
Abstract
Reliable reactions for the synthesis of two interesting anthracenetetrones have been identified and optimized. Both syntheses start from dihydroxy-9,10-anthraquinones and were selected for maximized efficiency and minimized workload. Work-up of all reactions can be achieved without column chromatography, which facilitates further scale-up. So far, both target compounds are considerably underexplored despite their promising molecular structure for use in devices and in organic synthesis, especially as building blocks for π-conjugated compounds. The crystal structure of 1,4,5,8-anthracentetrone is reported.
Graphical Abstract
Introduction
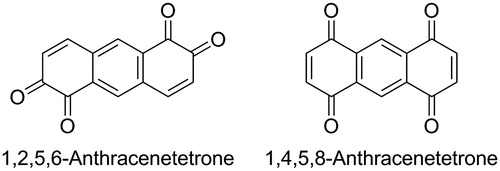
Anthracenetetrones are interesting compounds for technological applications and further conversion. For example, 1,4,9,10-anthracenetetrone (Scheme 1, top), which is currently the best investigated anthracenetetrone, was used to modify electrodes for supercapacitors,[Citation1] was applied as acceptor in charge-transfer complexes,[Citation2–4] it served as a precursor in the synthesis of antitumor compounds,[Citation5] and afforded push–pull chromophores in cycloaddition reactions with electron-rich alkynes.[Citation6] Investigations of this anthracenetetrone are facilitated by its facile synthesis in a single high-yielding reaction that uses low-cost 1,4-dihydroxy-9,10-anthraquinone as the starting material (Scheme 1, top).[Citation7] Different to other anthracenetetrones, also the crystal structure of this compound has been reported.[Citation8]
Scheme 1. Facile synthesis of 1,4,9,10-antracenetrone starting from 1,4-dihydroxy-9,10-anthraquinone (top), molecular structure of 1,2,5,6- and 1,4,5,8-anthracenetetrone (bottom).
In contrast, the synthesis of 1,2,5,6- and 1,4,5,8-anthracenetetrone (Scheme 1, bottom) is more challenging.[Citation9–12] This may explain why these compounds are less investigated despite the large number of available reactions for converting quinones, including our new conversion into cyanoarenes (demonstrated for 5,7,12,14-pentacenetetrone and a range of other para- and ortho-quinones).[Citation13–16] Beside other syntheses, the two tetrones are particularly interesting for the synthesis of π-conjugated small molecules and polymers, but they have been used in only a few reactions so far.
1,2,5,6-Anthracenetetrone was used for condensation with aromatic ortho-diamines, a reaction first demonstrated by Boldt.[Citation9] This reaction yields fully aromatic compounds by connecting the building blocks via pyrazine units. It has later been shown that the corresponding polycondensation with tetraamines yields fully conjugated ladder polymers.[Citation17] The polymers have not yet been tested in devices, but Bunz et al. recently optimized the condensation for larger ortho-diamines and tested the resulting compounds in organic light-emitting diodes.[Citation18] Regarding applications, environmentally benign methods for (poly)condensations, which afford highly crystalline materials, open up new opportunities.[Citation19,Citation20]
1,4,5,8-Anthracenetetrone, on the other hand, was used as a dienophile in double Diels–Alder cycloaddition reactions, which – depending on the diene – can yield macrocyclic compounds,[Citation21] substituted 5,7,12,14-pentacenetetrones,[Citation22] and pentacene precursors.[Citation23] The absorption and electrochemical properties of this tetrone, which is the most stable anthracenetetrone according to a computational study,[Citation24] were reported decades ago.[Citation11,Citation25] In a more recent study, the binding of this tetrone on graphene was computationally predicted to be the strongest of several electroactive compounds for green battery electrodes.[Citation26] Such strong binding benefits the cycling performance of batteries.
It was our aim to facilitate the use and investigation of 1,2,5,6- and 1,4,5,8-anthracenetetrone beyond computational studies by identifying and evaluating reliable synthetic routes to these tetrones, starting from dihydroxy-9,10-anthraquinones (as for the synthesis of 1,4,9,10-anthracenetetrone). The syntheses should be optimized regarding workload and efficiency.
Results and discussion
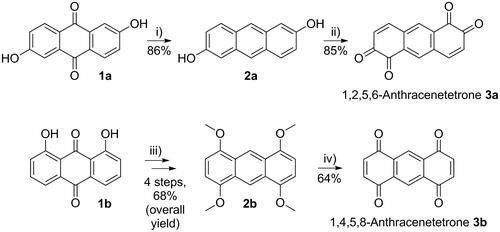
For the synthesis of 1,2,5,6-anthracenetetrone 3a, oxidation of 2,6-dihydroxyanthracene 2a (Scheme 2, top) using benzeneseleninic acid anhydride was reported to be superior to other reactions.[Citation10] Fortunately, the required precursor 2a can be efficiently prepared from 2,6-dihydroxy-9,10-anthraquinone 1a. Screening the literature for reliable protocols to minimize the workload and maximize the yields, we discovered a protocol reported by Guo et al.[Citation27] Following this protocol, we immediately achieved 86% yield (just 2% less than reported). There is no need for column chromatography or recrystallization to achieve high purity, which significantly reduces the workload.
Scheme 2. (i) Reduction of 2,6-dihydroxy-9,10-anthraquinone (1a) to 2,6-dihydroxyanthracene (2a). Conditions: NaBH4, 1 M Na2CO3 solution, under argon, RT, overnight. (ii) Oxidation to 1,2,5,6-anthracenetetrone (3a). Conditions: benzeneseleninic acid anhydride, dry THF, under argon, 50 °C, 3 h. (iii) Conversion of 1,8-dihydroxy-9,10-anthraquinone (1b) into 1,4,5,8-tetramethoxyanthracene (2b). Conditions: see Ref. [Citation28], and (iv) Oxidation to 1,4,5,8-anthracenetetrone 3b. Conditions: silica-supported ceric ammonium nitrate (CAN), CH2Cl2, RT, 1 h.
![Scheme 2. (i) Reduction of 2,6-dihydroxy-9,10-anthraquinone (1a) to 2,6-dihydroxyanthracene (2a). Conditions: NaBH4, 1 M Na2CO3 solution, under argon, RT, overnight. (ii) Oxidation to 1,2,5,6-anthracenetetrone (3a). Conditions: benzeneseleninic acid anhydride, dry THF, under argon, 50 °C, 3 h. (iii) Conversion of 1,8-dihydroxy-9,10-anthraquinone (1b) into 1,4,5,8-tetramethoxyanthracene (2b). Conditions: see Ref. [Citation28], and (iv) Oxidation to 1,4,5,8-anthracenetetrone 3b. Conditions: silica-supported ceric ammonium nitrate (CAN), CH2Cl2, RT, 1 h.](/cms/asset/cbe3ab5e-6ad3-47d7-8ce2-393f70798ac3/lsyc_a_1483027_sch0002_b.jpg)
In contrast to the synthesis of 2a, the subsequent oxidation to 3a required considerable optimization. Initially, we followed a protocol reported by Boldt et al.,[Citation10] which relies on crystallization of 3a from the reaction medium at room temperature and additionally requires extraction and crystallization steps to improve the yield to 71%. Unfortunately, we did not observe any crystallization of 3a at room temperature (also storing at −18 °C overnight just afforded small amounts of precipitate). As we wanted to simplify the protocol anyways, we decided to directly evaporate the reaction solvent as well as volatile by-products instead of storing the reaction for crystallization. The resulting solid was then recrystallized from 1,4-dioxane affording tetrone 3a as red needles. Rapid weathering prevented analysis of the crystal structure, but NMR spectroscopy confirmed the purity, except for some 1,4-dioxane, which disappears during storage or when heating the solid, as was observed before.[Citation9] Very good yields of 85% were obtained.
Our other target compound, 1,4,5,8-anthracenetetrone 3b, can be prepared by oxidation of 1,4,5,8-tetrahydroxyanthracene using freshly prepared silver oxide,[Citation29] but the yields of this reaction (and the five reactions needed to prepare the tetrahydroxy anthracene) were reported to be unsatisfactory by Miller et al.[Citation11] Instead, they suggest to oxidize 5,8-dimethoxy-1,4-anthraquinone, which they prepared in a sequence of seven reactions. Obviously, both routes do not comply with our aims of high yields and low workload.
Fortunately, tetrone 3b can also be prepared by oxidation of 1,4,5,8-tetramethoxyanthracene 2b (Scheme 2, bottom) using ceric ammonium nitrate (CAN),[Citation12] a reaction that seemed more promising to us despite the reported moderate yield of 45% and the not reported experimental details and characterization results. The required tetramethoxy anthracene 2b can be prepared from 1-bromo-2,5-dimethoxybenzene.[Citation30] The easy accessibility of this precursor certainly is an advantage of this reaction, but the low yield of 32% as well as the need for lithium 2,2,6,6-tetramethylpiperidide (LTMP) as the reagent are disadvantages that have to be considered. We decided to follow a different approach starting from low-cost 1,8-dihydroxy-9,10-anthraquinone 1b (Scheme 2, bottom), as this approach was reported to be useful also on a larger scale:
Starting from 1b, an overall yield of 68% was reported for the synthesis of 2b by methylation of the hydroxyl groups, subsequent reduction to 1,8-dimethyoxy anthracene, 2-fold bromination to 1,8-dibromo-4,5-dimethoxy anthracene, and final replacement of the bromo substituents by methoxy groups.[Citation28] Despite carrying out this reaction sequence on a 50% larger scale, we achieved exactly the same overall yield at the first try (lower yield for the methylation, higher yield for the reduction, similar yields for the two other steps). We followed the published protocol with slight adaptions: the crude methylation product was concentrated in vacuo and redissolved in CH2Cl2 before passing through a pad of silica, 2 N NaOH was used for washing the crude bromination product instead of 5% HCl, and 2b was triturated in boiling CH2Cl2 instead of room temperature. In contrast to the alternative synthesis of 2b, which uses 1-bromo-2,5-dimethoxybenzene as a precursor, none of the reactions required purification by column chromatography. Nevertheless, the synthesis using 1-bromo-2,5-dimethoxybenzene may be more convenient, if only small amounts of 2b are required.
For the final oxidation to target compound 3b, we developed a protocol based on a work using silica-supported CAN for the synthesis of benzoquinones.[Citation31] Besides other advantages, silica-supported CAN can be easily removed by filtration after the reaction. Furthermore, it allows for using CH2Cl2 as the solvent, which dissolves 2b better than the usual aqueous acetonitrile. The reaction was carried out simply by adding CH2Cl2 and 2b to freshly prepared silica-supported CAN and stirring for 1 h at room temperature. Facile work-up afforded crude 3b in quantitative yields. For removing impurities, the solid was recrystallized from 1,4-dioxane, which reduced the yield to 64%. Despite the loss during recrystallization, this is still significantly higher than reported before without experimental details.
In contrast to 3a, needles of 3b were suitable for structural characterization ( and Supplementary Material). In the solid state, 3b is essentially flat (largest distances from least-squares plane defined by the C-atoms: O2, 0.0385(9) Å and C3, 0.0183(10) Å). It crystallizes with one molecule of 1,4-dioxane in the space group P1¯. The asymmetric unit contains half of a formula unit (Z′ = 1/2), whereby 3b and 1,4-dioxane are located each on a center of inversion. No significant π–π stacking is observed. In addition to these results, we were also able to determine the crystal structure of the precursor 1,8-dibromo-4,5-dimethoxyanthracene, which has not yet been reported (in contrast to the crystal structure of the direct precursor 2b) and is provided as Supplementary Material.
Figure 1. Crystal structure of 3b·1,4-dioxane viewed down (a) [100] and (b) [010]. (c) The molecular structure of 3b. C (grey) and O (red) atoms are represented by ellipsoids drawn at the 50% probability levels; H atoms by white spheres of arbitrary radius.
![Figure 1. Crystal structure of 3b·1,4-dioxane viewed down (a) [100] and (b) [010]. (c) The molecular structure of 3b. C (grey) and O (red) atoms are represented by ellipsoids drawn at the 50% probability levels; H atoms by white spheres of arbitrary radius.](/cms/asset/5d68e1f9-605f-4cca-addd-7f48a2892f52/lsyc_a_1483027_f0001_c.jpg)
Conclusions
1,2,5,6- and 1,4,5,8-Anthracenetetrone 3a and 3b can be prepared in efficient reactions starting from dihydroxy-9,10-anthraquinones by following the presented synthetic routes. None of the reactions requires column chromatography for purification and the workload is generally low. For the synthesis of the precursor 2b, an alternative procedure is available, which may be preferred on a smaller scale. Both 3a and 3b form solvates when recrystallized from 1,4-dioxane, but the solvent can be removed easily. The developed synthetic routes greatly facilitate the exploration of tetrones 3a and 3b in devices and in the synthesis of π-conjugated small molecules and polymers.
Experimental
Reagents and solvents were purchased from commercial suppliers and used without further purification. Anhydrous solvents were prepared by filtration through drying columns (methanol, THF) or used as received from commercial suppliers (acetone). Reactions were monitored by thin-layer chromatography (Merck, silica gel 60 F254).
2,6-Dihydroxyanthracene 2a was prepared from 2,6-dihydroxy-9,10-anthraquinone 1a following a published protocol.[Citation27] The synthesis of 1,4,5,8-tetramethoxyanthracene 2b from 1,8-dihydroxy-9,10-anthraquinone 1b was also achieved following a published procedure, but with minor changes described above.[Citation28]
NMR spectra were recorded at 600 MHz for 1H and 151 MHz for 13C on a Bruker Avance III HD spectrometer. 1H NMR signals were assigned as suggested by MestReNova 12.0.1-20560 (Supplementary Material). For 13C NMR signals, the number of attached protons (C, CH) was assigned by attached proton test (APT) experiments.
Synthesis of 1,2,5,6-anthracenetetrone 3a
2,6-Dihydroxyanthracene 2a (263 mg, 1.25 mmol, 1.00 equiv.) was dissolved in 25 mL dry THF and purged with argon. The solution was then added in portions to a stirred solution of benzeneseleninic acid anhydride (900 mg, 2.50 mmol, 2.00 equiv.) in 50 mL dry THF under argon (in a three-necked round-bottom flask equipped with a reflux condenser with argon balloon and a thermometer). The reaction was heated to 50 °C for 3 h and was then allowed to cool to room temperature. After storing overnight, the solvent and volatile by-products were removed in vacuo using a rotary evaporator, which was placed in a fume hood to prevent inhalation of volatile selenium by-products. Recrystallization of the resulting solid from 1,4-dioxane afforded red needles (1,4-dioxane solvates), which were dried to afford pure tetrone 3a (252 mg, 1.06 mmol) in yields of 85%. 1H NMR (600 MHz, THF-d8): δ = 8.10 (s, 2H), 7.64 (d, J = 10.2 Hz, 2H), 6.44 (d, J = 10.2 Hz, 2H) ppm (in accordance with the literature[Citation18]). 1H NMR (600 MHz, CDCl3): δ = 8.13 (s, 2H), 7.57 (d, J = 10.2 Hz, 2H), 6.62 (d, J = 10.2 Hz, 2H) ppm. 13C NMR (151 MHz, CDCl3): δ = 179.5 (C), 177.9 (C), 143.3 (CH), 136.4 (C), 135.8 (C), 130.9 (CH), 130.1 (CH) ppm.
Synthesis of 1,4,5,8-anthracenetetrone 3b
Ceric ammonium nitrate (CAN) (6.03 g, 11.0 mmol, 5.50 equiv.) was dissolved in 6 mL water and added dropwise to 13 g silica (in a stirred 100 mL round-bottom flask equipped with a septum). The solid was stirred until a free-flowing yellow solid was obtained (about 30 min). 25 mL of CH2Cl2 was then added, followed by the addition of 1,4,5,8-tetramethoxyanthracene 2b (597 mg, 2.00 mmol, 1.00 equiv.) suspended in 35 mL CH2Cl2. Another 10 mL CH2Cl2 was used to transfer residues of 2b. The reaction stirred at room temperature for 1 h and was then filtered through a sintered glass funnel to remove the silica. The silica was washed with 200 mL CH2Cl2 and the combined solutions were washed with water and dried over sodium sulfate. The solvent was removed in vacuo and the solid residue was recrystallized from 1,4-dioxane, which afforded red needles (1,4-dioxane solvates). After drying, tetrone 3b (303 mg, 1.27 mmol) was obtained in yields of 64%. 1H NMR (600 MHz, CDCl3): δ = 8.82 (s, 2H), 7.14 (s, 4H) ppm (in accordance with the literature apart from reversed integrals[Citation11]). 13C NMR (151 MHz, CDCl3): δ = 183.3 (C), 139.4 (CH), 135.1 (C), 125.8 (CH) ppm.
Supplemental Material
Download PDF (994.6 KB)Acknowledgments
We thank Markus Lunzer and Andreas J. Morawietz for contributing to synthetic experiments. Brigitte Holzer and Christian Hametner are acknowledged for NMR measurements. The authors acknowledge the TU Wien University Library for financial support through its Open Access Funding Program.
Additional information
Funding
Related Research Data
References
- Isikli, S.; Díaz, R. J. Power Sources 2012, 206, 53–58. DOI:10.1016/j.jpowsour.2012.01.088.
- Kaul, B. B.; Yee, G. T. Polyhedron 2001, 20, 1757–1759. DOI:10.1016/S0277-5387(01)00685-4.
- Asahi, H.; Inabe, T. Synth. Met. 1995, 70, 1117–1118. DOI:10.1016/0379-6779(94)02780-3.
- Asahi, H.; Inabe, T. Chem. Mater. 1994, 6, 1875–1879. DOI:10.1021/cm00046a050.
- Schenck, L. W.; Kuna, K.; Frank, W.; Albert, A.; Asche, C.; Kucklaender, U. Bioorg. Med. Chem. 2006, 14, 3599–3614. DOI:10.1016/j.bmc.2006.01.026.
- Dengiz, C.; Swager, T. M. Synlett 2017, 28, 1427–1431. DOI:10.1055/s-0036-1588771.
- Cassis, R.; Valderrama, J. A. Synth. Commun. 1983, 13, 347–356. DOI:10.1080/00397918308066988.
- Kitamura, C.; Kawase, T. Acta Crystallogr., Sect. E 2013, 69, o1597. DOI:10.1107/S1600536813026342.
- Boldt, P. Chem. Ber. 1966, 99, 2322–2336. DOI:10.1002/cber.19660990731.
- Zippel, S.; Boldt, P. Synthesis 1997, 1997, 173–175. DOI:10.1055/s-1997-1171.
- Almlöf, J. E.; Feyereisen, M. W.; Jozefiak, T. H.; Miller, L. L. J. Am. Chem. Soc. 1990, 112, 1206–1214. DOI:10.1021/ja00159a049.
- Cory, R. M.; McPhail, C. L.; Dikmans, A. J. Tetrahedron Lett. 1993, 34, 7533–7536. DOI:10.1016/S0040-4039(00)60392-1.
- Glöcklhofer, F.; Lunzer, M.; Stöger, B.; Fröhlich, J. Chem. - Eur. J. 2016, 22, 5173–5180. DOI:10.1002/chem.201600004.
- Glöcklhofer, F.; Kautny, P.; Fritz, P.; Stöger, B.; Fröhlich, J. ChemPhotoChem 2017, 1, 51–55. DOI:10.1002/cptc.201600018.
- Glöcklhofer, F.; Morawietz, A. J.; Stöger, B.; Unterlass, M. M.; Fröhlich, J. ACS Omega 2017, 2, 1594–1600. DOI:10.1021/acsomega.7b00245.
- Glöcklhofer, F.; Petritz, A.; Karner, E.; Bojdys, M. J.; Stadlober, B.; Fröhlich, J.; Unterlass, M. M. J. Mater. Chem. C 2017, 5, 2603–2610. DOI:10.1039/c7tc00143f.
- Imai, K.; Kurihara, M.; Mathias, L.; Wittmann, J.; Alston, W. B.; Stille, J. K. Macromolecules 1973, 6, 158–162. DOI:10.1021/ma60032a002.
- Hahn, S.; Koser, S.; Hodecker, M.; Tverskoy, O.; Rominger, F.; Dreuw, A.; Bunz, U. H. F. Chem. - Eur. J. 2017, 23, 8148–8151. DOI:10.1002/chem.201701304.
- Baumgartner, B.; Bojdys, M. J.; Unterlass, M. M. Polym. Chem. 2014, 5, 3771–3776. DOI:10.1039/C4PY00263F.
- Baumgartner, B.; Svirkova, A.; Bintinger, J.; Hametner, C.; Marchetti-Deschmann, M.; Unterlass, M. M. Chem. Commun. 2017, 53, 1229–1232. DOI:10.1039/c6cc06567h.
- Cory, R. M.; McPhail, C. L.; Dikmans, A. J.; Vittal, J. J. Tetrahedron Lett. 1996, 37, 1983–1986. DOI:10.1016/0040-4039(96)00263-8.
- Bénard, C. P.; Geng, Z.; Heuft, M. A.; VanCrey, K.; Fallis, A. G. J. Org. Chem. 2007, 72, 7229–7236. DOI:10.1021/jo0709807.
- Chow, T. J.; Wu, C.-C.; Chuang, T.-H.; Hsieh, H.-H.; Huang, H.-H.; Synthesis and applications of soluble pentacene precursors and related compounds. U.S. Patent 8,277,903 B2, 2012.
- Nourmohammadian, F.; Salami, S. J. Mol. Struct.: THEOCHEM 2008, 861, 147–148. DOI:10.1016/j.theochem.2008.04.026.
- Fukuda, M.; Tajiri, A.; Oda, M.; Hatano, M. Bull. Chem. Soc. Jpn. 1983, 56, 592–596. DOI:10.1246/bcsj.56.592.
- Yu, Y.-X. ACS Appl. Mater. Interfaces 2014, 6, 16267–16275. DOI:10.1021/am504452a.
- Weidman, J. R.; Luo, S.; Zhang, Q.; Guo, R. Ind. Eng. Chem. Res. 2017, 56, 1868–1879. DOI:10.1021/acs.iecr.6b04946.
- Navale, T. S.; Rathore, R. Synthesis 2012, 44, 805–809. DOI:10.1055/s-0031-1289695.
- Boldt, P.; Vardakis, F. Angew. Chem., Int. Ed. 1965, 4, 1078–1078. DOI:10.1002/anie.196510782.
- Fitzgerald, J. J.; Drysdale, N. E.; Olofson, R. A. Synth. Commun. 1992, 22, 1807–1812. DOI:10.1080/00397919208020501.
- Hashmat Ali, M.; Niedbalski, M.; Bohnert, G.; Bryant, D. Synth. Commun. 2006, 36, 1751–1759. DOI:10.1080/00397910600616859.