
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A series of novel indoline-(thio)urea were designed and prepared using indoline(s) as a new platform and tested as organocatalysts in the Michael and Morita–Baylis–Hillman reactions. Most of the compounds were found to be very active catalysts although they did not promote the enantioselectivity. As agents for the conversion of thiocarbonyl compounds into carbonyl compounds, potentials of PIFA and DDQ were also displayed. Furthermore, DFT calculations rationalized the experimentally observed non-enantioselectivity of the catalysts.
GRAPHICAL ABSTRACT
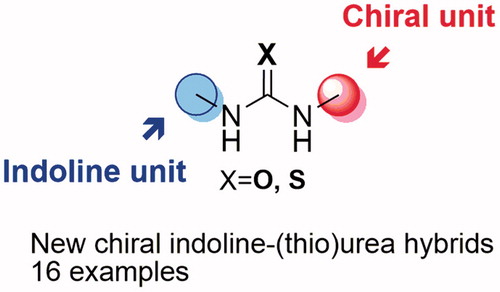
Introduction
Organocatalysis refers to the use of small organic molecules to catalyze organic transformations and has become a highly dynamic area in chemical research.[Citation1,Citation2] Metal-free catalysts or organocatalysts have been growing interest in the field of asymmetric catalysis over the past decades due to their notable advantages such as their affordability, safety, environmentally friendly or green chemistry, nontoxic nature, low cost, usually ease of use, water- and air-tolerant, and the easy structural modifications.[Citation2] Because of the absence of transition metals, organocatalysts provide the advantage of environmentally friendly chemistry or green chemistry, which is an essential criterion for scientific and industrial studies.[Citation3] The asymmetric synthesis of natural products and chiral drugs has become an important focus for synthetic organic chemists and industrial chemists.[Citation4] The efficiency and the scope of organocatalysis have been broadly increased since the rediscovery of proline-catalyzed aldol reactions.[Citation5] Frequently, organocatalysts are classified according to their binding capacity with the substrate through a covalent bond or noncovalent interactions such as hydrogen bonding and electrostatic cooperation.[Citation6] Various types of small organic molecules such as phosphoric acids,[Citation7] proline and its derivatives,[Citation8] cinchona derivatives,[Citation9] sulfones,[Citation10] (thio)urease,[Citation11] N-oxides,[Citation12] and amino acids[Citation13] have been employed as hydrogen bonding organocatalysts in various chemical reactions. In general, chiral (thio)urea-based molecules have found frequent applications as organocatalysts through hydrogen-bonding interactions to coordinate and activate H-bond accepting substrates in modern synthetic organic chemistry.[Citation11]
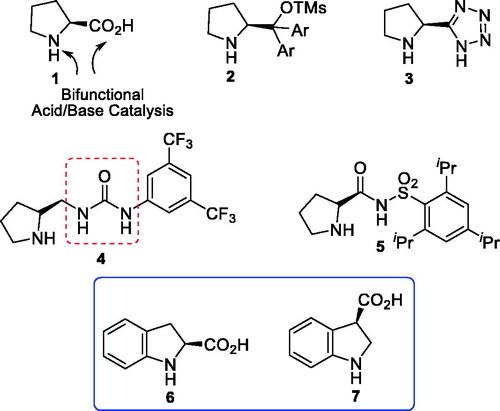
Cooperative catalysis with chiral bifunctional (thio)ureas for asymmetric organocatalysis has recently been a new and exciting strategy.[Citation11] Since (thio)ureas are a stronger H-bond donor, we were interested in exploring whether indoline-(thio)urea organocatalysts would perform or even better. Generally, many proline and pyrrolidine derivatives are well-known, versatile catalysts in organic synthesis (),[Citation8,Citation11] but only scattered examples of chiral indolines as organocatalysts are known. Indeed, only two indoline-carboxylic acids and a few of their derivatives have been used as organocatalyst until now.[Citation14] This encouraged us to synthesize and evaluate the new-type indoline-(thio)ureas as potential organocatalyst for the Michael addition and Morita–Baylis–Hillman reactions. We designed that appropriate combinations of (thio)urea and indoline in a chiral scaffold could result in a potential mono- and bifunctional organocatalysts. In this paper, we report the preliminary results of our investigations.
Figure 1. Some proline, pyrrolidine-type and indoline-type organocatalysts.
Results and discussions
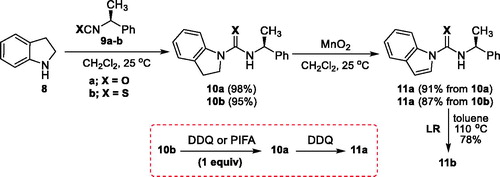
Initially, indoline (8) and (S)-1-phenylethyl isocyanate (9a) were employed for the synthesis of indoline-urea 10a by a known method (Scheme 1).[Citation15] Subsequent treatment of 10a with manganese dioxide gave indole-urea 11a in 91% yield. For the synthesis of thiourea analog, (S)-1-phenylethyl isothiocyanate (9b) reacted with 8 to afford thiourea 10b as shown in Scheme 1. Attempt to prepare indole-thiourea derivative 11b from 10b via a one-pot procedure involving the radical cascade and the manganese(IV) oxide (MnO2) oxidation resulted in the formation of indole-urea 11a as an unexpected product. The presented spectra explicitly confirm the structure of the synthesized urea 11a. In point of fact, chiral HPLC chromatogram of compound 11a shows that the optical purity and optical rotation were not compromised during the synthetic sequences. Because of unexpected oxidation of thiourea to urea during dehydrogenation of indoline to indole ring, we surveyed corresponding literature. According to this literature, a simple and efficient procedure for the mild conversion of thio-enolizable thioureas, thioamides end thiolactams to ureas, amides and lactams in good yields using active manganese dioxide were described.[Citation16] For this reaction, Sharma et alet al. suggested a probable mechanism involving the initial formation of carbodiimide, whereas Bhalerao et alet al. explained a mechanism via the participation of labile hydroxyl radicals to yield probably a cyclic intermediate that can undergo sulfur extrusion. Next, a variety of oxidants, phenyliodine bis(trifluoroacetate) (PIFA), and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) were screened in the reaction. The attempt to dehydrogenate indoline in thiourea 10b using a stoichiometric amount of DDQ gave an unexpected urea derivative 11a as a sole product instead of thiourea 11b. Indoline-urea 10a was oxidized with DDQ to afford the corresponding urea 11a. Also, the use of 1.1 equiv. of PIFA as an oxidant afforded urea 10a in 82% yield. When the reaction of 10a and PIFA under the conditions was carried out, only a trace amount of 11a was detected. After all unsuccessful efforts, the synthesis of thiourea 11b could be achieved by direct thionation of urea 11a with Lawesson’s reagent (LR) (reflux in toluene, 12 h; 78%) (Scheme 1).
Scheme 1. Synthesis of 11a/b from indoline (8).
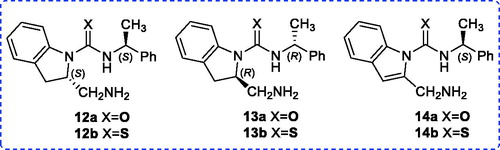
Next, our attention was focused on the synthesis of indoline-(enantiomers 12 and 13) and indole-(thio)ureas 14a/b in which there can potentially behave as a bifunctional catalyst (). To provide a cheaper synthesis cost, all test reactions were first carried out starting from a racemic compound 15. To access the diastereoisomers (dia-12/13) of enantiomers 12 and 13, esterification of indoline-2-carboxylic acid (rac-15) in the presence of thionyl chloride and methanol led to methyl ester rac-16 in 83% yield (Scheme 2).[Citation17]
Figure 2. Structures of another type-target indoline/indole-based chiral catalyst candidates.
Scheme 2. Attempts to synthesize rac-23/24.
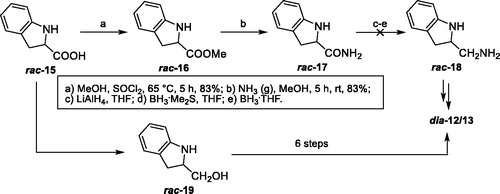
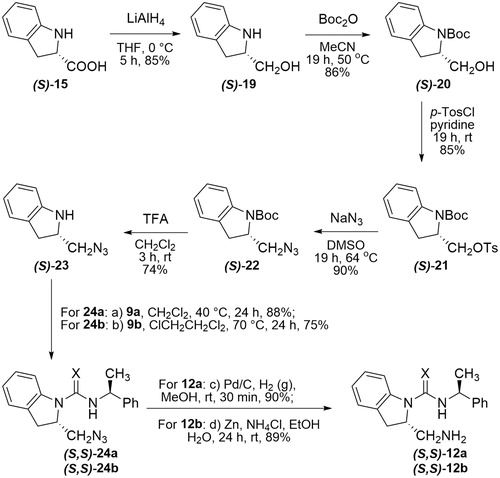
In next step, the methyl ester rac-16 could be converted to its amide rac-17 by treating with ammonia gas in methanol.[Citation18] Unfortunately, all attempts to reduce the corresponding amine rac-17 to amide rac-18 met with failure. We, therefore, changed our strategy and reached to the target molecules in seven steps involving reduction, N-tert-butoxycarbonyl (Boc) protection, tosylation, azidation, deprotection of Boc, addition, and reduction (Scheme 2). The enantiopure synthesis of the target (thio)ureas 12 was performed in seven steps (Scheme 3). As highlighted in Scheme 3, a lithium aluminum hydride reduction of (S)-(-)-indoline-2-carboxylic acid (S-15) delivered the desired alcohol (S)-19 in 85% yield.[Citation19] Later, alcohol (S)-19 reacted with di-tert-butyl dicarbonate (Boc2O) in acetonitrile to give the corresponding N-Boc protected derivative (S)-20 with good yields. N-Boc indoline (S)-20 bearing alcohol was then converted into tosylate (S)-21 using tosyl chloride. Displacement of the tosylate of (S)-21 with sodium azide in dimethyl sulfoxide (DMSO) at 64 °C gave the azide (S)-22 which was then subjected to deprotection of N-Boc with TFA to give the indoline (S)-23. The subsequent free amine (S)-23 reacted with (S)-1-phenylethyl isocyanate (9a) in methylene chloride under reflux to form urea (S,S)-24a. An identical procedure provided thiourea (S,S)-24b upon treatment with a free amine (S)-23 and (S)-1-phenylethyl isothiocyanate (9b) in refluxing 1,2-dichloroethane. The preparation of amine (S,S)-12a from the azide (S,S)-24a was conveniently achieved by Pd/C-catalyzed hydrogenation, whereas the reduction attempts of thiourea-azide (S,S)-24b using the same approach resulted in the poison of catalyst system. On the other hand, the reduction of azide moiety of (S,S)-24b to the amine (S,S)-12b was accomplished using Zn/NH4Cl as reducing agent in EtOH/water (3:1).[Citation20] The synthesis of (R,R)-13a/b was also achieved following the same protocols as explained earlier in Scheme 2.
Scheme 3. Synthesis of (S,S)-12a/b.
The attempts to oxidize were performed using diastereoisomeric compounds dia-12a and dia-24a (Scheme 4). However, the reaction of dia-12a with MnO2 (10 equiv.) in methylene chloride (CH2Cl2) gave only the unexpected fragmentation product (S)-N-(1-phenylethyl)-1H-indole-1-carboxamide (11a) in 78% yield (Scheme 4).
Scheme 4. Attempts to synthesize (S)-13a/14a and a proposed mechanism for the formation of 11a.
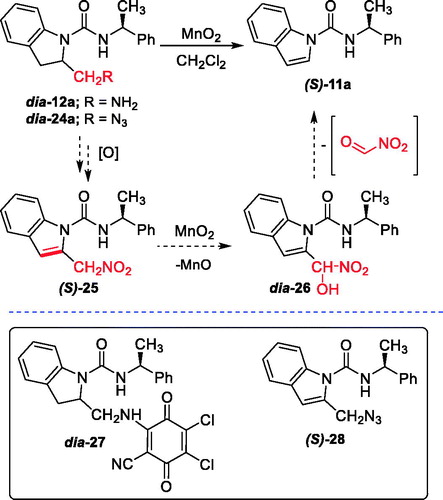
To our surprise, the treatment of dia-24a with manganese dioxide (10 equiv.) in methylene chloride at room temperature (rt) led to the unexpected formation of the fragmentation product 11a in nearly quantitative yield. A plausible mechanism to account for the outcome of the reaction is proposed in Scheme 4. The corresponding amine dia-12a (or azide dia-24a) may be initially oxidized by MnO2 to generate nitro compound (S)-25. Retro-aldol condensation followed by allylic oxidation to give 26 may then lead to the formation of 11a. Attempts on oxidation of dia-12a with 1 equiv. of DDQ in methylene chloride resulted in a complex mixture along with the formation of Michael adduct dia-27 in 10% yield (Scheme 4). The structure of dia-27 was especially confirmed by high-resolution mass spectrometry (HRMS), as well as nuclear magnetic resonance (NMR) spectroscopy (see Supporting Information). Unfortunately, when the azide dia-24a was treated with 1 equiv. of PIFA or DDQ, the desired oxidation product (S)-28 could be isolated in only 3.5–4% yield, along with substantial quantities of recovered starting material (88–90%) (Scheme 4). Since many oxidation attempts upon both dia-12a and dia-24a did not succeed to provide desired compounds, the synthesis of 14a/b was not achieved.
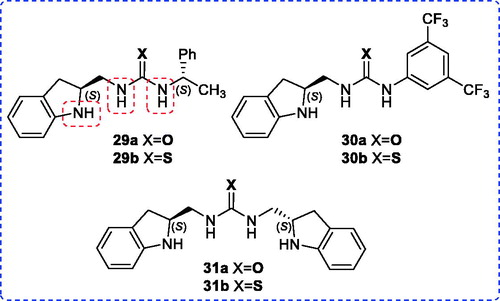
We also aimed the preparation of the new urea and thiourea derivatives 29–31 to determine their organocatalyst behaviors (). N-Boc-protected chiral amine 32 played a key role in synthesis. The organocatalyst 29a/b and 30a/b were readily synthesized by the reactions of indoline N-Boc-protected chiral amine 32 with the corresponding isocyanates and isothiocyanates 9a/b and 34a/b followed by deprotection step using TFA with high yields (Scheme 5).
Figure 3. Structures of target indoline-based chiral catalyst candidates.
Scheme 5. Synthesis of 29a/b and 30a/b.
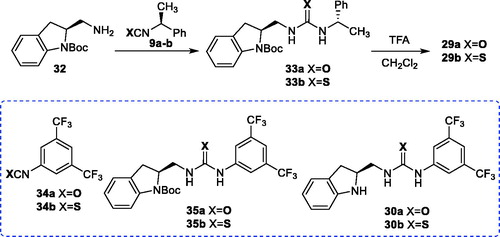
Symmetrical urea and thiourea 31a/b were synthesized in high yields via the reactions of 1,1-carbonyldiimidazole (37) and carbon disulfide with primary amine 36, which was obtained from deprotection of N-Boc group in 32, without incident, respectively (Scheme 6).[Citation21]
Scheme 6. Synthesis of 31a/b.
With a collection of all possible catalysts in hand after the synthesis, we also evaluated the catalytic effect of organocatalysts regarding reactivity and enantioselectivity. The catalytic performances were initially examined in the Michael addition of indole (38) to trans-β-nitrostyrene (39) as the model reaction. Reactions were performed using 10 mol% of 10, 11, 12, 13, and 29–31 in toluene at 25 and 50 °C for 12–48 h (, Entries 2–15). As shown in , the reactions of 38–39 were efficient but generated nearly racemic product (82–93% conversion, almost 1:1 enantiomeric ratio (er)).
Table 1. Catalyst screening for the asymmetric Michael addition of indole (38) to β-nitrostyrene (39).a
Structurally modified versions 10, 11, 12 and 13 were less efficient than 29–31, and all catalysts under study were ineffective in generating 40[Citation22] with significant enantioselectivity. Screening of the solvents and temperatures for catalysts 10a and 12a did not give expected positive results. We also examined the scope of the Michael acceptors and donors concerning the above reaction. The reaction using 2-methylindole as a Michael donor with catalyst 31a (20 mol%) in toluene at room temperature afforded the adduct 41 in 85% yield and in a racemic form ().[Citation23] The use of diethyl malonate for 48 h in toluene at both room temperature and 60 °C did not lead to product (cycloadduct 42) formation with these catalysts ().[Citation24] The treatments with Michael acceptors such as 2-cyclohexen-1-one, trans-chalcone, methyl trans-cinnamate and crotonaldehyde with indole and indoline in the presence of catalysts (20 mol%) in toluene did not result in any detectable reactions. Newly designed catalysts gave racemic products although with at least the same level of reactivity as catalysts reported in the literature.
Figure 4. Structures of Michael adducts 41–42.
Additive effects on asymmetric catalysis are reported very well.[Citation25] Therefore, we expected that the resulting enantiomeric ratio (er) would be higher using chiral and achiral Brønsted acids such as L-proline, benzoic acid, and TFA. The Michael additions of indole (38) to trans-β-nitrostyrene (39) were also examined using 10 mol% of 10, 12, 29, and 31 as a catalyst in toluene at 25 °C for 48 h for 48 h in the presence of L-proline, benzoic acid, and TFA. Acid co-catalysts did not also produce any positive effects on the yield or especially enantioselectivity of the transformation.
Next, the Morita–Baylis–Hillman reactions of 2-cyclohexen-1-one (43) and 4-(trifluoromethyl)benzaldehyde (44) and 4-nitrobenzaldehyde (45) in the presence of 30 mol% of 10, 12, 30, and 31 as organocatalysts and 4-dimethylaminopyridine (DMAP) as a nucleophile were examined without using the solvent (, Entries 2–9, 11–18). The expected allylic alcohols 46 and 47 were obtained in 83–96% yields although in racemic in all runs ().[Citation26,Citation27] Catalysts 31a/b afforded the best reaction yields. Also, the use of L-proline as co-catalyst could not promote the desired enantioselectivity for the corresponding reactions.
Table 2. Catalyst screening for the asymmetric Morita–Baylis–Hillman (MBH) reaction of 2-cyclohexen-1-one (43) with benzaldehydes (44 and 45).a
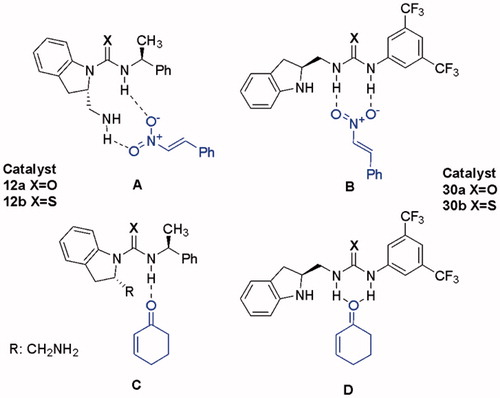
As a result of these studies, most of the novel indoline-(thio)urea hybrids were able to catalyze both Michael additions and Morita–Baylis–Hillman reactions selected as model reactions in high yields. However, catalysis did not proceed with high enantioselectivity. We believe that the driving force for the similar transformations is the interactions via single and dual hydrogen-bond between the electrophiles and the catalysts ().
Figure 5. Proposed activation modes between electrophiles and catalysts.
Based on extensive experimental studies, we hypothesized that the observed non-enantioselectivity for these reactions could be explained by the high conformational flexibility of the transition states, steric and electronic effects or the distance between the chiral unit and the reaction center, which lead to stereocontrol of the reaction products ().
Figure 6. Possible transition states for Michael addition reaction and MBH reaction.
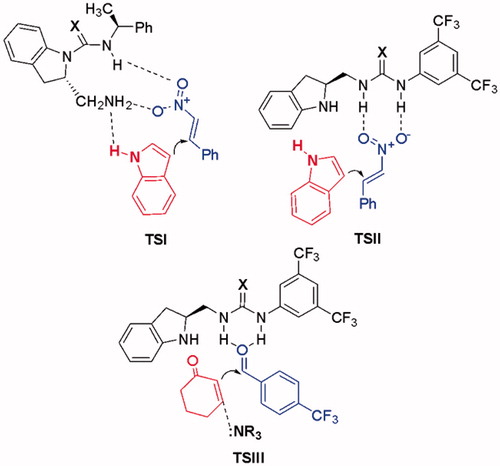
Since the catalysts 10 and 11 are not capable of double hydrogen bonding, transition state geometries have probably more flexibility (, transition state TSI). We especially hoped that the other structural motifs 30–31 could promote as bifunctional hydrogen-bond donor/Lewis base catalysts to afford enantioselective products. Unfortunately, they could not provide the expected enantioselectivity. Presumably, the steric hindrance and electronic effect produced by the benzene moiety in indoline structure plays a crucial role in stereo directing. While basicity of nitrogen in ring decreases due to delocalization into the benzene ring, the presence of benzene fragment can also result in unfavorable steric congestion or the large distance between the reaction center and the asymmetric center during the formation of transition states.
A Density Functional Theory (DFT) study was performed to assess the non-enantioselectivity of catalysts 12a and 30a towards the Michael addition of indole (38) to β-nitrostyrene (39) and to understand the catalyst’s effect on the reaction ().
The meta-hybrid GGA M06-2X[Citation28] was used for the optimizations due to the widely used functionally in organic systems including non-covalent interactions.[Citation29-32] All DFT calculations were performed at the M06-2X/6-31 + G(d,p) level of theory considering IEF-PCM[Citation33] (in toluene (ε = 2.3741)) as implemented in Gaussian 09.[Citation34] Since proton transfer is a fast process and carbon-carbon (C–C) bond formation is the rate-determining step (RDS), only C–C bond formation transition states (TSs) were used to compare the selectivity of the catalysts. Conformational spaces of catalysts 12a and 30a were thoroughly investigated, and only the TSs with the lowest activation barriers leading to R and S enantiomers were reported in . In this context, the uncatalyzed Michael addition reaction of indole (38) and β-nitrostyrene (39) were also modeled to elucidate energetic cost of the reaction, and the results were compared with the urea catalyzed Michael reaction (). Computed data indicates that the free energy of activation of the uncatalyzed Michael addition of 38 with 39 is significantly high (ΔG‡ = 31.5 kcal/mol) compared to the urea catalyzed Michael reaction (ΔG‡ ≈ 20 kcal/mol) confirming the experimentally high yields and the stabilizing effects of the novel catalysts ().
Figure 7. Free energy profile for 12a and 30a catalyzed and uncatalyzed Michael addition reaction of indole (38) to β-nitrostyrene (39). M06-2X/6-31 + G(d,p) with IEF-PCM in toluene; free energies in kcal/mol at 298 K and 1 atm. Results for TS-S, TS-12a-S and TS-30a-S are shown in parenthesis.
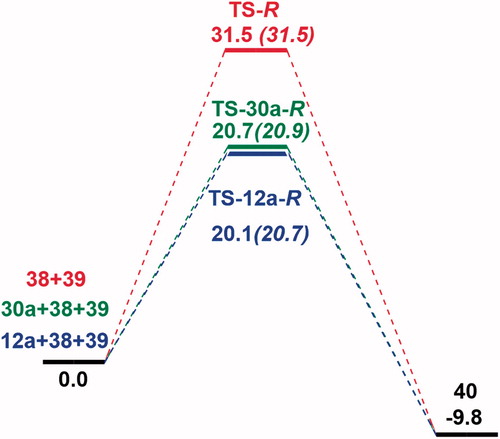
Figure 8. Optimized transition states and activation barriers (ΔG‡) for asymmetric Michael addition of 38 to 39 using catalyst 12a. M06-2X/6-31 + G(d,p) with IEF-PCM in toluene at 298 K and 1 atm, critical distances in Å.
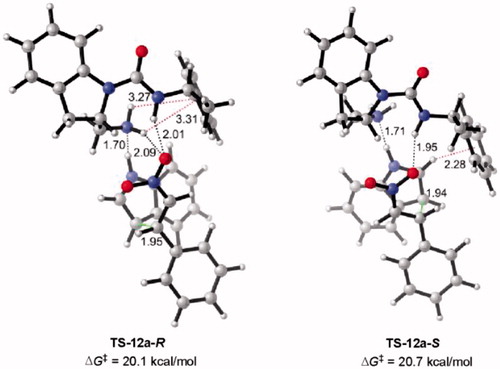
Figure 9. Optimized transition states and activation barriers (ΔG‡) for the Michael addition of 38 to 39 catalyzed by 30a. M06-2X/6-31 + G(d,p) with IEF-PCM in toluene at 298 K and 1 atm, critical distances in Å.
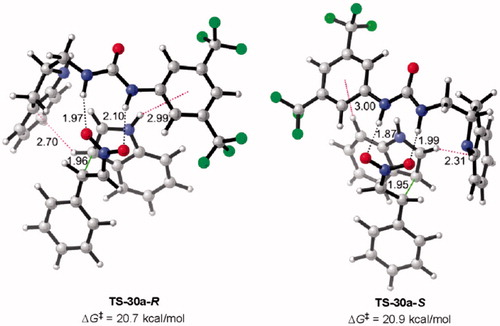
Along with the stabilization of TSs resulting from hydrogen-bonding interactions, there is also stabilizing T-shaped π-interactions (edge-to-face) as shown in .
In the case of TS-12a-R, intramolecular N–H···π interactions are coming from both H’s of amine and aromatic phenyl ring on 12a (NH···π distance =3.27 Å and 3.31 Å); and for TS-12a-S, CH···π stacking is shown between aromatic phenyl ring on 12a and indole (38) (CH···π distance =2.28 Å). However, the stabilization through H-bonding and T-shaped interactions are not selective, and similar interactions are preserved in TS-12a-R and TS-12a-S. Activation barriers for Si face (TS-12a-R, ΔG‡ = 20.1 kcal/mol) and Re face (TS-12a-S, ΔG‡ = 20.7 kcal/mol) attacks are nearly the same, validating the lack of enantioselectivity of 12a.
Similarly, illustrates how the urea NH’s activate the β-nitrostyrene (39) through bidentate hydrogen-bond interactions (N–H···O), as a result, indole (38) attacks 39 leading to product 40. Additionally, T-shaped π-interactions between the aromatic ring of 30a and 38 were identified. Electron clouds on aromatic rings of 30a stabilize 38 through CH-π stacking interactions (CH···π distance= 2.70 Å (TS-30a-R) and 2.31 Å and 3.00 Å (TS-30a-S)). Additionally, an N–H···π interaction between 30a and 38 also contributes to stabilization of indole 38 (TS-30a-S, NH···π distance = 2.99 Å).
The stacked TS structures (TS-30a-R and TS-30a-S) were favored in case of bidentate catalyst 30a. Moreover, shows that the activation barriers of Re and Si face attacks for 30a-catalyzed Michael reaction are approximately isoenergetic (ΔG‡ = 20.9 kcal/mol and ΔG‡ = 20.7 kcal/mol, respectively) confirming the lack of enantioselectivity of the catalysts, affording both R and S Michael products (40). Accordingly, intermolecular H-bonding interactions and T-shaped stacking interactions unselectively lower the activation barriers. In line with the experimental results, 12a and 30a catalysts display non-enantioselectivity towards the Michael addition of indole (38) to β-nitrostyrene (39).
Due to the high conformational flexibility of the catalysts, enantioselectivity was not achieved in the current study. To enhance enantioselectivity, more rigid and sterically crowded organocatalysts can be used by modifying the indoline moiety of the catalysts under study. To gain insight on the effect of the R1 group on the catalyst’s ability to select for the desired enantiomer, a phenyl substituted 48 is proposed as a selective catalyst for the Michael addition reaction ().
Figure 10. Proposed phenyl functionalized catalysts 48. M06-2X/6-31 + G(d,p) with IEF-PCM in toluene at 298 K and 1 atm.
Computed data in show that TS-48-S considerably lowers the activation barrier (by 2.7 kcal/mol) when compared to TS-48-R and could lead to higher experimental enantioselectivities.
Figure 11. Optimized TSs and relative free energies for asymmetric Michael addition of 38 to 39 using catalyst 48. M06-2X/6-31 + G(d,p) with IEF-PCM in toluene at 298 K and 1 atm, critical distances in Å.
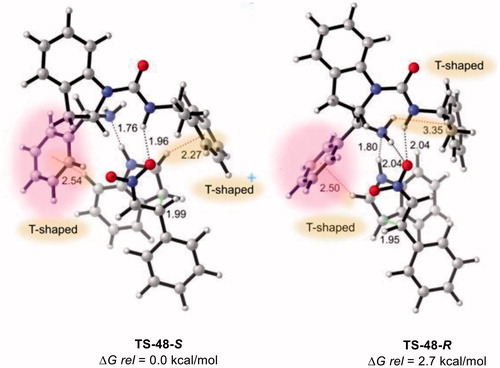
TS-48-S is closely packed compared to TS-48-R, which causes the stabilization of the TS leading to the S enantiomer. The β-nitrostyrene prefers monodentate stabilization of the urea N–H in TS-48-S, and the amine moiety on the indoline only stabilizes the NH of indole (N–H···N) through H-bonding interactions. Additionally, indole (38) is stabilized by both phenyl groups on the catalyst 48 via T-shaped π-interactions (CH···π distance= 2.54 Å and 2.27 Å). In case of TS-48-R, both amine moieties on the indoline and the urea N–H activate β-nitrostyrene (39) via H-bonding interactions. Similar to unmodified TS-12a-R, an intramolecular N–H···π interaction was identified (NH···π distance = 3.35 Å) in TS-48-R. However, indole (38) only interacts with the phenyl group (R1) through T-shaped π-interaction (CH···π distance= 2.50 Å) in comparison to TS-48-S. Si face approach of 38 leading to the R enantiomer (40-R) is hindered by the phenyl group on 48 compared to Re face, affording the S enantiomer (40-S). Consequently, computational data showed that Michael addition of 38 over the Re face of 39 selectively leads to the S enantiomer (40-S) via phenyl functionalization of the catalyst. In this context, it is proposed that the modification of R1 could induce higher experimental enantioselectivities.
Conclusion
In summary, we have developed the efficient synthesis of chiral (thio)urea organocatalysts derived from indolines and demonstrated that the new (thio)urea derivatives can catalyze the asymmetric Michael addition of indole with trans-β-nitrostyrene and Morita–Baylis–Hillman reaction of 2-cyclohexen-1-one (43) with benzaldehydes (44 and 45), giving high yields (up to 93% and 96%) but low enantioselectivities. Our results, however, indicate that PIFA and DDQ may use for an unusual conversion to urea from thiourea without loss of the optical activity. In the future, we aim the further investigation of these catalysts in other enantioselective transformations and hope to find more applications of these exciting molecules in asymmetric organocatalysis. DFT study provided remarkable insights into the lack of enantioselectivity for the novel catalysts confirming the experimental observations. Further efforts to improve the catalysts and the development of related modified catalysts are currently underway.
Experimental section
General experimental methods
All reagents and solvents were purchased from commercial suppliers and used without further purification. Column chromatography and thin-layer chromatography (TLC) were performed using silica gel 60 (70–230 Fluka) and silica gel 60 HF254 (Fluka), respectively. Melting points were carried out on a Buchi 539 capillary melting apparatus and uncorrected. Infrared spectra were recorded on a Mattson 1000 FT-IR spectrophotometer. 1H NMR and 13C NMR spectra were recorded on 400 (100)-MHz Varian and Bruker spectrometer and are reported in δ units with SiMe4 as the internal standard. Elemental analyses were carried out on a Leco CHNS-932 instrument. Enantiomeric purity for enantioselective syntheses was determined by chiral HPLC (Hewlett Packard 1200) analysis using an enantiopure stationary phase (Daicel Chiralcel OD, OJ), eluting with i-PrOH/hexane, and using UV detection at 254 nm. High-resolution mass spectrometry measurements were recorded on a Q-TOF mass spectrometer.
(S)-N-(1-Phenylethyl)indoline-1-carboxamide (10a)
To a stirred solution of (S)-(1-isocyanatoethyl)benzene (123.5 mg, 0.84 mmol) in CH2Cl2 (10 mL) was added indoline (100 mg, 0.84 mmol) and stirred at room temperature for 30 min. The solvent was removed under reduced pressure and the residue was recrystallized from CH2Cl2/hexane to give compound 10a as a white solid (219 mg, 98%) (Mp: 121–122 °C). 1H-NMR (400 MHz, CDCl3): δ 7.87 (d, J = 7.9 Hz, 1 H), 7.40–7.32 (m, 4 H), 7.29–7.23 (m, 1 H), 7.16–7.12 (m, 2 H), 6.91–6.88 (m, 1 H), 5.10 (p, J = 7.0 Hz, 1 H), 4.78 (bd, J = 7.0 Hz, 1 H), 3.96-3.89 (m, 2 H), 3.16 (t, J = 8.6 Hz, 2 H), 1.56 (d, J = 7.0 Hz, 3 H); 13C-NMR (100 MHz, CDCl3): δ 154.7, 144.2, 143.9, 130.5, 128.9, 127.8, 127.5, 126.4, 124.8, 122.0, 114.9, 50.0, 47.3, 28.0, 22.8. IR (CH2Cl2, cm−1): 3293, 3032, 2968, 2854, 1634, 1599, 1523, 1480, 1460, 1386, 1342, 1319, 1289, 1209, 1099, 919, 698. HPLC (OD column; hexane/2-propanol = 80/20; flow rate 0.8 mL/min; T = 25 °C; 254 nm): tR-(-) = 19.3 min. Enantiomeric rate: (+) = 0.0 and (−) = 100.0. = –40.0 (c = 0.25, CH2Cl2). Anal. Calcd for C17H18N2O: C, 76.66; H, 6.81; N, 10.52. Found: C, 76.60; H, 6.76; N, 10.60.
(S)-indolin-2-ylmethanol (S-19)19
To a stirred suspension of LiAlH4 (698 mg, 18.4 mmol) in THF (15 mL) under N2 at 0 °C, (S)-indoline-2-carboxylic acid (1 g, 6.1 mmol) solution in THF (20 mL) was added. The reaction mixture was stirred at 0 °C for 5 h. The reaction mixture was quenched with saturated ammonium chloride and then extracted with dichloromethane. The combined organic layers were washed with brine then dried over Na2SO4 and concentrated on rotary evaporator. The residue was purified using column chromatography (EtOAc/hexane = 6:4, 600 mL) to give desired alcohol as light brown solid (780 mg, 85%) (Mp: 65–66 °C). 1H-NMR (400 MHz, CDCl3): δ 7.05 (d, J = 7.5 Hz, =CH, 1 H), 6.99 (t, J = 7.5 Hz, =CH, 1 H), 6.70 (t, J = 7.5 Hz, =CH, 1 H), 6.59 (d, J = 7.5 Hz, =CH, 1 H), 3.97–3.90 (m, CH, 1 H), 3.70 (br, OH, 1 H), 3.62 (dd, J = 10.9, 3.8 Hz, A part of AB systems, CH2, 1 H), 3.49 (dd, J = 10.9, 7.1 Hz, B part of AB systems, CH2, 1H), 3.04 (dd, J = 15.8, 9.2 Hz, A part of AB systems, CH2, 1 H), 2.72 (dd, J = 15.8, 7.6 Hz, B part of AB systems, CH2, 1H); 13C-NMR (100 MHz, CDCl3): δ 150.5, 128.8, 127.5, 124.9, 119.2, 110.10, 65.4, 60.5, 32.1. IR (CH2Cl2, cm−1): 3328, 3047, 2918, 2845, 1608, 1485, 1398, 1337, 1317, 1246, 1155, 1039, 748. HPLC (OJ column; hexane/2-propanol = 80/20; flow rate 1.0 mL/min; T = 25 °C; 254 nm): tR-(+) = 9.4 min. Enantiomeric rate: (+) = 100.0 and (-) = 0.0. Anal. Calcd for C9H11NO: C, 72.46; H, 7.43; N, 9.39. Found: C, 72.44; H, 7.40; N, 9.35.
tert-Butyl (S)-2-((3-((S)-1-phenylethyl)ureido)methyl)indoline-1-carboxylate (33a)
To a solution of tert-butyl (S)-2-(aminomethyl)indoline-1-carboxylate (32) (400 mg, 1.6 mmol) in CH2Cl2 (15 mL) was added (S)-(1-isocyanatoethyl)benzene (237 mg, 1.6 mmol) and was stirred at room temperature for 4 h. The solvent was removed and crude product was purified using column chromatography respectively (EtOAc/hexane = 8:2, 300 mL) and (MeOH, 50 mL) to give 33a as a dark yellow oil (520 mg, 78%).1H-NMR (400 MHz, CDCl3): δ 7.41–7.18 (m, =CH, 6 H), 7.18–7.07 (m, =CH, 2 H), 6.94 (t, J = 7.4 Hz, =CH, 1 H), 5.07 (s, NH, 1 H), 4.78–4.73 (m, CH, 1 H), 4.48–4.47 (m, CH, 1 H), 3.49–3.36 (m, CH2, 1 H), 3.39–3.3.20 (m, CH2, 2 H), 2.75–2.70 (m, CH2, 1 H), 1.56 (s, CH3, 9 H), 1.41 (d, J = 6.1 Hz, CH3, 3 H). IR (CH2Cl2, cm−1): 3288, 3026, 2959, 2929, 2631, 2525, 1713, 1296, 1205, 1156, 1125, 1063, 1032, 965, 943, 922, 741, 697. Anal. Calcd for C23H29N3O3: C, 69.85; H, 7.39; N, 10.62. Found: C, 69.80; H, 7.34; N, 10.58.
Full experimental detail, 1H and 13C NMR spectra, HPLC traces, and optical rotation data. This material can be found via the “Supplementary Content” section of this article’s webpage.’
Supplemental Material
Download MS Word (3.7 MB)Additional information
Funding
Related Research Data
References
- (a) List, B. Chem. Rev. 2007, 107, 5413–5413. DOI: 10.1021/cr078412e. (b) Dalko, P. I.; Moisan, L. Angew. Chem. Int. Ed 2004, 43, 5138. DOI: 10.1002/anie.200400650. (c) Ma, J. A.; Cahard, D. Angew. Chem. Int. Ed. 2004, 43, 4566. DOI: 10.1002/anie.200300635.
- (a) Shibasaki, M.; Yoshikawa, N. Chem. Rev. 2002, 102, 2187. DOI: 10.1021/cr010297z. (b) Seayad, J.; List, B. Org. Biomol. Chem. 2005, 3, 719. DOI: 10.1039/b415217b.
- Takemoto, Y. Org. Biomol. Chem. 2005, 3, 4299. DOI: 10.1039/b511216h.
- (a) List, B.; Yang, J. W. Science 2006, 313, 1584. DOI: 10.1126/science.1131945. (b) Enders, D.; Grondal, C.; Hüttl, M. R. M. Angew. Chem. Int. Ed. 2007, 46, 1570. DOI: 10.1002/anie.200603129. (c) Pellissier, H. Tetrahedron 2007, 63, 9267. DOI: 10.1016/j.tet.2007.06.024. (d) MacMillan, D. W. Nature 2008, 455, 304. DOI: 10.1038/nature07367. (e) Dondoni, A.; Massi, A. Angew. Chem. Int. Ed. 2008, 47, 4638. DOI: 10.1002/anie.200704684.
- List, B.; Lerner, R. A.; Barbas, C. F. J. Am. Chem. Soc. 2000, 122, 2395. DOI: 10.1021/ja994280y.
- Alemán, J.; Cabrera, S. Chem. Soc. Rev. 2013, 42, 774. DOI: 10.1039/c2cs35380f.
- Connon, S. J. Angew. Chem. Int. Ed. Engl. 2006, 45, 3909. DOI: 10.1002/anie.200600529.
- (a) List, B. Tetrahedron 2002, 58, 5573. DOI: 10.1016/S0040-4020(02)00516-1. (b) Panday, S. K. Tetrahedron: Asymmetry 2011, 22, 1817. DOI: 10.1016/j.tetasy.2011.09.013. (c) Nguyen, T.-H.; Toffano, M.; Bournaud, C.; Vo-Thanh, G. Tetrahedron Lett. 2014, 55, 6377. DOI: 10.1016/j.tetlet.2014.10.004.
- (a) O'donnell, M. J. Acc. Chem. Res. 2004, 37, 506. DOI: 10.1021/ar0300625. (b) Lygo, B.; Andrews, B. I. Acc. Chem. Res. 2004, 37, 518. DOI: 10.1021/ar030058t. (c) Jew, S.-S.; Jeong, B.-S.; Yoo, M.-S.; Huh, H.; Park, H.-G. Chem. Commun. 2001, 1244. DOI: 10.1021/ar030058t.
- Alba, A.-N. R.; Companyó, X.; Rios, R. Chem. Soc. Rev. 2010, 39, 2018. DOI: 10.1039/b911852g.
- (a) Vachal, P.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 10012. DOI: 10.1021/ja027246j. (b) Connon, S. J. Chem. Commun. 2008, 2499. DOI: 10.1039/b719249e. (c) Koutoulogenis, G.; Kaplaneris, N.; Kokotos, C. G. Beilstein J. Org. Chem. 2016, 12, 462. DOI: 10.3762/bjoc.12.48.
- Liu, X.; Lin, L.; Feng, X. Acc. Chem. Res. 2011, 44, 574. DOI: 10.1021/ar200015s.
- Chai, Z.; Zhao, G. Catal. Sci. Technol. 2012, 2, 29. DOI: 10.1039/C1CY00347J.
- (a) Pietruszka, J.; Simon, R. C. Chem. Eur. J. 2010, 16, 14534. DOI: 10.1002/chem.201002130. (b) Pietruszka, J.; Simon, R. C. ChemCatChem 2010, 2, 505. DOI: 10.1002/cctc.201000041. (c) Kunz, R. K.; MacMillan, D. W. J. Am. Chem. Soc. 2005, 127, 3240. DOI: 10.1021/ja042774b.
- Wei, S.; Yalalov, D. A.; Tsogoeva, S. B.; Schmatz, S. Catal. Today 2007, 121, 151. DOI: 10.1016/j.cattod.2006.11.018.
- (a) T.; Sharma, N.; Sahni, A.; Lal, . BCSJ 1978, 51, 1245. DOI: 10.1246/bcsj.51.1245. (b) B. R.; Rani, M.; Rahman, U.; Bhalerao, Tetrahedron 1992, 48, 1953. DOI: 10.1016/S0040-4020(01)88517-3. (c) Corsaro, A.; Pistara, V. Tetrahedron 1998, 54, 15027. DOI: 10.1016/S0040-4020(98)00880-1.
- Alatorre-Santamaría, S.; Gotor-Fernández, V.; Gotor, V. Tetrahedron: Asymmetry 2010, 21, 2307. DOI: 10.1016/j.tetasy.2010.08.002.
- Nagata, R.; Tanno, N.; Kodo, T.; Ae, N.; Yamaguchi, H.; Nishimura, T.; Antoku, F.; Tatsuno, T.; Kato, T.; Tanaka, Y. J. Med. Chem. 1994, 37, 3956. DOI: 10.1021/jm00049a015.
- Brochu, J.-L.; Prakesch, M.; Enright, G. D.; Leek, D. M.; Arya, P. J. Comb. Chem. 2008, 10, 405. DOI: 10.1021/cc800036w.
- Lin, W.; Zhang, X.; He, Z.; Jin, Y.; Gong, L.; Mi, A. Synth. Commun. 2002, 32, 3279–3284. DOI: 10.1081/SCC-120014032.
- (a) Padiya, K. J.; Gavade, S.; Kardile, B.; Tiwari, M.; Bajare, S.; Mane, M.; Gaware, V.; Varghese, S.; Harel, D.; Kurhade, S. Org. Lett. 2012, 14, 2814. DOI: 10.1021/ol301009d. (b) Azizi, N.; Khajeh-Amiri, A.; Ghafuri, H.; Bolourtchian, M. Mol. Divers 2011, 15, 157. DOI: 10.1007/s11030-010-9236-7.
- Fan, Y.; Kass, S. R. J. Org. Chem. 2017, 82, 13288. DOI: 10.1021/acs.joc.7b02411.
- Mendez, I.; Rodriguez, R.; Polo, V.; Passarelli, V.; Lahoz, F. J.; Garcia-Orduna, P.; Carmona D. Chemistry 2016, 22, 11064. DOI: 10.1002/chem.201601301.
- Shubina, T. E.; Freund, M.; Schenker, S.; Clark, T.; Tsogoeva, S. B. Beilstein J Org Chem. 2012, 8, 1485. DOI: 10.3762/bjoc.8.168.
- Hong, L.; Sun, W.; Yang, D.; Li, G.; Wang, R. Chem. Rev. 2016, 116, 4006. DOI: 10.1021/acs.chemrev.5b00676.
- You, J.; Xu, J.; Verkade, J. G. Angew. Chem. 2003, 115, 5208. DOI: 10.1002/ange.200352233.
- Patra, A.; Batra, S.; Joshi, B. S.; Roy, R.; Kundu, B.; Bhaduri, A. P. J. Org. Chem. 2002, 67, 5783DOI: 10.1021/jo020199t.
- (a) Zhao, Y.; Truhlar, D. G. Theor Chem Account 2008, 120, 215. DOI: 10.1007/s00214-007-0310-x. (b) Zhao, Y.; Truhlar, D. G. Acc. Chem. Res. 2008, 41, 157. DOI: 10.1021/ar700111a.
- Dedeoglu, B.; Catak, S.; Yildirim, A.; Bolm, C.; Aviyente, V. ChemCatChem. 2015, 7, 4173. DOI: 10.1002/cctc.201500852.
- Catak, S.; Hemelsoet, K.; Hermosilla, L.; Waroquier, M.; Van Speybroeck, V. Chemistry 2011, 17, 12027. DOI: 10.1002/chem.201100712.
- Goossens, H.; Winne, J. M.; Wouters, S.; Hermosilla, L.; De Clercq, P. J.; Waroquier, M.; Van Speybroeck, V.; Catak, S. J. Org. Chem. 2015, 80, 2609. DOI: 10.1021/jo5027639.
- Catak, S.; D’hooghe, M.; De Kimpe, N.; Waroquier, M.; Van Speybroeck, V. J. Org. Chem. 2010, 75, 885. DOI: 10.1021/jo902493w.
- (a) Mennucci, B.; Tomasi, J. J. Chem. Phys. 1997, 106, 5151. DOI: 10.1063/1.473558. (b) Mennucci, B.; Cances, E.; Tomasi, J. J. Phys. Chem. B 1997, 101, 10506. DOI: 10.1021/jp971959k. (c) Tomasi, J.; Mennucci, B.; Cances, E. J. Mol. Struct. THEOCHEM 1999, 464, 211. DOI: 10.1016/S0166-1280(98)00553-3.
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; et al. Gaussian 09 Revision E.01. Gaussian, Inc.: Wallingford, CT, 2013.