablets (Found: C, 26.98; H, 0.75%. Calc. for C9H4OIClF6: C, 26.73; H, 1.00%). Mp 170–173 °C; lit.[ Citation 5 ] 166–168 °C; 1H NMR (270 MHz, CDCl3) δ H 8.09 (1H, d, J 8.3, Ar), 7.88–7.81 (1H, m, Ar), 7.74–7.72 (2H, m, Ar); 13C NMR (68 MHz, CDCl3) δ C 139.4, 133.9, 132.1, 131.7, 129.8 (septet, 2JCF 3), 128.6, 122.9 (q, 1JCF 290), 113.5; 19F NMR (282 MHz, CDCl3) δ F – 75.8; IR υmax (solid)/cm−1 1270 w, 1195 m, 1103 w, 969 w, 728 w, 645 s; CIMS m/z 406 (MH+, 37Cl, 32%), 404 (MH+, 35Cl, 100); HRMS: found (CI) MH+ 404.8964; C9H5OI35ClF6 requires M 404.8978; R f 0.59 (30% EtOAc in hexane).
1,1′-Oxy-bis(3,3-bis(trifluoromethyl)-3(1H)-1,2-benziodoxole) 4
In a round-bottomed flask, 21 (0.50 g; 1.2 mmol), was dissolved in CH2Cl2 (10 mL), and a solution of KOH (77 mg; 1.4 mmol) in water (1.3 mL) was added with stirring. Benzyltrimethylammonium chloride (20 mg; 0.07 mmol) was added as a PTC. After 15 min at rt, the CH2Cl2 layer decolorized, and stirring was continued for a further 2 h. The organic layer was separated and dried with MgSO4, and the solvent was removed to give a white solid (0.46 g; 97%). Purification by column chromatography (silica; 30% EtOAc in hexane as eluent) gave a white powder that was recrystallized from CH2Cl2 to give the title compound as colorless blocks (163 mg; 34%). Found: C, 28.51; H, 1.12%; calc. for C18H18O3I2F12:C, 28.67; H, 1.07%. Mp 240–241 °C; 1H NMR (100 MHz, DMSO-d) δ H 7.99–7.92 (4H, m, Ar), 7.80–7.70 (4H, m, Ar); 13C NMR (68 MHz, DMSO-d) δ C 133.9, 133.4, 131.6, 131.4, 129.5 (m), 128.6, 124.0 (q, 1JCF 290), 117.8; 19F NMR (282 MHz, DMSO-d) δ F – 75.8; IR υmax (solid)/cm−1 1261 m, 1185 s, 1142 m, 1130 m, 952 m, 730 m, 646 s; FABMS m/z 755 (MH+, 4%), 685 (100), 385 (55), 369 (35), 316 (63), 300 (39), 231 (75); HRMS: found (FAB) M+ 753.8400. C18H8O3I2F12 requires M 753.8371; R f 0.45 (30% EtOAc in hexane). Further elution afforded 1-hydroxy-3,3-bis(trifluoromethyl)-3(1H)-1,2-benziodoxole[ Citation 8 ] 7 as a white solid (270 mg; 57%). Found: C, 28.25; H, 1.18%; calc. for C9H5O2IF6:C, 28.00; H, 1.31%. Mp 242–246 °C; 1H NMR (270 MHz, DMSO-d 6) δ H 7.93–7.85 (2H, m, Ar), 7.74–7.65 (2H, m, Ar), 7.30 (1H, br s, OH interchangeable with D 2O); FABMS m/z 387 (MH+, 20%), 369 (14), 307 (19), 176 (28), 154 (100), 136 (84); R f 0.15 (30% EtOAc in hexane).
Preparation of 2-(N-(p-Toluenesulfonyl)imino)iodobenzylmethyl Ether 5
1,2-Iodobenzylmethyl Ether 8
Commercial 2-iodobenzyl alcohol (10.00 g, 42.7 mmol) was treated with sodium hydride (60% by weight in oil) (6.84 g, 170.9 mmol) in dry DMF (215 mL) at ambient temperature (1 h). The creamy reaction mass was treated with methyl iodide (11.2 mL, 179.3 mmol) and stirred at room temperature (4 h). The reaction was quenched with water, and the product was extracted into diethyl ether. Drying (MgSO4) and distillation gave (7.93 g, 72%). Bp 239–240 °C. 1H NMR (270 MHz, CDCl3) δ H 7.83–6.94 (4H, m, Ar), 4.46 (2H, s, CH 2 OMe), 3.48 (3H, s, OMe); 13C NMR (400 MHz, CDCl3) δ C 138.6, 137.4, 127.4, 126.6, 126.0, 124.0, 76.6, 56.8; R f 0.51 (10% diethyl ether in petrol).
2-(Bis-acetoxyiodo)-benzylmethyl Ether 9
Peracetic acid (37% by weight in AcOH, 50% activity) (6.63 g, 16.12 mmol) was added slowly to stirred 1,2-iodobenzylmethylether (2.00 g, 8.06 mmol) over 30 min at 30 °C. Effervescence was observed. The reaction was complete after 1 h by TLC. The reaction was chilled (∼ 0 °C), and water (30 mL) was added, forming a yellow solution. The product was extracted with chloroform (3 × 35 mL), and the combined organic layers was washed with water and dried (MgSO4). The solvent was removed, yielding the title product as a white solid (2.29 g, 78%). Mp 87–89 °C; 1H NMR (270 MHz, CDCl3) δ H 8.20 (1H, d, J = 7.9, Ar), 7.70–7.34 (3H, m, Ar), 4.73 (2H, s, CH 2 OMe) 3.47 (3H, s, OMe), 1.98 (6H, s, C(O)Me); 13C NMR (400 MHz, CDCl3) δ C 176.5, 139.5, 137.3, 132.6, 130.2, 129.6, 123.9, 76.7, 58.7, 20.3; IR νmax (CHCl3 solution)/cm−1 2933 m, 1713 s, 1647 s, 1365 s, 1275 s, 1046 m; R f 0.04 (10% diethyl ether in petrol). Compound 9 has appeared in the literature,[ Citation 6 ] but no data were presented.
2-(N-(p-toluenesulfonyl)imino)iodobenzylmethyl Ether 5
To a stirred solution of KOH (2.07 g; 37.57 mmol) in anhydrous MeOH (60 mL) under an inert atmosphere, p-toluenesulfonamide (2.57 g; 15.03 mmol) was added, and the solution was stirred for 30 min at rt. The reaction mixture was cooled to 0 °C, and 24 (5.50 g, 15.03 mmol) was added and left at 5 °C for 16 h. The resultant suspension was allowed to settle, and solvent was removed by filter canula. Recrystallization from anhydrous MeOH gave the title compound as a white powder (3.18 g; 51%). Mp 100–101 °C; 1H NMR (270 MHz, CDCl3) δ H 7.96–7.94 (1H, m, Ar), 7.84–7.81 (2H, m, Ar), 7.39–7.15 (5H, m, Ar), 4.64 (2H, s, CH 2OMe), 3.47 (3H, s, OMe), 2.35 (3H, s, ArMe); 13C NMR (68 MHz, CDCl3) δ C 141.5, 140.8, 135.5, 130.8, 130.1, 129.1, 128.6, 127.9, 126.7, 113.0, 74.3, 58.6, 21.4; IR υmax (CHCl3 solution)/cm−1 2960 w, 1566 w, 1454 w, 1128 w, 1135 s, 1082 s, 874 m.
General Procedure for the Direct Formation of Cyclic Sulfates and Sulfamidates from Olefins, Using Hypervalent Iodine Reagents
To a flame-dried Schlenk tube under an inert atmosphere, the hypervalent iodine compound (0.5 mmol) and anhydrous CH2Cl2 (2 mL) were added. The reaction mixture was stirred vigorously for 5 min until a free-flowing suspension was observed. The addition of SO3 · DMF (76 mg; 0.5 mmol) produced a yellow gum after 10 min at rt. The reaction mixture was cooled to 0 °C and alkene (0.5 mmol) added, after which time the solution was allowed to warm to rt and completion was determined by TLC analysis. Concentration of the reaction mixture in vacuo and purification by flash chromatography (silica; CH2Cl2 in hexanes as eluent) gave the pure cyclic sulfate or sulfamidate product.
Hexane-1,2-diol Cyclic Sulfate 10
Purification by filtration chromatography (over silica gel ∼1 g; 25% CH2Cl2/light petrol) to remove iodobenzene, then flushing with CH2Cl2, gave 10 (68 mg, 75%) as a colorless oil. Rf 0.65 (CH2Cl2). δ H (400 MHz, CDCl3): 5.02–4.95 (m, 1H; CHO); 4.71 (dd, 1H, J = 9.0, 6.0; CH 2α O); 4.34 (apparent t, 1H, J = 6.0; CH 2β O); 1.96–1.91 (m, 1H; CH 2α CHO); 1.83–1.75 (m, 1H; CH 2 β CHO); 1.53–1.31 (m, 4H; chain-CH 2); 0.94 (t, 3H, J = 6.7; Me). δ c (100 MHz, CDCl3): 83.0 (CH), 72.9 (CH2), 32.0, 26.7, 22.2, 13.8 (CH3). These data are in agreement with literature values.[ Citation 12 ]
5-Butyl-3-[(4-tolyl)sulfonyl]-1,2,3-oxathiazolidine 2,2-dioxide 11
Compound 11 was prepared from PhI=NTs (1.12 g, 3.0 mmol) in anhydrous dichloromethane (10 mL) using SO3 · DMF (460 mg, 3.0 mmol) and 1-hexene (0.31 mL, 2.5 mmol). Purification by flash chromatography (CH2Cl2) gave 11 (122 mg, 15%), which was isolated as a white solid; mp > 175 °C (dec.). 1H NMR (270 MHz, CDCl3) δ H 7.93 (2H, dd, J 8.0 and 1.0, Ar), 7.41 (2H, dd, J 8.0 1.0, Ar), 4.76–4.71 (1H, m, CHN), 3.97 (1H, dd, J 9.3 5.7, CH 2α O), 3.53 (1H, apparent t, J 9.1, CH 2 β O), 2.48 (3H, s, Me), 1.85–1.77 (1H, m, CH), 1.67–1.60 (1H, m, CH), 1.43–1.26 (6H, m, 3CH 2), 0.89 (3H, t, J 8.0, Me); 13C NMR (100 MHz, CDCl3) δ C 146.2, 133.0, 130.2, 128.8, 80.1, 52.1, 32.1, 26.6, 22.1, 21.8, 13.7. νmax (solid state)/cm−1: 1373 vs, 1202 s, 1169 vs, 1090 m, 1014 w, 937 m, 850 m, 815 m, 709 m, 694 m; HRMS [found (EI+) M+ 333.0714; C13H19NO5S2 requires 333.0705]. R f 0.72 (CH2Cl2).
4-Phenylbutane-1,2-diol Cyclic Sulfate 12
Preparation according to the general procedure using PhI=O and 4-phenyl butane (0.5 mmol) gave 12 as a colorless oil (85 mg; 75%). 1H NMR (270 MHz, CDCl3) δ H 7.36–7.18 (5H, m, Ar) 4.99–4.89 (1H, m, CHO), 4.63 (1H, dd, J 9.5 6.7, CH 2α O), 4.30 (1H, dd, J 9.5 9.0, CH 2β O),2.93–2.85 (1H, m, CH 2α CHO), 2.81–2.70 (1H, m, CH 2 β CHO), 2.37–2.25 (1H, m, PhCH 2α ), 2.10–1.97 (1H, m, PhCH 2β ); 13C NMR (100 MHz, CDCl3) δ C 139.1, 128.8, 128.4, 126.8, 81.8, 72.6, 33.9, 33.8; IR υmax (CHCl3 solution)/cm−1 3030 m, 2955 m, 1702 w, 1603 w, 1497 w, 1407 s, 1205 s, 1032 s, 822 m; HRMS: found (ES) MNa+ 251.0347; C10H12O4SNa requires M 251.0354; R f 0.31 (50% CH2Cl2 in hexane). These data are concordant with literature values.[ Citation 13 ]
5-(3-Phenylproyl)-3-[(4-tolyl)sulfonyl]-1,2,3-oxathiazolidine 2,2-Dioxide 13
Preparation according to general procedure using 5 and 4-phenylbutene (0.5 mmol) gave 13 as a colorless powder (37 mg; 23%); mp 130–132 °C; 1H NMR (400 MHz, CDCl3) δ H 7.90 (2H, d, J 8.4, Ar), 7.38 (2H, d, J 8.4, Ar), 7.32–7.13 (3H, m, Ar), 7.11 (2H, d, J 8.2, Ar), 4.72–4.66 (1H, m, CHO), 3.91 (1H, dd, J 9.3 5.7, CH 2α NTs), 3.53 (1H, apparent t, J 8.7, CH 2β NTs), 2.82–2.75 (1H, m, CH 2α CHO), 2.70–2.63 (1H, m, CH 2β CHO), 2.47 (3H, s, ArMe), 2.19–2.10 (1H, m, PhCH 2α ), 1.97–1.88 (1H, m, 1H; PhCH 2β ); 13C NMR (100 MHz, CDCl3) δ C 146.5, 139.3, 130.4, 129.1, 129.0, 128.9, 128.6, 127.0, 79.4, 52.2, 34.4, 31.0, 22.1; IR υmax solid/cm−1 1592 w, 1493 w, 1453 w, 1370 s, 1203 m, 1167 m, 1091 w, 1007 w, 929 m, 900 w, 814 s; EIMS m/z 278 (M-PhCH2CH2 +, 12%), 226 (Citation11), 91 (100) molecular ion not observed; R f 0.13 (50% CH2Cl2 in hexane).
Crystallographic Data
X-ray data for compounds 6, 4, and 5 are available from the Cambridge Crystallographic Database, citing CCDC-633375, CCDC-633375, and CCDC 699416, respectively. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
ACKNOWLEDGMENTS
Financial support from the European Union (EU) associated with the Ligbank® project (FP6-NMP3-C-2003-505267) and the COST (European Cooperation in the field of Scientific and Technical Research) program is acknowledged. One of us (A. N.) is grateful to GlaxoSmithKline for the provision of a studentship.
Related Research Data
REFERENCES
- (a) Tohma , H. ; Kita , Y. Hypervalent iodine reagents for the oxidation of alcohols and their application to complex molecule synthesis . Ad. Synth. Catal. 2004 , 346 , 111 – 124 ; (b) T. Wirth, T. (Ed.). Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis (Topics in Current Chemistry, 224); Springer-Verlag: Berlin, 2003 .
- Dess , D. B. ; Martin , J. C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones . J. Org. Chem. 1983 , 48 , 4155 – 4156 .
- Saltzman , H. ; Sharefkin , J. G. Iodosobenzene . Org. Synth. 1963 , 43 , 60 – 61 .
- Evans , D. A. ; Faul , M. M. ; Bilodeau , M. T. Copper-catalyzed aziridination of olefins by (N-(p-toluenesulfonyl)imino)phenyliodinane . J. Org. Chem. 1991 , 56 , 6744 – 6746 .
- Perozzi , E. F. ; Michalak , R. S. ; Figuli , G. D. ; Stevenson , W. H. ; Dess , D. B. ; Ross , M. R. ; Martin , J. C. Directed dilithiation of hexafluorocumyl alcohol—Formation of a reagent for the facile introduction of a stabilizing bidentate ligand in compounds of hypervalent sulfur (10-S-4), phosphorus (10-P-5), silicon (10-Si-5), and iodine (10-I-3). J. Org. Chem. 1981, 46, 1049–1053.
- Togo , H. ; Hoshina , Y. ; Muraki , T. ; Nakayama , H. ; Yokoyama , M. Study on radical amidation onto aromatic rings with (diacyloxyiodo)arenas . J. Org. Chem. 1998 , 63 , 5193 – 5200 .
- Braddock , D. C. ; Cansell , G. ; Hermitage , S. A. ; White , A. G. P. Bromoiodinanes with an I(III)–Br bond: Preparation, x-ray crystallography, and reactivity as electrophilic brominating agents . Chem. Commun. 2006 , 1442 – 1444 .
- Zhdankin , V. V. ; Kuehl , C. J. ; Krasutsky , A. P. ; Bolz , J. T. ; Simonsen , A. J. 1-(Organosulfonyloxy)-3(1H)-1,2-benziodoxoles: Preparation and reactions with alkynyltrimethylsilanes . J. Org. Chem. 1996 , 61 , 6547 – 6551 .
- Robinson , R. I. ; Woodward , S. Direct formation of cyclic sulfates utilising hypervalent iodine species and sulfur trioxide adducts . Tetrahedron Lett. 2003 , 44 , 1655 – 1657 .
- Cunningham , A. ; Mokal-Parekh , V. ; Wilson , C. ; Woodward , S. On the use of mixtures of organotin species for catalytic enantioselective ketone allylation—A detective story . Org. Biomol. Chem. 2004 , 741 – 748 .
- Woodward , S. In Transition Metals in Organic Synthesis , S. E. Gibson (Ed.), Oxford University Press : Oxford , 1997 ; pp. 1 – 34 ( see especially protocol 2 ).
- Zefirov , N. S. ; Sorokin , V. D. ; Zhdankin , V. V. ; Kozmin , A. S. Trivalent iodide sulfates: New reagents for olefin functioning into cyclic sulfates . Zhur. Org. Khim. 1986 , 22 , 450 – 452 .
- Zhao , H. ; Wu , Y. L. Chiral synthesis of yashabushiketol . Chin. Chem. Lett. 1994 , 5 , 367 .