
Abstract
Novel chiral menthol-based pyridyl nitrone ligands were synthesized and Au(I) coordination of the ligands gave chiral Au(I)–nitrone complexes. 1H NMR studies of the gold(I) coordination experiments with nitrone ligands afforded a convenient method for monitoring complex formation. The catalytic effect of Au(I)–nitrone complexes, shown to tune catalytic systems to produce uncommon products, was evaluated in [2 + 2 + 2] cyclotrimerization and [2 + 4] cyclodimerization reactions of non-terminal propargyl acetals. Alternative gold(I)-catalyzed [2 + 2], [2 + 4] and [3 + 4] cycloaddition reaction pathways of non-terminal propargyl acetals with imine substrates gave a diverse range of N-heterocyclic products. The present screening study demonstrates the potential and the versatility of non-terminal propargyl acetals in gold(I)-catalyzed cycloaddition reactions.
GRAPHICAL ABSTRACT
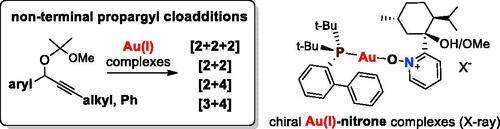
Introduction
The intensive development of gold chemistry the last decade has mainly focused on the discovery and understanding of new gold(I)-catalyzed reactions as well as the construction of new gold(I)-ligated complexes. We have contributed to the development of gold catalysis in organic synthesis in recent years by the development of a number of cycloaddition reactions based on the highly reactive terminal propargyl acetal substrates and a series of reactants, including nitrones.[Citation1–10]
We have previously studied the interesting dual aspect of nitrones in gold-catalyzed reactions, both as substrate in gold(I)-catalyzed [3 + 3] cycloaddition with propargyl acetals,[Citation5] but also as catalytic additives in gold(I)-catalyzed reactions, tuning the catalytic system to produce uncommon products. We have shown that gold(I)-nitrone complexes, either generated in situ or isolated prior to reaction, may catalyze unusual transformations, such as regio- and chemo-selective [2 + 2 + 2] cyclotrimerization of non-terminal 1,3-diaryl propargyl acetals 1 (Scheme 1).[Citation8,Citation10] The cyclohexylidene trimeric products 2a–c (up to 74% yield) were obtained as a mixture of three stereoisomers. Hydrolysis allowed isolation of 46% of the main trans,trans,cis-diastereomer.
Scheme 1. Gold(I)-nitrone-catalyzed [2 + 2+2] cyclotrimerization of diarylpropargyl acetal 1.
![Scheme 1. Gold(I)-nitrone-catalyzed [2 + 2+2] cyclotrimerization of diarylpropargyl acetal 1.](/cms/asset/643e3883-355b-4808-8dea-f880b63e11b3/lsyc_a_1731550_sch0001_b.jpg)
The cyclotrimerization approach demonstrates the ability of nitrones to tune the catalytic activity of Au(I) complexes and that unusual transformations may take place in the presence of Au(I)–nitrone complexes, which represent an interesting group of Au(I) catalyst with specific properties. Chiral nitrones have been used as Lewis bases to induce enantioselectivity in organic reactions[Citation11,Citation12] and would have the potential to act as chiral ligands and thereby afford enantioselective gold-catalyzed reactions.
Our previous results show that non-terminal propargyl substrates have ability to undergo uncommon cycloaddition reactions. Therefore, we have further studied the potential cycloaddition pathways which may take place with non-terminal propargyl acetals in the presence of either new prepared chiral Au(I)–nitrone complexes or conventional Au(I) catalysts.
Results and discussion
Synthesis of chiral nitrone ligands and Au(I)–nitrone complexes
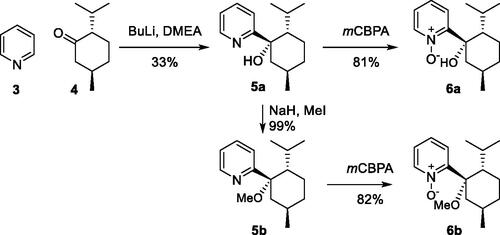
The chiral nitrone ligands 6a and 6b were synthesized via 2-pyridyl-menthol intermediate 5a, which was formed (33%, Scheme 2) by enantioselective addition of lithiated pyridine to (-)-menthone 4.[Citation9] The methyl-ether 5b was obtained in quantitative yields by methylation of alcohol 5a. Oxidation of pyridyl-menthol derivatives 5a and 5b gave the chiral nitrone ligands 6a and 6b in high yields (81–82%).
Scheme 2. Synthesis of chiral nitrone ligands 6a and 6b.
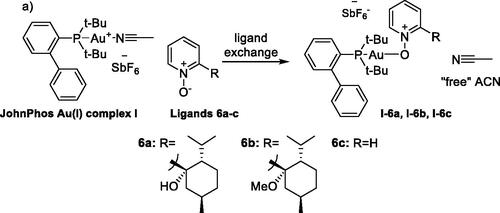
Prior to use in reactions, Au(I)–nitrone complexes I–6a, I–6b and I–6c were generated in situ by acetonitrile ligand exchange of the JohnPhos Au(I)–ACN complex I with the chiral pyridyl-nitrone ligands 6a and 6b as well as the non-substituted pyridine N-oxide 6c (Scheme 3; 1:2 ratio of complex I:nitrone 6).
Scheme 3. In situ generation of Au(I)–nitrone complexes (I–6a,b,c) by ligand exchange of JohnPhosAu(I)(ACN)SbF6 complex I with nitrones 6a–c.
1H NMR coordination studies
1H NMR analysis of gold(I) coordination experiments with nitrone ligands (), afforded a convenient method for monitoring Au(I) coordination (Scheme 4). The coordination abilities of pyridine-nitrones 6a and 6b to JohnPhosAu(I)SbF6 complex I to give chiral nitrone complexes I–6a and I–6b were clearly ascertained by comparing the changes in 1H NMR shifts going from pure nitrone (6a or 6b) and gold(I) complex I, respectively (), to 1:1 mixtures of the relevant nitrone (6a or 6b) and Au(I) complex I ().
Scheme 4. Coordination of nitrone 6a with JohnPhosAu(ACN)SbF6 I, shown as an equilibrium between [complex I + 6a] and [complex I–6a + “free” ACN].
![Scheme 4. Coordination of nitrone 6a with JohnPhosAu(ACN)SbF6 I, shown as an equilibrium between [complex I + 6a] and [complex I–6a + “free” ACN].](/cms/asset/da8e28b0-a497-45dc-8bca-5e05e035af77/lsyc_a_1731550_sch0004_b.jpg)
Figure 1. 1H NMR study of nitrones 6a and 6b coordination with JohnPhosAu(ACN)SbF6 I (1:1 in CDCl3).
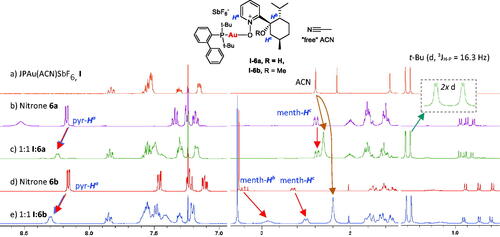
Formation of gold complex I–6b by coordination of nitrone ligand 6b with Au(I) complex I is clearly visible by 1H NMR (). All pyridine protons of nitrone 6b are deshielded, but the most significant increases in chemical shifts (Δδ1Hcoord = δ1Hcomplex – δ1Hligand) were observed for the pyr-Ha proton signal (Δδ1Hcoord 0.11 ppm). The opposite effect was seen for menthyl protons ROCCHb and ROCCHc2, which were strongly shielded (Δδ1Hcoord −0.29 ppm and −0.17 ppm, respectively). The up-field shift of the ligated ACN signal of Au(I)-complex I (from 2.39 ppm to 2.20 ppm; Δδ1Hcoord −0.19 ppm, ) is a characteristic proof of ACN de-coordination and ligand exchange to give non-ligated “free” acetonitrile.[Citation13] Also the aromatic protons of the phosphane ligand experienced minor deshielding effects.
Similar changes were observed for the I:6a mixture, indicating coordination of nitrone 6a, as well. However, a weaker observed effect on the Au(I)–ACN signal (), may indicate less efficient displacement of the ACN ligand and, hence, that nitrone 6a is less strongly coordinated to the Au(I) than nitrone 6b. A minor change of the t-Bu doublet (3JH–P = 16.3 Hz), which appears as two doublets in the I:6a mixture, indicates that diastereotopic t-Bu groups are formed by coordination of the chiral ligand (, expanded). Thus, the chiral nitrone 6a may have a greater steric effect on the phosphane ligand than nitrone 6b when coordinated to the Au(I) center.
Generally, the nitrone (6a and 6b) signals in the 1:1 mixtures are broader, indicating a dynamic ligand exchange (Scheme 4). Several observations confirm that nitrone 6b is more strongly bonded to the Au center than nitrone 6a, as the menthol proton signals are more strongly shielded and the ligated ACN peak is more affected for mixture I:6b compared to mixture I:6a. Intramolecular H-bonding in the non-methylated nitrone 6a unit (Scheme 4), shown in the crystal structure for I–6a (X-ray, ), provides one explanation for the weaker coordination of ligand 6a, compared to ligand 6b, as H-bonding is not possible in the methylated nitrone 6b
Figure 3. 1H NMR coordination study of nitrone 6a with Au(I) complex I with different I:6a ratios.
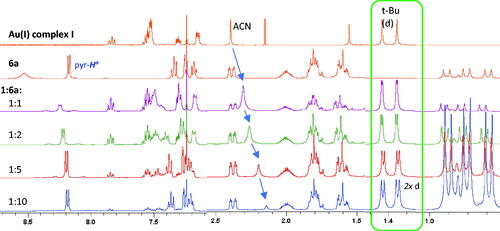
The Au(I) complexation of chiral nitrones 6a and 6b appears to be weaker compared to previously reported gold-nitrone complexes[Citation10] which gave sharp 1H NMR peaks for 1:1 mixtures of complex I and nitrones. The bulkiness of the chiral nitrones 6a and 6b may affect the ability to undergo coordination to gold(I). To get better understanding of the dynamics of the Au(I)–nitrone coordination and the equilibrium between coordinated and uncoordinated states (Scheme 4), further 1H NMR studies of complex I and nitrone 6a were carried out with increasing amounts of nitrone (ratio I:6a 1:1, 1:2, 1:5 and 1:10, and ).
Figure 2. Crystal structure (X-ray) of Au(I)–nitrone complex I–6a. Selected bond lengths (Å) and angles (°) in gold(I)-nitrone complexes I–6a, I–6c and I–6d are given.
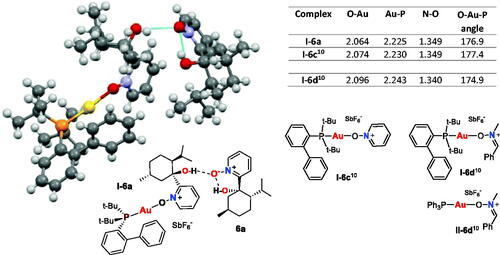
Table 1. Changes in 1H NMR peak shifts, Δδ1HcoordTable Footnotea (ppm), of mixtures with decreasing I:6a ratio.
A minor deshielding effect on the pyr-Ha proton was initially observed by a 1:1 ratio (, entry 1). By increasing to excess amount of ligand 6a, the presence of only one pyr-Ha doublet indicates a dynamic process, and not two distinct nitrone states. 1H NMR of the mixtures showed that the ACN methyl signal was strongly affected by increased amount of nitrone 6a, almost reaching the expected value of 2.10 ppm[Citation14] for “free” acetonitrile at 1:10 (I:6a) ratio (, entry 2). In contrast to ligand 6b (), ligand exchange of nitrone 6a with the ACN is less favored, as excess of nitrone appears necessary to promote full coordination to the gold center. By excess amount of ligand 6a, the phosphane tert-butyl doublet splits into two doublets with increased difference in chemical shift (Δδdiastereomer up to 0.009 ppm), demonstrating a significant diastereotopic effect by Au(I)–nitrone 2a coordination (, entry 4), as also seen to a lesser extent above ().
Similar 1H NMR coordination studies of nitrones 6a and 6b with the Au(I)-phosphane complex Ph3PAuSbF6 showed possible Au(I) coordination, in addition to decomposition of the phosphane or nitrone ligand. Coordination of Me3PAuSbF6 failed. The previously isolated Ph3PAu(I)–nitrone complex II–6d[Citation10] () was obtained in much lower yields (38%) than the corresponding JohnPhos-Au(I)–nitrone complex I–6d (74%), indicating lower coordination ability of the less electron-rich PPh3 ligand to give stable gold(I)-nitrone complexes.
Crystal structures (X-ray)
Crystalline complex I–6a was successfully obtained and the crystal structure was confirmed by X-ray analysis (), which showed the expected nearly linear nitrone-O-Au(I)-P coordination mode (176.9°). The deviation from linearity is less than for previously characterised[Citation10] complex I–6d, and slightly more than observed for pyridine-N-oxide complex I–6c, likely due to steric effects. The shorter O-Au bond length (2.064 Å) of chiral complex I–6a than for I–6c and benzaldimine nitrone I–6d, indicates that nitrone ligand 6a is more strongly bonded to the Au(I) center. This may be due to an electron donating effect of the menthol unit. The short N–O bond length of complex I–6a (1.349 Å) was similar to complex I–6c, indicating a N–O double bond character. The Au–P bond length (2.225 Å) in complex I–6a was comparable to that for complex I–6c, but shorter than for complex I–6d, likely due to the size of nitrone.
An uncoordinated nitrone 6a unit, also visible in the crystal structure (, right), enables Au(I)–nitrone coordination by hindering intramolecular hydrogen-bonding of ligand 6a, as the O-atom of the “free” nitrone unit forms an intermolecular H-bond to the OH group of the ligated nitrone 6a. The intramolecular H-bond within the uncoordinated nitrone also supports the theory that nitrone 6a has a poorer coordination ability to the Au center than MeO–nitrone 6b because of competing internal H-bonds.
Cycloaddition reactions of non-terminal propargyl acetals
The catalytic properties of the gold-nitrone complexes I–6a, I–6b and I–6c were evaluated in gold-catalyzed [2 + 2 + 2] cyclotrimerization[Citation8] and [2 + 4] cyclodimerisation of novel propargyl acetal 10 ().
Table 2. Gold(I)-catalyzed [2 + 2 + 2] cyclotrimerisation of propargyl acetal 1 to trimer 2.
Table 3. Gold(I)-catalyzed dimerization of propargl acetal 10.
Table 4. Cycloaddition reactions of (a) diene 14 with phenylbenzaldimine[Citation18] and (b) aryl-alkyl-propargyl acetals 10 and 10′ with imines 15a–d.
Catalytic effect of gold-nitrone complexes
[2 + 2 + 2] Cyclotrimerisation. The catalytic ability of complexes I–6a, I–6b and I–6c were tested in the gold(I)-nitronecatalysed [2 + 2 + 2] cyclotrimerization reaction of 1,3-diarylpropargyl acetals (1), which we have reported[Citation10] to give cyclohexylidene products 2. The presence of catalytic amounts of different nitrones was previously shown to be essential for successful selective cyclotrimerization, as Au(I) complex I gave no product 2 and complex product mixtures from diarylpropargyl acetal 1 in the absence of nitrone (, entry 1). A relevant hypothesis is that Au catalyst I is too active to afford selective trimerization, and that the reduced Au-catalyst activity by nitrone-Au coordination allows controlled chemoselective trimerization to take place.
The trimer 2 was obtained in moderate to good yields (40–73%) as mixtures of three diastereomers (2a-c, , entries 2–4). Complex I–6b gave highest yield (73%, entry 3), similar to the best yields reported[Citation8] for this reaction (74%, entry 5, nitrone 6c, counterion NTf2). Complex I–6a gave lower yield (40%, entry 2), possibly because nitrone 6a is more poorly coordinated to gold(I) than nitrone 6b, as discussed above. The success of the cyclotrimerisation reaction demands careful tuning of catalytic activity of the gold complex, and a stronger deactivation of the gold center than that provided by nitrone 6a appears to be necessary.
Some differences in the diastereoisomeric product distribution (2a–c) were observed. Complex I–6a gave highest stereoselectivity toward the main trans,trans,cis-diastereomer (55%, entry 2), while nitrone complex I–6b was slightly more selective toward isomer 2b than nitrone complex I–6c (35% vs 25%). The different isomer ratios could be attributed to a combination of factors, such as the bulk of the nitrones, as well as the strength of nitrone coordination to the gold center. Chiral HPLC separation of the three pairs of enantiomers in the product mixture for determination of possible % ee, was unsuccessful in our hands.
[2 + 4] Cyclodimerisation. As previous studies on non-terminal propargyl substrates only included phenyl-substituted propargyl acetals, such as diarylpropargyl substrate 1[Citation10] above, the reactivity of methyl-aryl-propargyl acetal 10 was explored. Catalytic amounts of both complexes I and I–6c gave full conversion of acetal 10, however, to different products; proposed to be the open and the cyclic dimeric products 11 (25%) and 12 (30%), respectively (). Both products were rather unstable.
The different dimerization reaction pathway of methyl-aryl-propargyl acetal 10 from the trimerization taking place with diaryl-propargyl substrate 1, may be explained by the proposed pathways for the formation of the octatriene 11 and cis,trans-cyclohexene 12 dimer products (Scheme 5). The trimerization of diaryl substrate 1 is proposed to proceed through [2 + 2 + 2] cycloaddition of allenic intermediates after gold-catalyzed 1,3-alkoxy rearrangement.[Citation10] In accordance with literature,[Citation8] the propargyl acetal 10 undergoes a gold-catalyzed 1,3-methoxy shift to give allenic intermediate 13. However, unlike the mechanism for diaryl-propargyl acetals, the methyl-substituted allene 13 may undergo proton shift and isomerization to give diene 14 (Scheme 5a). Such gold catalyzed transformations are reported[Citation15,Citation16] to be faster for electron-rich than electron-deficient allenes.[Citation17] Diene 14, formed by isomerization of allene 13 may subsequently undergo two alternative dimerization pathways; either with allene 13 or with a second unit of diene 14 to give the open and the cyclic dimers 11 and 12, respectively (Scheme 5b,c). NMR analysis of cyclohexene dimer 12 showed NOE correlations, such as between benzylic H3 and allylic H4 protons, which were used to establish the relative cis-trans stereochemistry of the diphenyl-vinyl-trisubstituted cyclohexene 12.
Scheme 5. Proposed pathways for (a) gold-catalyzed generation of allene 13 and diene 14 intermediates from propargyl acetal 10; (b) dimerization of diene 14 with allene 13 to give open dimer 11 and (c) dimerization of two units of diene 14 to give cyclic dimer 12.
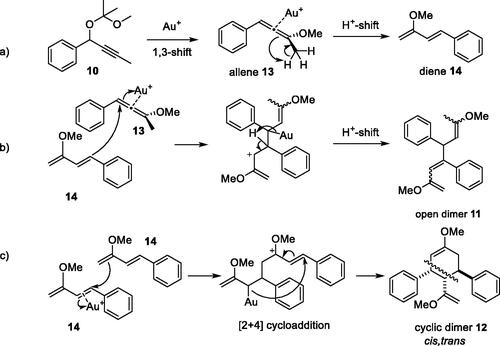
In contrast to Au(I) complex I, producing the open dimer 11, the results indicate that nitrone complex I–6c may have higher catalytic capacity for allene-to-diene isomerization (Scheme 5a), which enables dimerization of two units of diene 14 to give cyclic dimer 12 by a Diels-Alder-like [2 + 4] cycloaddition.
Cycloaddition studies of in situ generated intermediates from non-terminal propargyl acetals
Since alkyl-arylpropargyl acetal 10 seemed to generate allene 13 or diene 14 intermediates in the presence of Au(I) complexes, a series of possible Diels-Alder reactions were tested with substrate 10 and dienophiles. The reactions with alkene, alkyne, nitrile and carbonyl compounds failed to incorporate the dienophiles, and only dimers such as 11 and 12 (21–53%) were formed. However, as diene 14 is known to undergo hetero-Diels-Alder reactions with imines (48% yield of piperidine product 17a, ),[Citation18] the ability of gold(I) to catalyze possible cycloaddition reactions of imines with in situ generated allene 13 or diene 14 from alkyl-arylpropargyl acetal 10, was more promising. In fact, propargyl acetal 10 and aldimines 15a–d did undergo several cycloaddition reactions in the presence of Au(I) complex I with in situ generated intermediates 13 or 14 (). The polarized nitro-phenyl imines (15a and 15 b) mainly afforded the novel E,trans-benzylidene-diarylazetidine 16 (30–33%, entries 1 and 2), explained by the proposed [2 + 2] cycloaddition of imine and allene 13 (Scheme 6a). The more sterically hindered n-butyl-propargyl acetal 10′ gave similar product 16a′ in lower yield (<15%, entry 5). An analogous [2 + 2] cycloaddition is reported by the Fiksdahl group for dimerization of a diaryl propargyl ester to give a di-benzylidene-cyclobutyl product.[Citation8] Azetidines, such as products 16, are relatively stable; useful as biological scaffolds, reactive intermediates and chiral ligands.[Citation19,Citation20] They can be synthesized by several routes, commonly by intramolecular ring-closing, ring expansion or ring contraction, and a few intermolecular methods are known.[Citation19–21] Further chemical transformations of the azetidine moiety are useful in organic synthesis.[Citation20] A few copper-catalyzed [2 + 2] cycloadditions between allenes and imines are reported to give azetidines.[Citation20,Citation22] The presently reported gold(I)-catalyzed [2 + 2] cycloaddition of propargyl acetal 10 and imines 15a,b via in situ generated allene 13 represents a novel synthetic pathway to azetidines.
Scheme 6. Proposed mechanisms for formation of products 16–19.
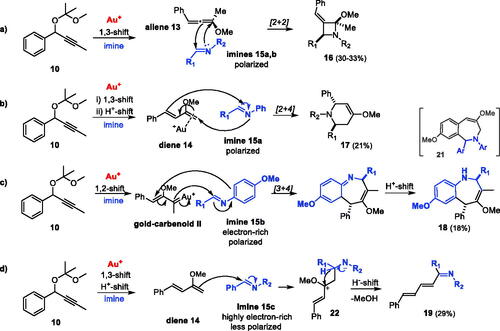
The reaction of polarized imine 15a with diene 14 also generated the [2 + 4] cycloaddition by-product 17b (21%, , entry 1 and Scheme 6b), while the more polarized and electron-rich 15 b followed an unusual competing [3 + 4] cycloaddition, involving the e-rich MeO-phenyl unit to give the benzazepine-type by-product 18 via an uncommon gold-carbenoid II (18%, , entry 2 and Scheme 6c). This product is similar to the [2 + 5] cycloaddition product 21 formed with imine and terminal aryl-propargyl acetals, reported in earlier studies,[Citation3] but the mechanism is different. The trans stereochemistry of heterocyclic products 16–18 was established from NMR NOE correlations between the respective substituents on the ring. The Z-configuration of 16 was based on NOE correlations between the relevant benzylidene protons and the R1-aryl and Me/MeO groups.
Reaction with the less polarized and more electron-rich methoxy-phenyl imine 15c followed a different reaction pathway than the cycloaddition seen for the polarized imine 15a (Scheme 6b). The main product was a fully conjugated E,E,E-pentadien-1-imine 19 (29%, , entry 3), which may be formed by diene 14 attack on imine 15c, followed by hydride shift and methanol elimination (Scheme 6d). The E,E,E-configuration of product 19 was assigned based on 1H NMR coupling constants and NOE correlations. Reaction with very electron-rich aldimine 15d gave no products, indicating that the polarization of the imine bond is important to enable nucleophilic attack on the imine.
Conclusions
Two novel chiral menthol-based pyridyl nitrones 6a and 6b were synthesized. Au(I)–nitrone complexes (I–6a and I–6b) were prepared by coordination of ligands 6a and 6b to JohnPhosAu(ACN)SbF6 complex I. 1H NMR coordination studies indicated that the new nitrones coordinated poorer to Au(I) than previously studied nitrones, probably due to the bulk of the chiral menthol-based substituent, as well as the competing intramolecular hydrogen-bonds in non-methylated ligand 6a.
The catalytic effect of Au(I)–nitrones 6a and 6b was evaluated in [2 + 2 + 2] cyclotrimerization and [2 + 4] cyclodimerization reactions. Complex I–6b gave highest yield (73%, entry 3), similar to the best yields reported previously (74%, nitrone 6c with counterion NTf2).[Citation8] The obtained diastereoisomeric ratio of the Au(I)–nitrone catalyzed cyclotrimerization products 2 from diaryl-propargyl substrate 1, was different than in previous studies,[Citation8] indicating that the nitrones have a special effect in this particular reaction. Chiral HPLC analysis of trimeric products 2a–c failed to give enantioseparation. Methyl-aryl-propargyl acetal 10 followed a different dimerization reaction pathway, based on in situ generated allene 13 and diene 14 intermediates, compared to the trimerization of diaryl-propargyl substrate 1. The Au(I) nitrone complex I–6c afforded the cyclic dimer 12 by a Diels-Alder-like [2 + 4] cycloaddition from methyl-aryl-propargyl 10, in contrast to Au(I) complex I, producing the open dimer 11.
Alternative pathways of non-terminal methyl-aryl propargyl acetal 10 were explored by testing the ability to undergo possible gold(I)-catalyzed cycloaddition reactions with different imines. Specific [2 + 2], [2 + 4] and [3 + 4] cycloaddition reactions of imines 15a–d with in situ generated allene 13 and diene 14 intermediates from acetal 10 gave a diverse range of N-heterocyclic products 16–19 (18–30%). Mechanistic discussions of the selective reaction pathways are presented.
The present screening study demonstrates the potential and the versatility of non-terminal propargyl acetals in gold(I) catalyzed cycloaddition reactions. Further optimization of reaction conditions, substrate variation and gold catalyst tuning may give improved yields and more efficient synthetic methods for preparation of selective cycloaddition products.
Experimental
(1S,2S,5R)-2-Isopropyl-5-methyl-1-(pyridin-2-yl)cyclohexanol (5a)
Butyllithium (2.0 M in cyclohexane, 2.9 mL, 5.8 mmol) under inert atmosphere was cooled to 0 °C and 2-(dimethylamino)ethanol (0.29 mL, 2.9 mmol) in dry heptane (2 mL) was added slowly. The mixture was stirred at the same temperature for 15 min. The mixture was then cooled to −78 °C and pyridine (0.12 mL, 1.5 mmol) in dry heptane (2 mL) was added. The mixture became bright orange over 30 mins, then (2S,5R)−2-isopropyl-5-methylcyclohexanone (0.50 mL, 2.9 mmol) in dry THF (5 mL) was added slowly. The mixture was stirred at this temperature for 1 h. The reaction was quenched with sat. aq. NH4Cl (25 mL) and extracted with DCM (3 × 25 mL), dried with brine and Na2SO4, filtered and evaporated in vacuo. Flash chromatography (1:1 DCM:pentane) gave the desired product as a colorless oil (114.6 mg, 33%). 1H and Citation13C NMR data were in accordance with published literature.[Citation9]
2-((1S,2S,5R)-2-Isopropyl-1-methoxy-5-methylcyclohexyl)pyridine (5 b)
Compound 5a (114.6 mg, 0.491 mmol) in dry THF (3 mL) was added dropwise to a suspension of NaH (93.8 mg, 3.91 mmol) in dry THF (12 mL). The mixture was stirred at r.t. for 30 mins then MeI (0.30 ml, 4.8 mmol) was added and the solution was stirred at r.t. for 3 h. The reaction was quenched carefully dropwise with sat. aq. NH4Cl (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic phases were dried with sat. aq. NaCl (20 mL), dried over Na2SO4, filtered and evaporated in vacuo. Flash chromatography (1:30 EtOAc:pentane) gave 5b as a colorless oil (119.6 mg, 99%). [α]20D = −51.1° (c 1.01, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 8.61 (ddd, 1H, J = 4.8/1.7/0.9 Hz, NCHAr), 7.65 (td, 1H, J = 7.8/1.8 Hz, CHAr), 7.45 (dt, 1H, J = 7.9/1.0 Hz, CHAr), 7.12 (ddd, 1H, J = 7.4/4.8/1.2 Hz, CHAr), 3.22 (s, 3 H, OCH3), 2.03–1.94 (m, 2 H, CH3OCCH2), 1.85–1.82 (m, 1H, CH3OCCH2CHCH2), 1.73–1.63 (m, 2 H, CH3CH and CH3OCCHCH2), 1.56–1.49 (m, 2 H, (CH3)2CHCH and CH3OCCHCH2), 1.29 (sepd, 1H, J = 6.9/1.5 Hz, (CH3)2CH), 1.11 (qd, 1H, J = 12.6/3.4 Hz, CH3OCCH2CHCH2), 0.96 (d, 3 H, J = 6.5 Hz, CH3), 0.87 (d, 3 H, J = 6.7 Hz, (CH3)2), 0.58 (d, 3 H, J = 6.9 Hz, (CH3)2); 13C NMR (100 MHz, CDCl3): δ ppm 164.0, 149.4, 135.3, 121.5, 121.2, 84.8, 51.7, 50.3, 39.9, 35.2, 28.0, 26.5, 23.6, 22.4, 21.2, 18.2; HRMS (ASAP) calcd for C16H26NO [M + H]+ 248.2014, found 248.2012.
2-((1S,2S,5R)-1-Hydroxy-2-isopropyl-5-methylcyclohexyl)pyridine 1-oxide (6a)
Compound 5a (95.0 mg, 0.41 mmol) was dissolved in dry DCM (2 mL) under inert atmosphere at 0 °C. 3-chlorobenzoperoxoic acid (210.3 mg, 0.938 mmol) was added. The reaction mixture was stirred at 0 °C for 5 min then the ice bath was removed and the reaction was allowed to come to r.t. overnight (17.5 h). The reaction mixture was diluted with DCM (10 mL) and washed with aq. KOH (6 N, 3 × 5 mL) and the organic layer was dried over Na2SO4, filtered and the solvent evaporated in vacuo. Flash chromatography (1:1 EtOAc:pentane) gave 6a as a colorless oil (82.0 mg, 81%). [α]20D = −79.4° (c 1.01, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ ppm 8.58 (br s, 1H, OH), 8.20 (dd, 1H, J = 6.5/0.8 Hz, CHAr), 7.38 (td, 1H, J = 7.8/1.3 Hz, CHAr), 7.28 (dd, 1H, J = 8.2/1.8 Hz, CHAr), 7.21 (td, 1H, J = 6.9/2.0 Hz, 1H, CHAr), 2.43–2.39 (m, 1H, CH2COH), 2.09–1.96 (m, 1H, CHCOH), 1.87–1.77 (m, 3 H, CH2CHCOH and CH2CH2CHCOH and CH(CH3)2), 1.66–1.59 (m, 2 H, CHCH(CH3)2 and CH2CH2CHCOH), 0.98 (d, 3 H, J = 6.7 Hz, CH(CH3)2), 0.93 (d, 3 H, J = 7.0 Hz, CH(CH3)2), 0.87 (d, 3 H, J = 6.7 Hz, CHCH3); 13C NMR (100 MHz, CDCl3): δ ppm 155.1 (CAr), 141.3 (CHAr), 127.8 (CHAr), 123.7 (CHAr), 123.6 (CHAr), 79.6 (COH), 47.8 (CHCH(CH3)2), 46.6 (CH2COH), 35.0 (CH2CHCOH), 27.8 (CH(CH3)2), 27.2 (CHCH3), 23.8 (CH(CH3)2), 22.0 (CHCH3), 21.1 (CH2CH2CHCOH), 18.7 (CH(CH3)2); HRMS (ASAP) calcd for C15H24NO2 [M + H]+ 250.1807, found 250.1809.
General procedure for gold(I)-catalyzed reaction of propargyl acetal 10 and imines 15a-d
JohnPhosAu(ACN)SbF6 (4.5 µmol) was dissolved in DCM (0.5 mL) and a mixture of acetal 10 (46 µmol) and the appropriate imine (0.138 mmol) in DCM (1 mL) was added. The reaction mixture was stirred at r.t. for 4 h, then the reaction was quenched with NEt3 and solvent removed in vacuo. Product isolation and purification was done by silica chromatography (EtOAc:pentane).
Supporting information: Additional Supporting information with Full experimental detail, 1H and 13 C NMR spectra, may be found online in the Supplementary material.
Supplemental Material
Download MS Word (1.8 MB)References
- Sperger, C. A.; Tungen, J. E.; Fiksdahl, A. Gold(I)-Catalyzed Reactions of Propargyl Esters with Vinyl Derivatives. Eur. J. Org. Chem. 2011, 2011, 3719–3722. DOI: 10.1002/ejoc.201100291.
- Iqbal, N.; Sperger, C. A.; Fiksdahl, A. Gold(I)-Catalysed Alkene Cycloaddition Reactions of Propargyl Acetals. Eur. J. Org. Chem. 2013, 2013, 907–914. DOI: 10.1002/ejoc.201201328.
- Iqbal, N.; Fiksdahl, A. Gold(I)-Catalyzed Benz[c]Azepin-4-ol Synthesis by Intermolecular [5 + 2] Cycloaddition. J. Org. Chem. 2013, 78, 7885–7895. DOI: 10.1021/jo401075n.
- Siah, H.-S. M.; Kaur, M.; Iqbal, N.; Fiksdahl, A. Gold(I)-Catalysed Tandem Cyclisation of Propargyl Acetals and Vinyl Esters. Eur. J. Org. Chem. 2014, 2014, 1727–1740. DOI: 10.1002/ejoc.201301674.
- Evjen, S.; Fiksdahl, A. Gold(I)-Catalysed [3 + 3] Cycloaddition of Propargyl Acetals and Nitrones. Tetrahedron 2016, 72, 3270–3276. DOI: 10.1016/j.tet.2016.04.058.
- Evjen, S.; Fiksdahl, A. Gold(I)-Catalysed Azepine Synthesis from Propargyl Acetals and Aryl Azides. Eur. J. Org. Chem. 2016, 2016, 2858–2863. DOI: 10.1002/ejoc.201600319.
- Siah, H.-S. M.; Hogsnes, M. C.; Iqbal, N.; Fiksdahl, A. Gold(I)-Catalysed Tandem Cyclization of Propargyl Acetals and Alkynes. Tetrahedron 2016, 72, 1058–1068. DOI: 10.1016/j.tet.2015.12.080.
- Jónsson, H. F.; Evjen, S.; Fiksdahl, A. Gold(I)-Catalyzed [2 + 2+2] Cyclotrimerization of 1,3-Diarylpropargyl Acetals. Org. Lett. 2017, 19, 2202–2205. DOI: 10.1021/acs.orglett.7b00411.
- Reiersølmoen, A. C.; Østrem, E.; Fiksdahl, A. Gold(III)-Catalysed Cis-to-Trans Cyclopropyl Isomerization. Eur. J. Org. Chem. 2018, 2018, 3317–3325. DOI: 10.1002/ejoc.201800419.
- Jónsson, H. F.; Fiksdahl, A. Studies on Gold–Nitrone Systems. Dalton Trans. 2019, 48, 142–149. DOI: 10.1039/C8DT04254C.
- Ulč, J.; Nečas, D.; Koukal, P.; Havlíček, V.; Tošner, Z.; Hybelbauerová, S.; Kotora, M. Chiral Unsymmetrically Substituted Bipyridine N,N′-Dioxides as Catalysts for the Allylation of Aldehydes. Eur. J. Org. Chem. 2018, 2018, 5109–5116. DOI: 10.1002/ejoc.201800485.
- Reep, C.; Morgante, P.; Peverati, R.; Takenaka, N. Axial-Chiral Biisoquinoline N,N′-Dioxides Bearing Polar Aromatic C–H Bonds as Catalysts in Sakurai-Hosomi-Denmark Allylation. Org. Lett. 2018, 20, 5757–5761. DOI: 10.1021/acs.orglett.8b02457.
- Siah, H.-S. M.; Fiksdahl, A. Dual-Gold(I)-Generated Trifluoromethylation of Terminal Alkynes with Togni’s Reagent. J. Fluorine Chem. 2017, 197, 24–33. DOI: 10.1016/j.jfluchem.2017.01.004.
- Fulmer, G. R.; Miller, A. J. M.; Sherden, N. H.; Gottlieb, H. E.; Nudelman, A.; Stoltz, B. M.; Bercaw, J. E.; Goldberg, K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. DOI: 10.1021/om100106e.
- Brown, T. J.; Robertson, B. D.; Widenhoefer, R. A. Synthesis and X-Ray Crystal Structure of a Cationic Gold (I) π-(1,3-Diene) Complex Generated via Isomerization of a Gold π-Allene Complex. J. Organomet. Chem. 2014, 758, 25–28. DOI: 10.1016/j.jorganchem.2014.01.030.
- Jones, A. C. Gold π-Complexes as Model Intermediates in Gold Catalysis. In Homogeneous Gold Catalysis; Slaughter, L. M., Ed. Springer International Publishing: Cham, 2015; pp 133–165.
- Li, H.; Harris, R. J.; Nakafuku, K.; Widenhoefer, R. A. Kinetics and Mechanism of Allene Racemization Catalyzed by a Gold N-Heterocyclic Carbene Complex. Organometallics 2016, 35, 2242–2248. DOI: 10.1021/acs.organomet.6b00307.
- Takasu, K.; Shindoh, N.; Tokuyama, H.; Ihara, M. Catalytic Imino Diels-Alder Reaction by Triflic Imide and Its Application to One-Pot Synthesis from Three Components. Tetrahedron 2006, 62, 11900–11907. DOI: 10.1016/j.tet.2006.09.092.
- Mehra, V.; Lumb, I.; Anand, A.; Kumar, V. Recent Advances in Synthetic Facets of Immensely Reactive Azetidines. RSC Adv. 2017, 7, 45763–45783. DOI: 10.1039/C7RA08884A.
- Takizawa, S.; Arteaga, F. A.; Yoshida, Y.; Suzuki, M.; Sasai, H. Organocatalyzed Formal [2 + 2] Cycloaddition of Ketimines with Allenoates: Facile Access to Azetidines with a Chiral Tetrasubstituted Carbon Stereogenic Center. Org. Lett. 2013, 15, 4142–4145. DOI: 10.1021/ol401817q.
- Łysek, R.; Furman, B.; Kałuża, Z.; Frelek, J.; Suwińska, K.; Urbańczyk-Lipkowska, Z.; Chmielewski, M. [2 + 2] Cycloaddition of Chlorosulfonyl Isocyanate to Allenyl-Sugar Ethers. Tetrahedron: Asymmetry 2000, 11, 3131–3150. DOI: 10.1016/S0957-4166(00)00260-3.
- Akiyama, T.; Daidouji, K.; Fuchibe, K. Cu(I)-Catalyzed Enantioselective [2 + 2] Cycloaddition of 1-Methoxyallenylsilane with α-Imino Ester: Chiral Synthesis of α,β-Unsaturated Acylsilanes. Org. Lett. 2003, 5, 3691–3693. DOI: 10.1021/ol0353621.