
Abstract
Possibilities for the mono- and diesterification of phenylphosphonic acid were evaluated considering the microwave(MW)-assisted direct esterification, and the alkylating esterification. It was found that regarding the monoesterification, the reaction with 15-fold alcohol excess in the presence of [bmim][BF4] additive utilizing MWs is superior than the approach by alkylation. At the same time, for the conversion of the monoester intermediate to the diester, the reaction with alkyl halides in the presence of triethylamine as the base, again under MW irradiation, was found to be the method of choice. Phosphonates with both identical and different alkoxy groups were made available.
GRAPHICAL ABSTRACT
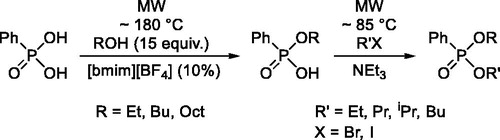
Introduction
Phosphinic and phosphonic derivatives, especially the corresponding phosphinates and phosphonates may be important intermediates in organophosphorus chemistry. The “classical” method for their synthesis involves the reaction of phosphinic chlorides and phosphonic dichlorides with alcohols.[Citation1-5] However, the use of P-chlorides cannot be regarded “green”, and the introduction of the P=O group via the chloro derivatives is not atomic efficient. Keglevich and Kiss were who elaborated the microwave(MW)-assisted direct esterification of phosphinic acids.[Citation6,Citation7] To eliminate the shortcoming meant by the relatively high temperature (≥200 °C) required, the procedure was refined further. It was found that certain ionic liquid (IL) additives had a beneficial effect allowing a lower reaction temperature, shorter reaction time and complete conversions.[Citation8] As regards the preparation of dialkyl phenylphosphonates, a three-step procedure starting from phenyl-H-phosphinic acid (I) was developed (Scheme 1). According to this, phosphinic acid I was subjected to MW-assisted direct esterification, the alkyl phenyl-H-phosphinates (II) so obtained were converted to the corresponding phenylphosphonic-acid-esters (III) by oxidation using meta-chloroperbenzoic acid, and finally monoesters III were esterified utilizing MWs.[Citation9]
Scheme 1. Earlier method for the synthesis of dialkyl phenylphosphonates.[Citation9]
![Scheme 1. Earlier method for the synthesis of dialkyl phenylphosphonates.[Citation9]](/cms/asset/48d31163-0b24-4260-aa04-6a1d95fe03f8/lsyc_a_1637894_sch0001_b.jpg)
However, we were not satisfied with this method, since the oxidation step was critical, as the purification of monoester III could not be solved perfectly, and the overall yield of the 3-step procedure was rather poor. As another possibility, we wished to start from phenylphosphonic acid. The preliminary experiments carried out in the presence of [bmim] ionic liquids as the catalyst were encouraging in respect of monoesterification.[Citation2] For this, we wished to develop a generally applicable method for the conversion of phenylphosphonic acid to dialkyl phenylphosphonates.
Results and discussion
Esterification of phenylphosphonic acid
MW-assisted direct esterification of phenylphosphonic acid with alcohols
First, we wished to overview and extend our MW-assisted method elaborated for the monoesterification of phenylphosphonic acid. It was found that irradiating the mixture of phenylphosphonic acid and 15 equivalents of butanol or octanol at 200/220 °C for 4 h, the conversions were not complete, and the corresponding monoesters (2a and 2c, respectively) were the main components (92/75%) beside the diesters (3a and 3c) formed as minor by-products (, entries 1 and 5).[Citation9] According to our recent finding, the use of 10% of [bmim][BF4] allowed complete conversion at a lower reaction temperature of 180 °C after a shorter reaction time of 45 min (, entries 2 and 6). In these cases, the monoesterifications were more selective, and products 2a and 2c were isolated in a yield of 82% and 90%, respectively.[Citation10] Our procedure seemed to be of a more general value, as it worked also for the monoesterification with ethanol. Volatility of EtOH allowed a maximum reaction temperature of 165 °C, and after an irradiation of 8 h in the presence of the ionic liquid additive, ethyl ester 2b was obtained in a yield of 70% (, entry 4). The comparative experiment carried out in the absence of the additive led to a low conversion of 30% (, entry 3).
Table 1. Direct esterification of phenylphosphonic acid with different alcohols under MW conditions.
O-Alkylation of phenylphosphonic acid
Then, the alkylating esterification was evaluated as another alternative. Phenylphosphonic acid was reacted with 1 equivalent of butyl bromide in the presence of triethylamine under MW irradiation. However, both at 80 °C for 4 h, and at 100 °C for 2 h, the conversion remained incomplete (58/62%), and the selectivity of the formation of the monoester (2a) was only 76/61%, as 24/39% of the dibutylester (3a) was also formed (, entries 1 and 3). The comparative thermal experiment led to a lower conversion of 47% (, entry 2). In the solid–liquid phase transfer catalytic (PTC) version applying K2CO3 as the base, the conversion was even lower (7%) (, entry 4).
Table 2. Attempted monoalkylation of phenylphosphonic acid with butyl bromide under different conditions.
It was envisaged that the use of BuBr in excess may promote the formation of dibutyl phenylphosphonate (3a). For this, the alkylation of phenylphosphonic acid was performed at 100 °C for 4 h using 2.2 equivalents of BuBr in the presence of TEA on MW irradiation. Sure enough, the expected diester (3a) was the major component (68%) together with 32% of the monoester (2a), but the conversion was not complete (, entry 1). Elevating the temperature to 120 and 150 °C did not help (, entries 2 and 3). However, a drastic increase of the molar ratio of the phosphonic acid (1) and BuBr to 1:5 led to a complete conversion, and to a better selectivity of 83% for the dibutylester (3a) that was isolated in a yield of 69% (, entry 4). The phase transfer catalytic version applying K2CO3 as the base was not successful, as the conversion was rather low (11%), moreover the monoester (2a) was the major product (, entry 5).
Table 3. Attempted dialkylation of phenylphosphonic acid with butyl bromide under different conditions.
Esterification of phenylphosphonic monoesters
It was shown that the monoesters of phenylphosphonic acid (2) may be best synthesized by MW-assisted direct esterification in the presence of [bmim][BF4] as the catalyst. It was a question for us, if the monoesters (2) prepared can be esterified further to afford the diesters (3).
MW-assisted direct esterification of phenylphosphonic acid monoesters
The first series of the diesterification attempts involved MW-assisted direct esterification of the monoesters (2). Without an IL additive, the MW-promoted reaction of the corresponding PhP(O)(OR)(OH) with 15-fold ROH took place in only low conversions. Esterification of monobutylester 2a with BuOH at 180 or 220 °C for 6 h led to a conversion of 7% and 17%, respectively (, entries 1 and 2), while the similar reaction of the monooctyl derivative (2c) with octanol at 200, 220 and 235 °C proceeded with conversions of 22%, 27% and 28%, respectively (, entries 5–7). Repetitions in the presence of 10% of [bmim][BF4] were more successful at 220 °C for 6 h, and at 235 °C for 3 h, but the conversions of 45% and 72% still meant incomplete transformations (, entries 3 and 8) that could not be increased. The use of [bmim][PF6] instead of [bmim][BF4] was not helpful (, entries 4 and 9). It can be seen that the monoesters (2) cannot be converted efficiently to the diesters (3) by MW-assisted direct esterification.
Table 4. Direct esterification of phenylphosphonic monoesters.
Alkylating esterification of phenylphosphonic monoesters
The other option was to try the monoester (2) → diester (3) conversion by O-alkylation. Hence, the monoesters (2) were first reacted with the corresponding alkyl halides in the presence of triethylamine and in the absence of any solvent at 85 °C for 30 min under MW irradiation. The alkylation of half-esters 2a, 2b and 2c with BuBr, EtI and OctBr furnished diesters 3a, 3b and 3c in yields of 80%, 92%, and 72%, respectively (, entries 1, 3 and 5). The alternative protocol applying K2CO3 and tetrabutylammonium chloride (TEBAC) in acetonitrile as the solvent provided the diesters (3a–c) in somewhat lower yields of 58–76% (, entries 2, 4 and 6). Worth of mentioning that the comparative thermal experiment resulted in ester 3a in a lower yield of 57% (, entry 1/footnote b). It is concluded that the MW-assisted alkylating esterification is more advantageous for the conversion of the monoesters (2) to the diesters (3), than the MW-promoted direct esterification.
Table 5. Synthesis of dialkyl phenylphosphonates by the alkylation of phenylphosphonic monoesters.
Finally, we aimed at the synthesis of phenylphosphonates with different alkyl groups. In the first series of experiments, the monobutyl ester of phenylphosphonic acid 2a was reacted with EtI, PrBr and iPrBr in the presence of triethylamine without any solvent at 85 °C. After a 0.5 h’s irradiation, work-up and purification, the corresponding phosphonates with two different alkyl groups (3d, 3e and 3f) were obtained in yields of 71%, 68%, and 48%, respectively (, entries 1, 3 and 5). The similar alkylation of the monoethyl ester of phenylphosphonic acid 2b with PrBr, iPrBr and BuBr almost as above, furnished diesters 3g, 3h and 3d, in yields of 72%, 54%, and 72%, respectively (, entries 7, 9 and 11). The reaction with the hindered iPrBr required a longer reaction time of 1 h. In the second round, monoesters 2a and 2b were alkylated in the presence of K2CO3 and TEBAC in acetonitrile at 100 °C for 1 h under MW. After work-up and chromatography, the solid–liquid accomplishments provided the mixed diesters 3d–h in yields of 37–67% (, entries 2, 4, 6, 8, 10 and 12). The preparations in homogeneous phase were more efficient in all cases than the heterogeneous-phase syntheses. It is noteworthy that no matter if the butyl ethyl diester (3d) was prepared from the monobutyl ester (2a) or from the monoethylester (2b), the outcomes were comparable (See , entry 1 vs. entry 11, and entry 2 vs. entry 12).
Table 6. Synthesis of phenylphosphonates with different alkyl groups by the alkylation of phenylphosphonic monoesters.
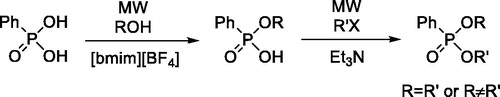
Taking into account all experiences described above, the 2-step conversion including a MW-assisted direct esterification in the first step, and an alkylating esterification in the second step seems to be the optimum solution for the stepwise esterification of phenylphosphonic acid (Scheme 2).
Scheme 2. The new protocol elaborated for the synthesis of dialkyl phenylphosphonates.
Conclusions
The possibilities for the conversion of phenylphosphonic diacid to the monoester PhP(O)(OR)(OH) and to the dialkyl phosphonates were explored. Principally, both the monoesterification, and the esterification of the monoester may be performed either by MW-assisted reaction with an alcohol, or by alkylation with an alkyl halide. The latter step may be achieved in homogeneous or heterogeneous phase using triethylamine or K2CO3 as the base, respectively. It was found that while for the monoesterification, the MW-assisted and [bmim][BF4]-catalyzed direct esterification is the method of choice, the conversion of the monoester to the dialkyl phosphonate may be performed best by alkylation in the homogeneous phase. Phosphonates with both identical and different alkyl groups could be prepared by the novel 2-step protocol.
Experimental
General procedure for the direct esterification of phenylphosphonic acid (1)
A mixture of 0.10 g (0.63 mmol) of phenylphosphonic acid (1) and 9.45 mmol of an alcohol (0.55 mL of ethanol, 0.87 mL of n-butanol, or 1.5 mL of n-octanol) was measured in a sealed tube, and irradiated in the MW reactor applying 50–150 W irradiation in the presence of 0.012 mL (0.060 mmol) [bmim][BF4] at the temperatures and for the times shown in . (The pressure developed was in the range of 1–18 bar.) The excess of alcohol was removed under reduced pressure, and the residue purified by column chromatography (silica gel/ethyl acetate) to afford products 2a–c in yields shown in .
General procedure for the alkylating esterification of phenylphosphonic acid monoesters (2)
To 0.75 mmol of phenylphosphonic acid monoalkyl ester (2a: 0.16 g, 2b: 0.14 g, 2c: 0.20 g) was added 0.75 mmol of an alkyl halide (0.060 mL of ethyl iodide, 0.068 mL of propyl bromide, 0.070 mL of isopropyl bromide, 0.081 mL of butyl bromide, or 0.13 mL of octyl bromide) and 0.12 mL (0.83 mmol) of triethylamine. The contents of the closed vial were irradiated in the MW reactor at 10–20 W. The resulting mixture was filtered, and removal of the volatile components provided the crude product that was purified by column chromatography (Brockmann I type neutral aluminum oxide/ethyl acetate–hexane) to afford diesters 3a–h in purities of >99% as colorless oils (, entries 1, 3 and 5, as well as , entries 1, 3, 5, 7, 9 and 11).
Esters 3d–h are new compounds, while derivatives 2a–c and 3a–c have been described in the literature.[Citation11-15] See the Supplementary Information for the characterization data of products 2a–c and 3a–h.
Supplemental Material
Download PDF (1 MB)Additional information
Funding
Related Research Data
References
- Quin, L. D. A Guide to Organophosphorus Chemistry; Wiley: New York, 2000. ISBN: 978-0-471-31824-8.
- Kiss, N. Z.; Keglevich, G. Methods for the Preparation of Phosphinates and Phosphonates with a Focus on Recent Advances. In Organophosphorus Chemistry – Novel Developments; Keglevich, G., Ed.; De Gruyter: Berlin, 2018; pp 35–52.
- Kiss, N. Z.; Keglevich, G. An Overview of the Synthesis of Phosphinates and Phosphinic Amides. Curr. Org. Chem. 2014, 18, 2673–2690. doi:10.2174/1385272819666140829011741.
- Barton, D., Ollis, W. D. (Eds.). Comprehensive Organic Chemistry, Vol. 2, Sutherland, I. O. (Vol. Ed.); Pergamon: Oxford, 1979. ISBN: 978–0080213149.
- Kosolapoff, G. M.; Maier, L. Phosphonic acids and derivatives, In Organic Phosphorus Compounds; Kosolapoff, G. M.; Maier, L., Eds.; Wiley-Interscience: New York, 1973; Vol. 6, Ch. 18, p. 1. ISBN: 978–0471504467.
- Keglevich, G.; Kiss, N. Z.; Mucsi, Z.; Jablonkai, E.; Balint, E. The Synthesis of Phosphinates: Traditional versus Green Chemical Approaches. Green Proc. Synth. 2014, 3, 103–110. doi:10.1515/gps-2013-0106.
- Kiss, N. Z.; Bottger, E.; Drahos, L.; Keglevich, G. Microwave-Assisted Direct Esterification of Cyclic Phosphinic Acids. Heteroatom. Chem. 2013, 24, 283–288. doi:10.1002/hc.21092.
- Kiss, N. Z.; Keglevich, G. Microwave-Assisted Direct Esterification of Cyclic Phosphinic Acids in the Presence of Ionic Liquids. Tetrahedron Lett. 2016, 57, 971–974. doi:10.1016/j.tetlet.2016.01.044.
- Kiss, N. Z.; Mucsi, Z.; Böttger, É.; Drahos, L.; Keglevich, G. A Three-Step Conversion of Phenyl-1H-Phosphinic Acid to Dialkyl Phenylphosphonates Including Two Microwave-Assisted Direct Esterification Steps. Curr. Org. Synthesis 2014, 11, 767–772. doi:10.2174/1570179410666131212231130.
- Kiss, N. Z.; Keglevich, G. Direct Esterification of Phosphinic and Phosphonic Acids Enhanced by Ionic Liquid Additives. Pure Appl. Chem. 2019, 91, 59–65. doi:10.1515/pac-2018-1008.
- Crenshaw, M. D. Synthesis of Alkyl- and Arylphosphonic Acid Monoesters by Direct Esterification of Dibasic Phosphonic Acids in the Presence of an Arsonic Acid Catalyst. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 1509–1516. doi:10.1080/10426500490464032.
- Szabó, A.; Jászay, Z. M.; Töke, L.; Petneházy, I. Interesting By-Products in the Synthesis of Chiral a-Aminophosphinates from Enantiopure Sulfinimines. Tetrahedron Lett. 2004, 45, 1991–1994. doi:10.1016/j.tetlet.2003.12.151.
- Jablonkai, E.; Keglevich, G. P-Ligand-Free, Microwave-Assisted Variation of the Hirao Reaction under Solvent-Free Conditions; the P–C Coupling Reaction of > P(O)H Species and Bromoarenes. Tetrahedron Lett. 2013, 54, 4185–4188. doi:10.1016/j.tetlet.2013.05.111.
- Chen, T.-H.; Reddy, D. M.; Lee, C.-F. A Palladium-Catalyzed Oxidative Cross-Coupling Reaction between Aryl Pinacol Boronates and H-Phosphonates in Ethanol. RSC Adv. 2017, 7, 30214–30220. doi:10.1039/C7RA04619G.
- Iranpoor, N.; Firouzabadi, H.; Moghadam, K. R.; Motavalli, S. First Reusable Ligand-Free Palladium Catalyzed C–P Bond Formation of Aryl Halides with Trialkylphosphites in Neat Water. RSC Adv. 2014, 4, 55732–55737. doi:10.1039/C4RA07680J.