
Abstract
UV curing of photopolymerizable monomers, like (meth)acrylates, has been utilized for coatings for more than half a century and more recently in further developed areas such as tissue engineering. However, these monomers have major disadvantages, e.g., high irritancy and cytotoxicity, which leads to limited use in tissue engineering regarding health issues. Vinyl esters (VE) and vinyl carbonates (VC) can compete with (meth)acrylates in terms of material properties and have significantly lower toxicity, but lack in cost efficient synthesis methods. The purpose of this communication is to establish new pathways to overcome this drawback. It was shown that VEs can be synthesized either by vinyloxy trimethylsilane or by acetaldehyde in excellent yields. Moreover, a new method to synthesize vinyl chloroformate as precursor for VCs in lab scale was evolved by a catalyzed reaction of vinyloxy trimethylsilane with a phosgene solution. Finally, the cytotoxicity tests showed auspicious results.
Graphical Abstract
Introduction
Since more than a half-century, UV curing of photopolymerizable monomers has been used for decorative and protective coatings. Especially (meth)acrylates are state-of-the-art monomers in photopolymerization of such coatings and printing inks.[Citation1-3] Moreover, they are also widely utilized in further advanced fields, such as 3 D-printing or tissue engineering, owing to their large variety of commercially available building blocks, fast curing rates, and good storage stability.[Citation4] Nevertheless, these monomers have major drawbacks, as for instance cytotoxicity caused by the monomer itself and also their toxic and irritating degradation products, which lead to a limited applicability in respect to health and environmental issues.[Citation5] For that reason, vinyl ester (VE) and vinyl carbonate (VC) monomers were evaluated recently.[Citation5–10] These compounds exhibit a significant lower toxicity, good storage stability, similar or even better mechanical properties of the final polymer and sufficient reactivity toward radical polymerization.[Citation5–7,Citation9] Lower toxicity can be assigned to the hydrolytic degradation of crosslinked VEs and VCs to nontoxic and FDA-approved poly(vinyl alcohol).[Citation6]
However, only a few building blocks of these monomers are commercially available due to toxic, costly or ineffective synthesis methods. Typically, VE monomers are synthesized by transesterification of carboxylic acids with vinyl acetate in the presence of a catalyst. The applied catalyst is based on toxic mercury (II) salts, whereas nucleophilic groups can deactivate the Hg+-intermediate.[Citation11] Thus, palladium (II) salts are used, as they are less toxic and stable toward nucleophiles. Still, they are expensive and hard to recycle.[Citation12] Another synthetic pathway is based on the use of phenyl selenium ethanol as a precursor.[Citation13] However, the poor atom efficiency and multistep reaction disfavors this method.
The preparation of VCs also lacks cheap and efficient synthesis pathways. The most common method uses vinyl chloroformate (VOCl) reacting with an alcohol in a modified Einhorn acylation, as here chloroformate replaces acyl chloride.[Citation14] Moreover, pyridine is utilized as catalyst and acid scavenger.[Citation15,Citation16] The vinyl chloroformate pathway is favorable regarding quantitative yields and an easy work-up procedure. Nonetheless, over the last years, the number of suppliers has been steadily decreasing. Consequently, the price has increased tremendously. There are two commercial processes in industrial scale available.[Citation17] One method is based on the pyrolysis of ethylene glycol bis(chloroformate), which is synthesized by acylation of ethylene glycol with phosgene.[Citation18] The more recent two-step reaction involves phosgene together with mercury bisacetaldehyde.[Citation19] In summary, it is required to establish new methods, or to refurb the state-of-art synthesis of VE and VC monomers, respectively.
The purpose of this communication is to present a more convenient, more economic and safer pathway to obtain VE in good yields that offer versatile possibilities for these biocompatible and photopolymerizable monomers. Additionally, the synthesis of VOCl is improved by excluding mercury educts and facilitate the processability in lab scale.
Results and discussion
Vinyl esters from vinyloxy trimethylsilane (1)
To overcome the shortages of the available synthetic pathways generating VE monomers, two strategies were evaluated (Schemes 1 and 2).
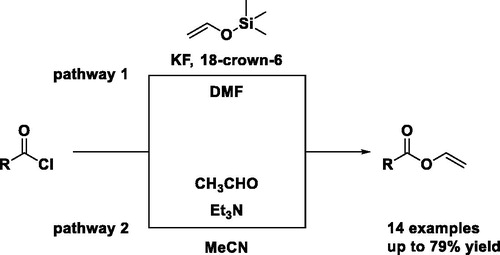
As shown in Scheme 1, the equilibrium of acetaldehyde can be shifted by the amine base triethylamine into the temporarily formed vinyl alcohol and can be captured by trimethylsilyl chloride (TMS-Cl) to obtain vinyloxy trimethylsilane (1). Subsequently, 1 can be reacted with acyl chlorides in the presence of potassium fluoride (KF) and 18-crown-6 ether to obtain VEs.
Scheme 1. Pathway 1 for the synthesis of VE monomers.
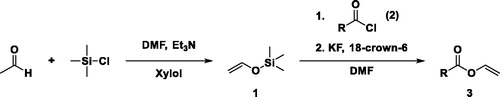
Scheme 2. Pathway 2 for the synthesis of VEs.
In detail, the vinyl donor vinyloxy trimethylsilane (1) was synthesized according to the procedure of Olofson et al.[Citation20] with some refinements to compile a more convenient process. Following to the tautomeric base-catalyzed shift of acetaldehyde to vinyl alcohol, the emerging intermediate was caught with TMS-Cl to obtain 1 and the accruing hydrochloric acid was neutralized in situ by a tertiary amine. In more detail, an excess of dry acetaldehyde (2.5 equiv.) was slowly dripped into the mixture of dry triethylamine (Et3N) (1.25 equiv.), dry N,N’-dimethylformamide (DMF) and distilled TMS-Cl (1 equiv.). After the work-up procedure, 59% of the theoretical yield of a colorless oil could be obtained. Noteworthy, the usage of 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) as enolization agent did not lead to formation of the desired product 1. Contrary to this approach, Olofson et al.[Citation20] stated that if acetaldehyde was directly dripped into the mixture, vinyloxy trimethylsilane (1) could be obtained only in yields of 15–20%. Therefore, they slowly distilled acetaldehyde into the mixture with the distillation outlet below the surface of the mixture to obtain 62% theoretical yield. Thereby, it is possible that the reaction solution shoots back into the acetaldehyde flask due to underinflation causing enormous dangerous reaction heat. However, with our straightforward strategy no complex reaction apparatus is required to synthesize 1 in good yields and thus, underinflation can be excluded.
Similar to the work of Olofson et al.,[Citation20] where thiocarbonates were synthesized using 1 as precursor, we used 1 to obtain vinyl esters. Therefore, different acyl chlorides (2a–c) were reacted with 1 to obtain the aromatic VE vinyl benzoate (3a), the aliphatic VE vinyl decanoate (3b) and the difunctional VE divinyl adipate (3c) (). In general, 1.5 equiv. of 1 was dropped into a mixture of 1 equiv. freshly distilled acyl chloride (2a, b or c), 5 equiv. dry potassium fluoride (KF), 0.1 equiv. dry 18-crown-6 ether in dry DMF. 18-crown-6-ether was used as phase transfer agent between the organic phase and the insoluble salt KF. Additionally, the 18-crown-6 ether is designed to catch the potassium cation. Consequently, the fluoride anion gets more nucleophilic due to the increased distance to the potassium cation. Thus, the fluoride anion attaches to the silicon of 1 and forms a pentavalent intermediate. Based on the increased nucleophilicity of this intermediate the lone pair of the oxygen of 1 attacks the carbonyl carbon of the acyl chloride (2) (Scheme 3).[Citation21] The energetically more stable bond between silicon and fluoride, as compared to the bond between silicon and oxygen, causes the bond of silicon and oxygen to break. Simultaneous, the fluoride anion performs partially an ion exchange with the chloride of the acyl chloride (2) to obtain the corresponding more reactive fluoroformate.[Citation22] It is important to use at least 3 equiv. of KF in case of traces of remaining water present to scavenge possibly created fluoric acid to potassium bifluoride.[Citation23] Noteworthy, the aprotic polar solvent DMF improves the SN2-reaction, where the desired vinyl ester (3) and the side product trimethylsilyl fluoride are formed.[Citation24]
Scheme 3. Mechanism of pathway 1.
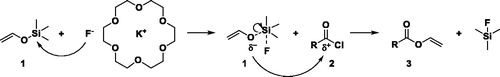
Table 1. Universality of pathway 1.
The reaction control was carried out by thin layer chromatography (TLC). After an aqueous work-up procedure, the crude mixture was purified by column chromatography to afford colorless liquid 3a in 69% yield. To demonstrate the versatility of precursor 1 aliphatic VEs 3b and 3c were synthesized, as well, utilizing the procedure for 3a. Freshly distilled decanal chloride was used to obtain 78% yield of 3b. The difunctional VE 3c could be isolated in mediocre yields of 48% with adipoyl chloride as a starting material ().
Vinyl esters directly from acetaldehyde (5)
The variety of carboxylic acids applied in the synthesis of VEs is widespread. Thus, the corresponding acyl chlorides could be interesting for the synthesis of VEs in combination with vinyloxy trimethylsilane (1). Nonetheless, this reaction lacks in atom efficiency for industrial scale owing to the protecting group. Therefore, pathway 2 was established (Scheme 2) which overcomes this drawback of route 1.
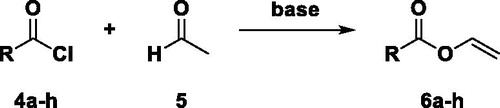
This pathway is based on the tautomeric vinyl alcohol - acetaldehyde equilibrium, whereby the vinyl alcohol intermediate should perform an Einhorn acylation[Citation14] with an acyl chloride. Sladkov et al. described a method for the acylation of the enol form of acetaldehyde.[Citation25] This is the only one known in literature, in which acetaldehyde reacts with acyl chloride in an Einhorn acylation[Citation14] to numerous different VEs.
We tried to confirm the work of Sladkov et al. using pyridine as enolization agent and vinyl benzoate (6a) in a model reaction.[Citation25] After several attempts, no product could be obtained, as found by others.[Citation26] Before heating the reaction to 45 °C, the proton nuclear magnetic resonance (1H-NMR) measurements and TLC only showed educts. After 18 h at 45 °C, the brown reaction mixture consisted of several aldol reaction products. Thus, the base was changed to Et3N. The first attempt using DMF and a molar ratio of 1 equiv. benzoyl chloride (4a), 1.25 equiv. acetaldehyde (5) and 2.5 equiv. Et3N led to a crude yield of 55% of 6a. The yield was determined based on the 1H-NMR spectrum.
To optimize the yields of this system, different solvents were tested. In this test series, a molar ratio of 1 mol 4a, 1.25 mol acetaldehyde (5) and 2.50 mol Et3N was used. With acetonitrile (MeCN) an 11% higher conversion of 4a–6a was achieved after one day at 45 °C, compared to the reaction in bulk with 58% yield. Moreover, the reaction in bulk is difficult to handle and due to overheating during the reaction, polymerization could occur. The same reaction conditions in N,N’-dimethylformamide (DMF) led to a conversion of 55%, in dichloromethane to 35%, in tetrahydrofuran to 15%, in toluene and xylene to 10%. It is necessary to use an aprotic solvent, otherwise the acyl chloride hydrolyzes, and it is important to keep an eye on the dielectric constant (ε) of the solvent. As shown in , the order of the yield is in accordance with the order of ε. The utilization of MeCN resulted in the highest yields with 69%. Therefore, MeCN was used for further experiments.
Table 2. Optimization of the model reaction for pathway 2.
Another crucial factor is the molar ratio of the educts. Different molar ratios of 4a, 5 and Et3N were examined to elaborate the optimum conditions for this reaction. The reaction was performed in a sealed penicillin flask using MeCN at 45 °C for 24 h. At first, the ratio of acetaldehyde 5 was increased compared to the acid chloride 4a and triethylamine using 1:3:4 for 4a:5:Et3N (Entry 8 in ). However, this did not lead to an increase in yield. Thus, the ratio was changed to 1:2:5 for 4a:5:Et3N with an increased content of the tertiary amine using MeCN as solvent. With these conditions, the highest yield, namely 79%, was achieved after one day. Hence, an increased amount of acetaldehyde 5 and Et3N increases the conversion of the acid chloride 4a. Summarized, there is a positive coherence between molar ratio and conversion.
In addition, the versatility of this type of reaction in respect to the used base was extended by using KF and 18-crown-6 ether under the same conditions as in pathway 1, but with acetaldehyde instead of 1. Consequently, changing the base caused an at least 20% lower yield. Finally, 6a was synthesized with optimized conditions in an isolated yield of 79% using Et3N as tertiary amine base, MeCN as solvent and a molar ratio of 1 equiv. acyl chloride (4a), 2 equiv. acetaldehyde (5), and 5 equiv. triethylamine.
Based on these optimized conditions, a variety of different acyl chlorides () were used to prove the generality of pathway 2 for the synthesis of VEs. Firstly, simple aliphatic acyl chlorides, such as acetyl chloride (4b), propionyl chloride (4c), acryloyl chloride (4d) and butyryl chloride (4e), were reacted in the same manner as 4a. After 24 h reaction time the mixture turned dark red brown and solidified. A crude 1H-NMR sample showed no vinyl peaks in any of these attempts. Furthermore, the work-up procedure led to a mixture of different aldol reaction products. Two more complex acyl chlorides, namely oxalyl chloride (4f) and chloroacetyl chloride (4g), were tested in the same way as described before. Similar to the simple aliphatic acyl chlorides, the 1H-NMR spectra showed only different aldol products.
Table 3. Universality of pathway 2.
It is known from literature,[Citation29] that the α-CH of acyl chlorides has a pKa value of 16, which is close to the pKa value of acetaldehyde (pKa 17). These similar acidities could explain the resulting mixture of aldol products. To prove this hypothesis, pivaloyl chloride (4h), lacking an α-CH, was reacted with acetaldehyde. Indeed, the crude 1H-NMR spectrum showed formed pivaloyl VE and is in accordance with the literature shifts of pivaloyl VE.[Citation30]
In addition, this provides evidence that the mechanism of pathway 1 depends on the pentavalent intermediate. A formed enolate would undergo a fast tautomerization to the corresponding aldehyde. Based on these findings di- and trifunctional aromatic acyl chlorides were investigated, as well, because functionality is necessary to build different polymer networks and structures. Here, divinyl terephthalate (6i) was synthesized using 4 equiv. of 5 and 10 equiv. of Et3N in MeCN. After purification by column chromatography, a white solid could be obtained in good yields of 58%.
Furthermore, it was tried to use KF in 18-crown-6-ether, which enolizes acetaldehyde and nucleophilically attacks the carbonyl C of the acyl chloride.[Citation31] Compared to the reaction utilizing Et3N, this led only to mediocre yields of 26% of 6i. In addition, divinyl isophthalate (6j) was synthesized in consonance with the procedure of 6i to investigate the influence of the steric differences of the distinct conformations of the phthalic acyl chloride. Similar to 6i the enolization using Et3N led to a significant higher yield of 42% than the pathway using the enolization effect of the naked F- anion, which achieved a yield of only 20%. In summary, steric hindrance caused lower yields independent on the enolization methods applied. Following this, trivinyl trimesate (6k) was synthesized using 6 equiv. of 5 and 15 equiv. of Et3N in MeCN. After purification by column chromatography, a white solid could be obtained in good yields of 72%.
In order to verify the expected toxicity of vinyl esters,[Citation6,Citation9,Citation10] 6a was tested exemplarily on mouse fibroblast cell line L929 and compared with the corresponding (meth)acrylates. The cytotoxicity of VE was 4 times lower than of methacrylates and even one order of magnitude lower than of acrylates, which emphasizes the potential of VEs (see Supporting Info).
Vinyl carbonates
The synthesis of vinyl carbonates, another low toxic monomer, should be investigated based on the precursor vinyl chloroformate (7).
For the common synthesis of 7 in lab scale, phosgene is reacted with mercury acetaldehyde.[Citation32] However, organic mercury compounds are highly toxic and mercury itself is accumulated in the body over years and causes mercury poising.[Citation33] As an alternative, a synthetic route to avoid mercury was established by Bausch & Lomb Inc. for the synthesis of 7 on an industrial level.[Citation34] This reaction was slightly modified to make it accessible for lab scale and to enhance safety issues.
In detail, 1.4 equiv. of the protected vinyl alcohol 1 was reacted with 1 equiv. of a 20 wt% phosgene solution in toluene with acetonitrile as solvent in the presence of 0.0075 equiv. of palladium acetate to obtain 7 instead of the industrial route[Citation34] using gaseous phosgene in adiponitrile (Scheme 4). During this catalyzed reaction, the double bond of 1 coordinates on the surface of the Pd catalyst. Then, TMS reacts with acetate and the enolate tautomerizes to the keto state. A nucleophilic attack onto the carbonyl C of phosgene and an additional H-elimination lead to the desired product. The catalyst is regenerated by reacting TMS-acetate with chloride of phosgene. Here, it is necessary to avoid traces of water to prevent phosgene from hydrolysis.
Scheme 4. Synthesis of vinyl chloroformate.
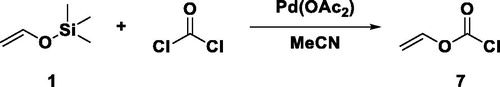
It was not possible to obtain pure 7. In the course of the distillation via vigreux column, the solvent MeCN formed an azeotrope with toluene with a boiling point close to the one of 7. Nevertheless, the obtained toluene/MeCN solution contained 10% of 7, which is equivalent to 55% of the theoretical yield. Moreover, the residual solvent does not disturb the subsequent reaction with an alcohol (8) due to higher volatility of the solvent, as compared to the resulting product (9).
For testing the applicability of 7 in this pathway, different substrates based on difunctional alcohols were reacted with 7 in the presence of pyridine in an Einhorn acylation.[Citation14] Firstly, polyethylene glycol (PEG) (8a), widely used in biomedical applications,[Citation35] was reacted with 2.1 equiv. of 7 to obtain PEG divinyl carbonate (9a) in a nearly quantitative yield after purification by flash chromatography. The steric more enhanced polypropylene glycol (PPG) divinyl carbonate (9b) was synthesized in the same manner and reacted with full conversion of PPG (8b) to the desired product. To show a variety within this monomer class ethoxylated bisphenol A (8c) was modified with a divinyl carbonate group to obtain ethoxylated bisphenol A divinyl carbonate (9c) in high yields ().
Table 4. Synthesis of different VCs.
Finally, to evaluate the biocompatibility of VCs as shown in other publications,[Citation36,Citation37] 9a was tested exemplarily on mouse fibroblast cell line L929 and compared with the corresponding (meth)acrylates. It was observed that VC is as cytotoxic as the methacrylate and one order of magnitude lower than the acrylate, which emphasizes the potential of VCs to replace acrylates (see Supporting Info).
Conclusion
In this communication, it is demonstrated that mono- and multifunctional aromatic VEs can be prepared by simply reacting an acyl chloride with acetaldehyde using triethylamine as enolization agent. However, this synthetic route is not versatile enough to obtain aliphatic VEs. Pathway 1, using vinyloxy trimethylsilane and acyl chlorides in the presence of KF and 18-crown-6-ether to obtain VEs, could be used as an alternative synthesis route to receive mono- and difunctional aliphatic VE. Furthermore, this communication also evidenced a successful mercury-free synthesis of vinyl chloroformate in lab scale and subsequent reaction with an alcohol to obtain VCs. Both, VE and VC showed significantly lower cytotoxicity compared to the corresponding acrylate, and VE was even 4 times less cytotoxic than the corresponding methacrylate. This protocol demonstrates an innovative, cheap and facile synthesis for low toxic monomers applied in photopolymerization.[Citation5-10] Most recently, this class of polymers has been used in highly developed fields of medicinal chemistry, and it will enhance the future of 3 D printing in the field of tissue engineering.[Citation38]
Experimental part
1H-NMR- and 13C-NMR-spectra were measured either on a BRUKER Avance DRX-400 FTNMR spectrometer at 400 MHz for 1H-NMR and at 100 MHz for 13C-NMR or on a BRUKER AC-E-200 FTNMR spectrometer at 200 MHz for 1H-NMR and 50 MHz for 13C-NMR. Chemical shifts (δ) are reported in ppm (s = singulett, d = duplet, t = triplett, q = quartett, quint = quintet, m = multiplet) and coupling constants (J) in Hz. Deutero-chloroform (CDCl3) and deuterated dimethyl sulfoxid (d6-DMSO) from the company Eurisotop were used as solvents and referenced to the solvent peak on literature data.[Citation39] The grade of deuteration was at least 99.8%. For thin layer chromatography (TLC) aluminum foils, coated with silicagel 60 F254 from the company Merck were applied. Column chromatography was performed with a glass column was packed with silicagel 60 µm from the distributor ROTH. Microanalyses were performed on PerkinElmer Series II CHNS/O 2400 elemental analyzer.
General procedure for compounds 3a-c
All used glassware was dried and flushed with argon. A 20 mL sealed glass vial was charged with a solution of distilled acyl chloride (2 mmol) in 5 mL dry DMF, dry KF (10 mmol), dry 18-crown-6-ether (0.2 mmol) and finally bubbled with argon. The mixture was heated to 40–50 °C, vinyloxy-trimethylsilane (3 mmol) was slowly added, stirred for 20 h, and meanwhile the solution turned red-brownish. The reaction progress was monitored by TLC, whereas butanol was used to react with the residual acyl chloride for monitoring the conversion. The solution was diluted in 20 mL ethyl acetate, washed with water (2 × 40 mL), brine (2 × 40 mL), and dried over Na2SO4. The organic phase was filtered and the solvent was removed under reduced pressure. For further purification, the red-brownish liquid was cleaned via column chromatography to obtain a colorless oil.
Vinyl benzoate (3a)[Citation40]
Colorless liquid. Yield: 0.2 g (69%). 1H-NMR (400 MHz, CDCl3): δ (ppm) = 8.08 (2 H, d, J = 8.0 Hz, o-aromatic), 7.60–7.35 (4 H, m, m-, p-aromatic, CH2=CH), 5.05 (1 H, dd, J = 13.9, 1.5 Hz, trans-CH2=), 4.68 (1 H, dd, J = 6.3, 1.5 Hz, cis-CH2=). 13C-NMR (50 MHz, DMSO) δ (ppm) = 163.52 (C=O), 141.40 (CH2=CH), 133.55 (aromatic), 129.94 (aromatic), 128.90 (aromatic), 128.49 (aromatic), 98.13 (CH2=).
General procedure for compounds 6a-k
All used glassware was dried and flushed with argon. A 20 mL sealed glass vial was charged with distilled acyl chloride (2 mmol), dried Et3N (10 mmol for 6a–h/20 mmol for 6i–j/30 mmol for 6k) and stirred in 5 mL dry acetonitrile at room temperature under argon. Afterwards the reaction mixture was cooled with an ice bath and dried acetaldehyde (4 mmol for 6a–h/8 mmol for 6i–j/12 mmol for 6k) was dropped slowly to this mixture. The solution was heated to 40–50 °C and stirred for 24 h, while it turned orange brown. The reaction progress was monitored via TLC, whereas butanol was used to react with the residual acid chloride for monitoring the conversion. Furthermore, the mixture was diluted in 20 mL ethyl acetate, triethylamine was neutralized with sat. NH4Cl (2 × 20 mL), washed with and water (2 × 40 mL). Before evaporating the solvent, the organic layer was firstly dried with brine (2 × 40 mL) and then over Na2SO4. The organic phase was filtered and concentrated under reduced pressure. For further purification, the brownish liquid was cleaned via column chromatography.
Vinyl benzoate (6a)[Citation40]
Colorless liquid. Yield 0.23 g (79%). 1H-NMR (400 MHz, CDCl3): δ (ppm) = 8.08 (2 H, d, J = 8.0 Hz, o-aromatic), 7.60–7.35 (4 H, m, m-, p-aromatic, CH2=CH), 5.05 (1 H, dd, J = 13.9, 1.5 Hz, trans-CH2=), 4.68 (1 H, dd, J = 6.3, 1.5 Hz, cis-CH2=). 13C-NMR (50 MHz, DMSO) δ (ppm) = 163.52 (C=O), 141.40 (CH2=CH), 133.55 (aromatic), 129.94 (aromatic), 128.90 (aromatic), 128.49 (aromatic), 98.13 (CH2=).
General procedure for compounds 9a-c
A round bottom flask equipped with a stirrer bar was charged with dried alcohol (7 mmol), dry 50 mL DCM and pyridine (14.7 mmol) and cooled by an ice bath. Afterwards vinyl chloroformate (14.7 mmol) was slowly dropped to the reaction mixture, warranting the reaction temperature does not increase. To ensure, that both functional groups are reacted, an excess of vinyl chloroformate is necessary. Meanwhile, the reaction progress was monitored by 1H-NMR spectroscopy and via IR. After stirring overnight, the residual vinyl chloroformate was quenched with water, inhibitor was added and concentrated in high vacuo. Finally, the product was purified by flash column chromatography with acetone as solvent to obtain the desired product.
Polyethyleneglycol divinyl carbonate (9a)
Slightly yellow viscous oil. Yield: 4.1 g 95%. 1H-NMR (200 MHz, DMSO): δ (ppm) = 7.04 (2 H, dd, J = 6.17 Hz, J = 13.9 Hz, 2x CH2=CH–), 4.92 (2 H, dd, J = 1.78 Hz, J = 13.9 Hz, 2x trans-CH2=), 4.67 (2 H, dd, J = 2.01, J = 6.17, 2x cis-CH2=), 4.27 (4 H, t, J = 4.6 Hz, 2x–O–CH2–CH2–O–C = O), 3.64 (4 H, t, J = 4.6 Hz, 2x –O–CH2–CH2–O–C=O), 3.51 (48 H, m, 12x –O–CH2–CH2–O–). 13C-NMR (50 MHz, DMSO) δ (ppm) = 152.07 (C=O), 142.73 (CH2=CH), 98.24 (CH2=CH), 69.73–67.52 (CH2). Anal. Calcd. for C33.6H61.2O18.8 (the sum formula is depending on the results of the OH-titration): C, 52.68; H, 8.05. Found: C, 52.40; H, 8.09.
Full experimental details, spectroscopic data, biological data, tables, and figures can be found via the “Supplementary Content” section of this article’s webpage.
Supplemental Material
Download MS Word (292.1 KB)Acknowledgements
The authors kindly thank Viktor Savic for his valuable comments.
Additional information
Funding
References
- Schuster, M.; Turecek, C.; Kaiser, B.; Stampfl, J.; Liska, R.; Varga, F. Evaluation of Biocompatible Photopolymers I: Photoreactivity and Mechanical Properties of Reactive Diluents. J. Macromol. Sci. A 2007, 44, 547–557. DOI: 10.1080/10601320701235958.
- Schuster, M.; Turecek, C.; Mateos, A.; Stampfl, J.; Liska, R.; Varga, F. Evaluation of Biocompatible Photopolymers II: further Reactive Diluents. Monatsh. Chem. 2007, 138, 261–268. DOI: 10.1007/s00706-007-0609-2.
- Fouassier, J.-P. Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications; Hanser: New York, 1995.
- Nichol, J. W.; Koshy, S. T.; Bae, H.; Hwang, C. M.; Yamanlar, S.; Khademhosseini, A. Cell-Laden Microengineered Gelatin Methacrylate Hydrogels. Biomaterials 2010, 31, 5536–5544. DOI: 10.1016/j.biomaterials.2010.03.064.
- Heller, C.; Schwentenwein, M.; Russmüller, G.; Koch, T.; Moser, D.; Schopper, C.; Varga, F.; Stampfl, J.; Liska, R. Vinylcarbonates and Vinylcarbamates: Biocompatible Monomers for Radical Photopolymerization. J. Polym. Sci. A Polym. Chem. 2011, 49, 650–661. DOI: 10.1002/pola.24476.
- Mautner, A.; Steinbauer, B.; Orman, S.; Russmüller, G.; Macfelda, K.; Koch, T.; Stampfl, J.; Liska, R. Tough Photopolymers Based on Vinyl Esters for Biomedical Applications. J. Polym. Sci. A Polym. Chem. 2016, 54, 1987–1997. DOI: 10.1002/pola.28065.
- Mautner, A.; Qin, X.; Wutzel, H.; Ligon, S. C.; Kapeller, B.; Moser, D.; Russmueller, G.; Stampfl, J.; Liska, R. Thiol‐Ene Photopolymerization for Efficient Curing of Vinyl Esters. J. Polym. Sci. A Polym. Chem. 2013, 51, 203–212. DOI: 10.1002/pola.26365.
- Husar, B.; Liska, R. Vinyl Carbonates, Vinyl Carbamates, and Related Monomers: Synthesis, Polymerization, and Application. Chem. Soc. Rev. 2012, 41, 2395–2405. DOI: 10.1039/c1cs15232g.
- Husar, B.; Heller, C.; Schwentenwein, M.; Mautner, A.; Varga, F.; Koch, T.; Stampfl, J.; Liska, R. Biomaterials Based on Low Cytotoxic Vinyl Esters for Bone Replacement Application. J. Polym. Sci. A Polym. Chem. 2011, 49, 4927–4934. DOI: 10.1002/pola.24933.
- Heller, C.; Schwentenwein, M.; Russmueller, G.; Varga, F.; Stampfl, J.; Liska, R. Vinyl Esters: Low Cytotoxicity Monomers for the Fabrication of Biocompatible 3D Scaffolds by Lithography Based Additive Manufacturing. J. Polym. Sci. A Polym. Chem. 2009, 47, 6941–6954. DOI: 10.1002/pola.23734.
- Sabel, A.; Smidt, J.; Jira, R.; Prigge, H. Exchange of Vinyl and Other Unsaturated Groups between Esters and Carboxylic Acids Catalyzed by Platinum Metal Salts. Chem. Ber. 1969, 102, 2939–2950. &. DOI: 10.1002/cber.19691020908.
- Lobell, M.; Schneider, M. P. Synthesis of Hydroxycarboxylic Acid Vinyl Esters. Synthesis 1994, 1994, 375–377. DOI: 10.1055/s-1994-25479.
- Weinhouse, M. I.; Janda, K. D. A New Methodology for the Preparation of Vinyl Esters. Synthesis 1993, 1993, 81–83. DOI: 10.1055/s-1993-25802.
- Wang, Z. Einhorn Acylation. In Comprehensive Organic Name Reactions and Reagents; Wang, Z., Ed.; Wiley: New York, 2010; pp. 967–970.
- Mori, M.; Nakanishi, M.; Kajishima, D.; Sato, Y. A Novel and General Synthetic Pathway to Strychnos Indole Alkaloids: Total Syntheses of (-)-Tubifoline, (-)-Dehydrotubifoline, and (-)-Strychnine Using Palladium-Catalyzed Asymmetric Allylic Substitution. J. Am. Chem. Soc. 2003, 125, 9801–9807. DOI: 10.1021/ja029382u.
- Pozo, M.; Gotor, V. Chiral Carbamates through an Enzymatic Alkoxycarbonylation Reaction. Tetrahedron 1993, 49, 4321–4326. DOI: 10.1016/S0040-4020(01)85748-3.
- Kreutzberger, C. B. A.; Olofson, R. A.; Buszek, K. R. Vinyl Chloroformate. In Encyclopedia of Reagents for Organic Synthesis; Paquette, L. A., Ed.; Wiley: New York, 2001.
- Kung, F. E. Unsaturated Chlorocarbonates and Method of Preparation; Google Patents, 1945.
- Malfroot, T.; Piteau, M. Industrial Synthesis of Vinyl Chloroformates; Societe Nationale des Poudres et Explosifs: France, 1979; p. 9.
- Olofson, R. A.; Cuomo, J. A Useful Route to Alkenyl s-Phenyl Thiocarbonates: Reagents for the Introduction of the Enyloxycarbonyl Moiety in Synthesis. J. Org. Chem. 1980, 45, 2538–2541. DOI: 10.1021/jo01300a066.
- Rendler, S.; Oestreich, M. Hypervalent Silicon as a Reactive Site in Selective Bond-Forming Processes. Synthesis 2005, 2005, 1727–1747. DOI: 10.1055/s-2005-869949.
- Lee, I.; Shim, C. S.; Chung, S. Y.; Kim, H. Y.; Lee, H. W. Cross-Interaction Constants as a Measure of the Transition-State Structure. Part 1. The Degree of Bond Formation in Nucleophilic Substitution Reactions. J. Chem. Soc, Perkin Trans. 2 1988, 1988, 1919–1923. DOI: 10.1039/p29880001919.
- Olofson, R. A.; Dang Vu, A.; Morrison, D. S.; De Cusati, P. F. Simple One-Step Preparations of Vinylic Carbonates from Aldehydes. J. Org. Chem. 1990, 55, 1–3. DOI: 10.1021/jo00288a001.
- Dehmlow, E. V.; Fastabend, U.; Keßler, M. A One-Pot Synthesis of Trimethylsilyl Fluoride. Synthesis 1988, 1988, 996–997. DOI: 10.1055/s-1988-27783.
- Sladkov, A. M.; Petrov, G. S. Acylation of the Enol Form of Acetaldehyde. Zh. Obshch. Khim. 1954, 24(3), 450–454.
- Yakubovich, A. Y.; Razumovskii, V. V.; Vostrukhina, Z. N.; Rozenshtein, S. M. Syntheses of Vinyl Monomers. Zh. Obshch. Khim. 1958, 28(7), 1930–1936.
- Luo, C. J.; Stride, E.; Edirisinghe, M. Mapping the Influence of Solubility and Dielectric Constant on Electrospinning Polycaprolactone Solutions. Macromolecules 2012, 45, 4669–4680. DOI: 10.1021/ma300656u.
- Richards, T. W.; Shipley, J. The Dielectric Constants of Typical Aliphatic and Aromatic Hydrocarbons, Cyclohexane, Cyclohexanone, and Cyclohexanol. J. Am. Chem. Soc. 1919, 41, 2002–2012. DOI: 10.1021/ja02233a017.
- McMurry, J. Organic Chemistry; Brooks/Cole Cengage Learning: Australia, 2012.
- Kim, J. K.; Caserio, M. C. Acyl Transfer Reactions in the Gas Phase. Ion-Molecule Chemistry of Vinyl Acetate. J. Am. Chem. Soc. 1982, 104, 4624–4629. DOI: 10.1021/ja00381a020.
- Olofson, R. A.; Cuomo, J. A Regiospecific and Stereospecific Route to Enol Carbonates and Carbamates: Closer Look at a “Naked Anion”. Tetrahedron Lett. 1980, 21, 819–822. DOI: 10.1016/S0040-4039(00)71514-0.
- Piteau, M. D. A.; Malfroot, T. A. Isopropenyl Chloroformate and Isopropenyl and Vinyl Chlorothioformates; Societe Nationale des Poudres et Explosifs: France, 1978; p. 23.
- Wren, C. D. A Review of Metal Accumulation and Toxicity in Wild Mammals: I. Environ. Res. 1986, 40, 210–244. DOI: 10.1016/S0013-9351(86)80098-6.
- Nunez, I. M.; Seelye, D. E. Process for Preparing Vinyl Chloroformate; Bausch & Lomb Incorporated: New York, 2013; p. 33. Chemical Indexing Equivalent to 157:493128 (US).
- Lin, C.-C.; Anseth, K. S. PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine. Pharm. Res. 2009, 26, 631–643. DOI: 10.1007/s11095-008-9801-2.
- Mautner, A.; Steinbauer, B.; Russmüller, G.; Lieber, R.; Koch, T.; Stampfl, J.; Liska, R. Vinyl Carbonate Photopolymers with Improved Mechanical Properties for Biomedical Applications. Des. Monomers Polym. 2016, 19, 437–444. DOI: 10.1080/15685551.2016.1169378.
- Mautner, A.; Qin, X.; Kapeller, B.; Russmueller, G.; Koch, T.; Stampfl, J.; Liska, R. Efficient Curing of Vinyl Carbonates by Thiol-Ene Polymerization. Macromol. Rapid Commun. 2012, 33, 2046–2052. DOI: 10.1002/marc.201200502.
- Orman, S.; Hofstetter, C.; Aksu, A.; Reinauer, F.; Liska, R.; Baudis, S. Toughness Enhancers for Bone Scaffold Materials Based on Biocompatible Photopolymers. J. Polym. Sci. A Polym. Chem. 2019, 57, 110–119. DOI: 10.1002/pola.29273.
- Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. DOI: 10.1021/jo971176v.
- Ishihara, K.; Nakajima, N. Structural Aspects of Acylated Plant Pigments: Stabilization of Flavonoid Glucosides and Interpretation of Their Functions. J. Mol. Catal. B Enzym. 2003, 23, 411–417. DOI: 10.1016/S1381-1177(03)00106-1.