Abstract
Aniline undergoes regioselective trichlorination by N-chlorosuccinimide (NCS) in acetonitrile in good yield. The product 2,4,6-trichoroaniline was converted into 1,3,5-trichlorobenzene by reduction of its diazonium salt. Reaction of the methyl carbamate of aniline with NCS gave only the 2,4-dichlorophenyl carbamate.
The synthesis of 2,4,6-trichloroaniline (1) has almost[ Citation 1 ] invariably been accomplished by chlorination of aniline with chlorine[2–9] or sulfuryl chloride.[ Citation 7 , Citation 10 , Citation 11 ] Although the latter reactions typically provide 1 in high yield, these reagents require special handling because of their highly corrosive nature. In addition, chlorine and, especially, sulfur dioxide emissions must be strictly controlled.[ Citation 12 ]
More convenient reagents for aromatic chlorinations are N-chloro reagents such as N-chlorosuccinimide (NCS). The chlorination of aniline and substituted anilines with N-chloro reagents, including NCS, has been studied.[ Citation13–15 ] The main concern was determining the ortho/para regioselectivity of monochlorination. However, it was hinted that further chlorination was possible as small amounts of 2,4-dichloroaniline impurity had also formed.[ Citation 15 , Citation 16 ] Further literature study found that 4-bromoaniline and 3,5-difluoro-4-iodoaniline had been shown to undergo dichlorination at the 2,6-position in chloroform with NCS or N-chloro-2,4-dichloroacetanilide.[ Citation 17 , Citation 18 ] Thus, it appeared aniline might undergo trichlorination with N-chloro reagents under the appropriate conditions.
Initial conditions for the trichlorination of aniline were based on the recent work of Manka and Kaszynski.[ Citation 18 ] Treating aniline with 3 equivalents of NCS in chloroform in the presence of 1 equivalent of trifluoroacetic acid gave 1 by thin-layer chromatographic (TLC) analysis. The other major component of the mixture was 2,4-dichloroaniline. This partial success was hampered by the fact that the reaction was dark purple, presumably from oxidation; no isolation of the products was attempted.
Nickson and Roche-Dolson studied the reaction of NCS upon deactivated anilines.[ Citation 19 ] They found that acetonitrile was the solvent of choice for monochlorination of, for example, 2- or 4-nitroaniline. It seemed reasonable to assume that both 4-chloroaniline and 2,4-dichloroaniline might also behave as deactivated anilines. Reacting these two compounds with NCS (1 and 2 equivalents, respectively) in refluxing acetonitrile both gave 1. Subsequently, aniline was treated in the same way with 3 equivalents NCS and, remarkably, 1 was isolated in 88% yield (Scheme ). Pouring the reaction mixture into water caused precipitation of crude 1 as a purple solid, while the by-product succinimide was washed away. Crude 1 was readily purified by treatment with charcoal followed by filtration through silica gel.
Scheme 1 Reagents and conditions: a) N-chlorosuccinimide, MeCN, reflux.
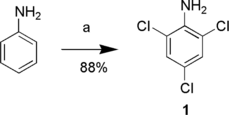
During preparations of larger batches of 1 by this procedure, it was discovered that addition of the third equivalent of NCS is accompanied by a strong exotherm. The solution adopted was to check the reaction by proton nuclear magnetic resonance and TLC after the second equivalent of NCS had been added. Typically, after 1 h of reflux, the formation of 2,4-dichloroaniline was complete, the heating was shut off, and the last equivalent of NCS was added portionwise to control the exotherm.
A cheap and more efficient substitute for NCS is trichloroisocyanuric acid (TCICA).[ Citation 20 ] Juenge et al. obtained a low yield of 2- and 4-chloroanilines from reaction of TCICA with aniline in carbon tetrachloride.[ Citation 21 ] Preliminary experiments showed that 1 is formed by reaction of aniline with TCICA in acetonitrile. The reaction was conducted at 0 °C butwas accompanied by a much greater degree of colored by-product formation than with NCS.
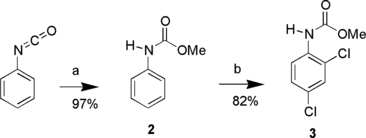
It was believed that the free amine of aniline and/or its reaction products undergoes oxidation to colored by-products. The methyl carbamate of aniline (2) was synthesized from phenyl isocyanate and methanol. Refluxing 2 with NCS in acetonitrile was essentially a colorless reaction (Scheme ). But even with 5 equivalents of NCS, chlorination only went as far as methyl 2,4-dichlorophenylcarbamate (3). Apparently, carbamate is more reactive than amide, as Zanka and Kubota[ Citation 22 ] only isolated 4-chlorophenylacetamide when acetanilide was reacted with 3 to 5 equivalents NCS in 2-propanol.
Scheme 2 Reagents and conditions: a) MeOH, reflux; b) N-chlorosuccinimide, MeCN, reflux.
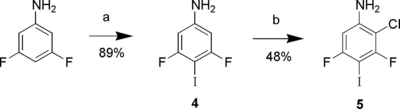
In the study by Manka and Kaszynski,[ Citation 18 ] they sought to prepare 2,6-dichloro-3,5-difluoro-4-iodoaniline by chlorination of 3,5-difluoro-4-iodoaniline (4). Chlorination of the latter with 2 equivalents of NCS in refluxing acetonitrile gave the unreported mono-chlorinated product (5) (Scheme ). Further study of conditions (longer reaction time, more NCS) that might provide the dichlorinated product was not pursued.
Scheme 3 Reagents and conditions: a) N-iodosuccinimide, MeCN; b) N-chlorosuccinimide, MeCN, reflux.
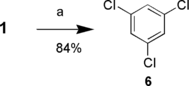
The deamination of 1 to prepare 1,3,5-trichlorobenzene (6) was investigated.[ Citation23–27 ] The recent conditions of Mehilal et al.[ Citation 9 ] were used with slight modification (Scheme ). After diazotization of 1 in aqueous sulfuric acid, hexanes were added, making a two-phase reaction mixture. This solved the problem of foaming, because nitrogen is generated during the addition of the reducing solution, as well as dissolving 1, as it precipitated from the acid phase. In addition, it was found that aqueous sodium hypophosphite was a suitable replacement for aqueous hypophorus acid in the reduction step. The yield of the deamination was 97%.
Scheme 4 Reagents and conditions: a) 1. 70% H2SO4, NaNO2, 0 °C; b) 2. hexanes, NaH2PO2, H2O, 0 °C to rt.
In conclusion, 2,4,6-trichloroaniline (1) can be synthesized readily from aniline and N-chlorosuccinimide. This reaction, coupled with the facile diazotization/deamination sequence, makes a convenient laboratory preparation of 1,3,5-trichlorobenzene (6).
EXPERIMENTAL
The melting points were collected on an electrothermal capillary melting-point apparatus and are not corrected. All NMR data were collected on a Bruker Avance II 300-MHz spectrometer (1H at 300 MHz, 13C at 75 MHz, 19F at 282 MHz). NMR data (free induction decays) were processed using NUTS software from Acorn NMR (Livermore, CA). All 1H, 13C, and 19F spectra are referenced to solvent, tetramethylsilane or fluorotrichloromethane. Aniline 99.5% ACS reagent, N-chlorosuccinimide (NCS) 98%, N-iodosuccinimide (NCI), acetonitrile (MeCN) 99.5% ACS reagent, phenyl isocyanate 98%, and sodium hypophosphite hydrate were purchased from Sigma-Aldrich (Milwaukee). Silica gel (32–63 µm, 60 Å) was purchased from Scientific Adsorbents Inc. (Atlanta). All other reagents were obtained commercially and used as received. Elemental analyses were made by Atlantic Microlab, Inc. (Norcross, GA).
2,4,6-Trichloroaniline (1)
A 5-L, two-necked, round-bottomed flask equipped with magnetic stirbar was charged with 50 g freshly distilled aniline (0.54 mol) in 1 L MeCN. One neck was equipped with a reflux condenser and N2 purge, and the second neck was glass stoppered. The solution was brought to reflux. N-Chlorosuccimide (86 g, 1 equiv) was added through the glass-stoppered neck in one portion. The color of the reaction became purple-brown. After 1 h, a second addition of 86 g NCS (1 equiv) was made in one portion After 1 h, NMR of the reaction mixture showed only traces of NCS remained in addition to formation of primarily 2,4-dichloroaniline. The final addition of 86 g NCS (1 equiv) was made in eight ∼10-g portions, as the reaction occurs with a vigorous exotherm. After the addition, TLC showed the reaction was complete. The mixture was cooled to rt and poured into 3 L vigorously stirred H2O. After stirring 1 h, the light purple solid was collected on a coarse-porosity glass frit, washed with 1 L H2O, and air-dried on the frit for 2 h. The crude was dissolved in 1 L Et2O and washed with 500 mL H2O and 500 mL brine. The organic layer was dried with anhydrous MgSO4 and filtered. The filtrate was treated with 11 g Darco G-60 for 1 h. The mixture was filtered through diatomaceous earth, and the solvent was rotary evaporated. The red-brown solid was dissolved in 1 L hexanes. The solution was filtered through 300 g SiO2 on a medium-porosity glass frit that removed most of the color. The filtrates were rotary evaporated, leaving 111.6 g of the title compound as a pale yellow solid that was >97% by 1H NMR (88%). Recrystallization from EtOH gave the title compound in analytically pure form as soft, colorless needles. Mp 68–70 °C [lit.[ Citation 4 ] 77.5 °C]. δ H (CDCl3): 7.13 (s, 2H), 4.36 (bs, NH2); δ C (CDCl3): 139.24, 127.81, 122.06, 119.92. Elemental analysis calculated for C6H4Cl3N: C, 36.68; H, 2.05; N, 7.13. Found: C, 36.87; H, 1.93; N, 7.07.
Methyl Phenylcarbamate (2)
A 100-mL, round-bottomed flask equipped with magnetic stirbar was charged with 50 mL anhydrous MeOH. With vigorous stirring, 5 g phenyl isocyanate (0.04 mol) was added in one portion. The mixture was refluxed for 30 min, and the solvent was rotary evaporated. The remaining colorless liquid spontaneously crystallized. The colorless crystals were slurried with 20 mL hexanes and filtered (6.1 g, 97%). Mp 37–39 °C [lit.[ Citation 28 ] 47 °C]. δH (CDCl3): 7.45–7.36 (m, 2H), 7.36–7.26 (t, J = 7.4 Hz, 2H), 7.07 (t, J = 7.4 Hz, 1H), 6.82 (bs, NH), 3.78 (s, 3H); δC (CDCl3): 154.33, 138.08, 129.21, 123.64, 118.99, 52.48. Elemental analysis calculated for C8H9NO2: C, 63.56; H, 6.00; N, 9.27. Found: C, 63.46; H, 6.01; N, 9.28.
Methyl 2,4-Dichlorophenylcarbamate (3)
A 100-mL, round-bottomed flask equipped with magnetic stirbar was charged with 1.51 g 2 (0.01 mol), 6.65 g NCS (0.05 mol, 5 equiv), and 20 mL MeCN. The mixture was refluxed for 12 h. After cooling, the mixture was poured into 250 mL 5% Na2SO3, and a solid precipitated. The solid was filtered on a coarse-porosity glass frit and washed well with water. After air drying, the crude was recrystallized from hexanes to give the title compound as colorless needles (1.8 g, 82%). Mp 58–62 °C. δH (CDCl3): 8.12 (d, J = 8.9 Hz, 1H), 7.35 (d, J = 2.2 Hz, 1H), 7.24 (dd, J = 8.9 and 2.4 Hz, 1H), 7.09 (bs, NH), 3.80 (s, 3H); δC (CDCl3): 153.67, 133.72, 128.92, 128.39, 128.12, 122.70, 120.81, 52.88. Elemental analysis calculated for C8H7Cl2NO2: C, 43.66; H, 3.21; N, 6.37. Found: C, 43.84; H, 3.09; N, 6.32.
3,5-Difluoro-4-iodoaniline (4)
A 100-mL, round-bottomed flask equipped with magnetic stirbar was charged with 2.43 g 3,5-difluoroaniline (18.8 mmol) in 30 mL MeCN. The mixture was cooled in an ice bath, and 4.24 g NIS (18.8 mmol, 1 equiv) were added all at once. The cooling bath was removed, and the mixture was stirred at rt for 30 min. After this time, TLC showed the reaction was complete. The mixture was poured into a mixture of 200 mg Na2SO3 dissolved in 200 mL H2O. The mixture was extracted twice with 50-mL portions of Et2O. The organic phases were collected and washed with 50 mL H2O followed by 50 mL brine. After drying with anhydrous MgSO4, the solvent was evaporated, leaving 4.3 g of a tan solid (89%). The title compound was obtained as colorless needles by recrystallization from heptane. Mp 104–106 °C (lit.[ Citation 29 ] 112–114 °C). δ H (CDCl3): 6.26 (d, J = 8.1 Hz, 1H), 3.95 (bs, NH2); δ C (CDCl3): 163.36 (dd, 1 J CF = 243 Hz and 3 J CF = 8.7 Hz), 149.46 (t, 3 J CF = 13.0 Hz), 98.51 (dd, 2 J CF = 28.1 and 4 J CF = 2.4 Hz), 55.18 (t, 2 J CF = 30.3 Hz). Elemental analysis calculated for C6H4F2IN: C, 28.26; H, 1.58; N, 5.49. Found: C, 28.10; H, 1.42; N, 5.44.
2-Chloro-3,5-difluoro-4-iodoaniline (5)
A 100-mL, round-bottomed flask equipped with magnetic stirbar was charged with 2.43 g 4 (16.9 mmol), 4.5 g NCS (33.7 mmol, 2 equiv), and 50 mL MeCN. The mixture was refluxed for 18 h, then poured into a mixture of 1 g Na2SO3 dissolved in 200 mL H2O. The mixture was extracted twice with 50-mL portions of Et2O. The organic phases were collected and washed with 50 mL H2O followed by 50 mL brine. After drying with anhydrous MgSO4, the solvent was evaporated, leaving 4.46 g of a tan solid. The title compound was obtained as clear tan needles by recrystallization from heptane (2.36 g, 48%). Mp 67–70 °C. δH (CDCl3): 6.39 (dd, 3 J HF = 9.2 Hz and 5 J HF = 1.9 Hz, 1H), 4.37 (bs, NH2); δC (CDCl3): 161.21 (dd, 1 J CF = 243.7 Hz and 3 J CF = 8.1 Hz), 158.59 (dd, 1 J CF = 243.7 Hz and 3 J CF = 8.8 Hz), 145.50 (dd, 3 J CF = 13.0 Hz and 3 J CF = 4.4 Hz), 102.46 (dd, 2 J CF = 23.0 Hz and 4 J CF = 4.0 Hz), 97.97 (dd, 3 J CF = 29.0 Hz and 4 J CF = 2.7 Hz), 55.35 (dd, 2 J CF = 32.7 Hz and 2 J CF = 30.4 Hz); δF (CDCl3): −92.92 (t, 5 J FH and 4 J FF = 2.0 Hz, 5-F), −96.07 (dd, 3 J FH = 9.3 Hz and 4 J FF = 3.1 Hz, 3-F). Elemental analysis calculated for C6H3ClF2IN: C, 24.90; H, 1.04; N, 4.84. Found: C, 25.10; H, 1.09; N, 4.88.
1,3,5-Trichlorobenzene (6)
A 500-mL, round-bottomed flask equipped with magnetic stirbar was charged with 55 mL 70% H2SO4 and stirred, then 11 g 1 (0.056 mol) were added portionwise. After all the solid was added, the mixture was allowed to stir until all the solids dissolved, forming an orange solution. The mixture was then placed in an ice bath to stir, and a solution of 7.7 g NaNO2 (0.112 mol, 2 equiv) in 11 mL H2O was added dropwise over 30 min. After the addition, the mixture was stirred for 15 min. Cold hexanes were added to prevent foaming and to dissolve the product. An aqueous solution of sodium hypophosphite was added dropwise. The evolution of N2 occurred shortly afterward. The color gradually changed from orange to colorless. After stirring for 30 min, the ice bath was removed, and the mixture was stirred at rt for several hours. The phases were split, and the aqueous layer was further extracted with hexanes. The combined extracts were washed with H2O, saturated NaHCO3, and then brine. After drying over MgSO4 and filtration, the filtrate was filtered through SiO2 to remove the color. The colorless filtrates were rotary evaporated, leaving the crude product as a white solid. The entire mixture was poured onto 250 mL H2O, and the solids were collected on a coarse-porosity glass frit and washed with copious H2O. After air drying on the frit, the crude solid weighed 8.5 g (84%). Mp 57–60 °C [lit.[ Citation 9 ] 62 °C] δ H (CDCl3): 7.28; δ C (CDCl3): 135.79, 127.45. Elemental analysis calculated for C6H3Cl3: C, 39.72; H, 1.67. Found: C, 39.97; H, 1.68.
ACKNOWLEDGMENTS
The financial support of Timothy I. Mahoney (NAWCWD Pilot Plant) is gratefully acknowledged. Thanks to Juanita K. Morton (NAWCWD) for assistance in purchasing the hypophosporous acid. Thanks to Ann M. Moorehead, Cynthia M. Kitchens, and the staff of the NAWC Technical Library (China Lake) for obtaining Ref. 9.
REFERENCES
- Muathen , H. A. Oxidative halogenation of aromatic compounds with metal halides and sodium bismuthate . Helv. Chim. Acta. 2003 , 86 , 164 – 168 .
- Hofmann , A. W. Metamorphosen des indigo's: Erzeugung organischer basen, welche chlor und brom enthalten . Ann. 1845 , 53 , 1 – 57 .
- Meyer , V. ; Sudborough , J. J. Weiteres über die esterbildung aromatischer säuren . Ber. 1894 , 27 , 3146 – 3153 .
- Sudborough , J. J. Chlorination of aniline . J. Chem. Soc. 1894 , 65 , 1028 – 1031 .
- Chattaway , F. D. ; Irving , H. 2:4:6-Trichloroaniline . J. Chem. Soc. 1933 , 142 – 143 .
- Orloff , H. D. ; Napolitano , J. P. Preparation of 2,4,6-trichloroaniline . U.S. Patent 2,675,409, Apr. 13, 1954 .
- Werner , F. ; Roxo , B. ; Mannes , K. ; Trescher , V. Process for the preparation of 2,4,6-trichloroaniline . U.S. Patent 4,447,647, May 8, 1984 .
- Kim , C.-U. ; Jin , H.-J. ; Lee , S.-B. ; Lee , J.-M. A study on the synthesis of 2,4,6-trichloroaniline by chlorination of aniline . Hwahak Konghak 1990 , 28 , 230 – 236 .
- Mehilal ; Salunke , R. B. ; Agrawal , J. P. A newly developed synthesis of 1,3,5-trichlorobenzene (syn. TCB) from aniline . Indian J. Chem., Sect. B. 2002 , 41 , 604 – 607 .
- Wenghöffer , L. Ueber das verhalten von sulfurylchlorid und aethylschwefelsäurechlorid gegen anilin und anilide . J. Prakt. Chem. 1877 , 16 , 448 – 466 .
- Eller , W. ; Klemm , L. Über die einwirkung von sulfurylchlorid auf aromatische amine . Ber. 1922 , 55 , 217 – 224 .
- U.S. Clean Air Act. Public Law 101–549, 1990 .
- Chao , T. H. ; Cipriani , L. P. The chlorination of N,N-dimethylaniline . J. Org. Chem. 1961 , 26 , 1079 – 1081 .
- Searle , N. E. ; Cupery , H. E. Synthesis of carbon-14 labeled 3-(p-chlorophenyl)-1,1-dimethylurea . J. Org. Chem. 1954 , 19 , 1622 – 1627 .
- Neale , R. S. ; Schepers , R. G. ; Walsh , M. R. The chlorinations of reactive anilines . J. Org. Chem. 1964 , 29 , 3390 – 3393 .
- Lindsay Smith , J. R. ; McKeer , L. C. ; Taylor , J. M. Highly selective aromatic chlorinations, part 2: The chlorination of substituted phenols, anisoles, anilines, and related compounds with N-chloroamines in acidic solution . J. Chem. Soc. Perkin Trans. 2 , 1988 , 2 , 385 – 391 .
- Reed , W. W. ; Orton , K. J. P. The wandering of bromines in the chlorination of bromoanilines . J. Chem. Soc. 1907 , 91 , 1543 – 1554 .
- Manka , J. T. ; Kaszynski , P. Synthesis and thiolation of 1,3-difluoro-2,4,6-trihaloanilines and benzenes. J. Flourine Chem. 2003, 124, 39–43.
- Nickson , T. E. ; Roche-Dolson , C. A. A convenient procedure for the chlorination of deactivated anilines . Synthesis 1985 , 669 – 670 .
- Tilstam , U. ; Weinmann , H. Trichloroisocyanuric acid: A safe and efficient oxidant . Org. Process Res. Dev. 2002 , 6 , 384 – 393 .
- Juenge , E. C. ; Beal , D. A. ; Duncan , W. P. Chlorination of aromatic systems with trichloroisocyanuric acid under polar and free-radical conditions . J.Org. Chem. 1970 , 35 , 719 – 722 .
- Zanka , A. ; Kubota , A. Practical and efficient chlorination of deactivated anilines and anilides with NCS in 2-propanol . Synlett 1999 , 1984 – 1986 .
- Jackson , C. L. ; Lamar , W. R. On certain derivatives of trichlorodinitrobenzol . Am. Chem. J. 1896 , 18 , 664 – 685 .
- Doyle , M. P. ; Dellaria , J. F. , Jr. ; Siegfried , B. ; Bishop , S. W. Reductive deamination of arylamines by alkyl nitrites in N,N-dimethylformamide: A direct conversion of arylamines to aromatic hydrocarbons . J. Org. Chem. 1977 , 42 , 3494 – 3498 .
- Lahoti , R. J. ; Parameswaran , V. ; Wagle , D. R. Reduction of aryldiazonium salts to arenes . Indian J. Chem., Sect. B 1981 , 20 , 767 – 769 .
- Wassmundt , F. W. ; Kiesman , W. F. Efficient catalysis of hydrodediazoniations in dimethylformamide . J. Org. Chem. 1995 , 60 , 1713 – 1719 .
- Mitsuhashi , H. ; Kawakami , T. ; Suzuki , H. A mild one-pot deamination of aromatic amines bearing electron-withdrawing groups: Calcium hypophosphite as a dediazonation reagent in nonaqueous media . Tetrahedron Lett. 2000 , 41 , 5567 – 5569 .
- Hentschel , W. Ueberführung von carbanilsäureäther in amidobenzoësäure . Ber. 1885 , 18 , 977 – 981 .
- Tomcufcik , A. S. ; Seeger , D. R. The synthesis of the 3′-fluoro and 3′,5′-difluoro derivatives of 4-amino-4-deoxy-N10-methylpteroylglutamic acid (methotrexate) . J. Org. Chem. 1961 , 26 , 3351 – 3356 .