
Abstract
We report the attempted development of a photoredox-catalyzed α-C–H alkylation reaction of aliphatic amine derivatives, using N-tosylhydrazones as radical alkylating partners. The original intention was to intercept α-aminoalkyl radical intermediates with N-tosylhydrazones, followed by the expulsion of a sulfonyl radical by β-scission to generate N-H diazene species. Facile denitrogenation of these intermediates would remove all traces of the hydrazone moiety and provide a net C–H alkylation process. However, our plans were derailed by issues with the low reactivity of N-tosylhydrazones toward intermolecular capture by nucleophilic radicals, and several unexpected side reactions. Our findings, though unsuccessful, do serve to identify challenges for future researchers attempting to develop similar transformations.
Graphical Abstract

Introduction
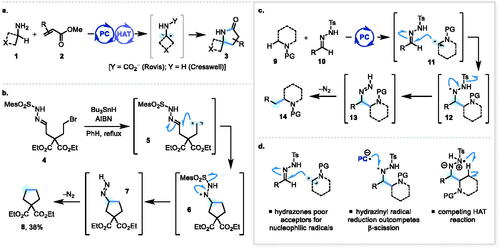
The centrality of aliphatic amines in drug discovery[Citation1] and the ever-increasing drive to expand chemical space in this area has spurred the design of new and creative approaches to these important building blocks.[Citation2] As part of this effort, chemists have made significant strides in the area of C–H functionalization of aliphatic amines, addressing C–H bonds α- and β- to nitrogen, as well as more remote C–H bonds.[Citation3] For example, we recently reported a powerful new approach for the catalytic α-C–H alkylation of primary amines 1,[Citation4] using photoredox catalysis[Citation5] in conjunction with hydrogen atom transfer (HAT) catalysis[Citation6] (e.g., 1 + 2 → 3, Scheme 1a). Given the profound effect that the introduction of simple alkyl groups—including methyl groups[Citation7]—can have upon the physicochemical properties of drugs, we were motivated to develop an α-C–H alkylation reaction for alkylamines that could introduce such motifs without the need for transition metal co-catalysis.[Citation3a,b] We were inspired by reports utilizing N-tosylhydrazones as alkylating agents for either alkyllithium reagents[Citation8] or alkyl radicals,[Citation9] in each case relying upon denitrogenation of transient diazenes[Citation10] to enable a “traceless” C–C bond-forming process (e.g., 4 → 8 from reference[Citation9c], Scheme 1b). Our intended reaction design was to harness established photoredox protocols for the generation of α-amino radicals 11[Citation11] from N-protected aliphatic amines 9 and intercept these radicals with N-tosylhydrazones 10. A subsequent β-scission[Citation12] event from hydrazinyl radical 12 would expel a sulfonyl radical (to oxidatively turnover the photoredox catalyst) and furnish N–H diazenes 13. Subsequent denitrogenative decomposition of these species would give the desired α-alkylated amines 14 (Scheme 1c). However, a number of reactivity problems and unexpected side reactions conspired to derail our ambitions, including poor radicophilicity of N-tosylhydrazones toward intermolecular capture by highly nucleophilic α-amino radicals, competitive SET reduction of the putative hydrazinyl radical intermediate 12, and a suspected intramolecular HAT process possible with cyclic amine substrates (Scheme 1d).
Scheme 1. (a) Prior art: Catalytic γ-lactam synthesis from unprotected 1° alkylamines; (b) Inspiration: traceless alkyl-alkyl cross-coupling using N-tosylhydrazones; (c) Original aim of this work: metal-free α-C–H alkylation of amines; (d) Problems and side reactions.
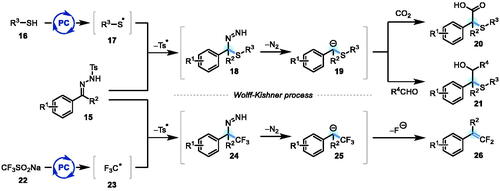
As the studies described herein were nearing completion, König et al. reported the successful execution of a very similar concept to our intended “traceless” alkylation, albeit with electrophilic thiyl (RS•) or trifluoromethyl (F3C•) radicals in lieu of nucleophilic α-amino radicals (Scheme 2).[Citation9a] In this instance, the N-tosylhydrazones are displaying nucleophilic reactivity[Citation10] at carbon, in contrast to the electrophilic behavior[Citation8,Citation9] we have envisioned. The successful β-scission step to expel a sulfonyl radical, leading to diazenes 18 and 24 as key intermediates, is also noteworthy, and contrasts with our own findings (vide infra).
Scheme 2. König’s merger of photoredox catalysis with Wolff-Kishner-type reactivity of N-tosylhydrazones.
Results and discussion
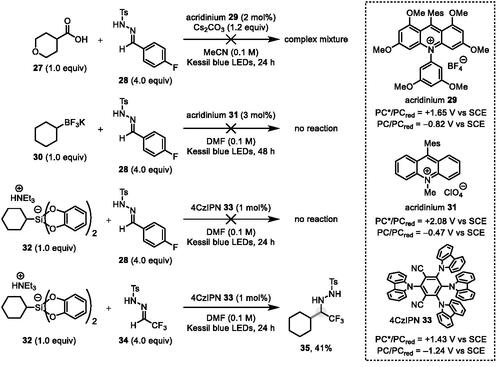
Our investigations commenced to add photogenerated simple alkyl radicals to N-tosylhydrazone 28 (derived from 4-fluorobenzaldehyde), using the para-fluoro group as a spectroscopic handle for 19F NMR (Scheme 3). Initial attempts with acridinium photocatalysts employing carboxylic acids (e.g., 27) or trifluoroborates (i.e., 30) were unsuccessful, with only a complex mixture generated in the former case and no reaction in the latter. Control experiments verified that hydrazone 28 is sensitive to extensive decomposition on exposure to Cs2CO3, even in the absence of light, likely via generation of the corresponding diazo species. We next tested the easily oxidized bis(catecholato)silicate 32 (Ep/2red = +0.69 V vs. SCE in DMF)[Citation13] as an alkyl radical precursor, and 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) 33 [E½ (PC*/PC•–) = +1.43 V vs. SCE in MeCN][Citation14] as the photoredox catalyst. Again, with N-tosylhydrazone 28, no reactivity was observed, but with the much more activated hydrazone 34 (derived from trifluoroacetaldehyde), a successful reaction ensued to give α-CF3 hydrazide 35 in 41% isolated yield (albeit alongside several other, unidentified minor products). This result suggests that simple aromatic N-tosylhydrazones like 28 are not sufficiently electrophilic to efficiently capture nucleophilic radicals,[Citation9a] but that strong electron-withdrawing groups on the hydrazone carbon (as in 34) can remedy this situation. Unfortunately, this result also indicated that our originally conceived β-scission[Citation12] to expel a sulfonyl radical and generate a transient N-H diazene is not operative in this case and that the hydrazinyl radical generated after hydrazone addition is rapidly quenched via SET reduction by the reduced photocatalyst [E½ (PC/PC–•) = −1.24 V vs. SCE in MeCN]. We also attempted to use an α-CF3 hydrazone substituted with a 4-(trifluoromethyl)phenylsulfonyl group instead of a Ts group, but the photoredox reaction with 32 was significantly messier and lower-yielding than that with hydrazone 34, and the product could not be isolated by chromatography without extensive decomposition.
Scheme 3. Attempts at photocatalytic alkyl radical addition to N-tosylhydrazones.
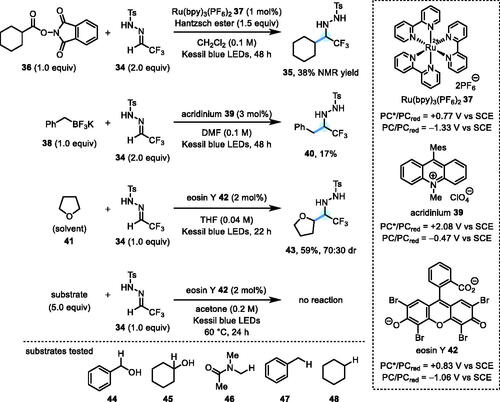
We next investigated some other alkyl radical precursors[Citation15] with α-CF3 hydrazone 34 as the acceptor, in the hope that we might identify an instance in which the β-scission step might be competitive with reduction of the N-centred radical (Scheme 4). With redox-active ester 36 as the radical precursor, a set of reaction conditions previously reported for photocatalytic conjugate additions[Citation15a] gave α-CF3 hydrazide 35 in 38% NMR yield, but no sign of the desired alkyl-alkyl cross-coupling product (via a β-scission and denitrogenation pathway analogous to that in Scheme 1c). The outcome was similar for benzylic trifluoroborate 38, from which the α-CF3 hydrazide 40 was isolated in a 17% yield. In both cases, the crude product mixtures were complex and difficult to deconvolute via 19F NMR, with several unidentified, minor fluorinated species also evident. We also attempted the reaction of tetrahydrofuran 41 (as the solvent and radical precursor) with N-tosylhydrazone 34, using eosin Y 42 as a photo-HAT catalyst.[Citation15b] In this case also, an α-CF3 hydrazide 43 was isolated in 59% yield (70:30 dr), but again with no sign of products resulting from a putative N-H diazene. Unfortunately, this process did not prove to be general with other substrates (44–48), all of which gave no reaction, despite being used in a 5-fold excess, relative to 34.[Citation15b]
Scheme 4. Further studies using other alkyl radical precursors.
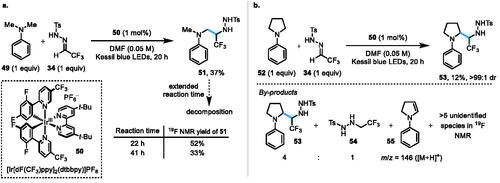
Given that there is precedent for the controlled decomposition of N-tosyl hydrazides by a Wolff-Kishner-type pathway,[Citation9a,b] we reasoned that our original goal of a “traceless” α-C–H alkylation process for aliphatic amines may still be within reach, albeit as a two-step process (i.e., via an N-tosyl hydrazide intermediate). Thus, we next investigated the reactivity of N-tosylhydrazone 34 with N,N-dimethylaniline 49—a known precursor to α-amino radicals via SET oxidation-deprotonation.[Citation11] Using an iridium photocatalyst (50) previously employed with N-Ph amines,[Citation16] the coupling of N,N-dimethylaniline 49 (E1/2 = +0.74 V vs. SCE)[Citation17] with N-tosylhydrazone 34 generated hydrazide 51 in 37% isolated yield (Scheme 5a). Minor unidentified by-products were observed by 19F NMR, alongside remaining hydrazone starting material 34. However, allowing the reaction to proceed until all of the starting hydrazone 34 had been consumed (41 h) led to a decrease in product 51 yield, suggesting that 51 may be decomposing under the conditions. Moreover, on repetition of this reaction, the yield of 51 and the extent of decomposition varied considerably. Attempts to stem the decomposition by varying both the photocatalyst and the reaction solvent (i.e., MeOH, MeCN, CH2Cl2) were fruitless. Moreover, employing a large excess of hydrazone 34 (8.0 equiv.) conferred no benefit, providing an NMR yield of 30% of 51 after 20 h. Concurrently, the reaction of N-Ph pyrrolidine 52 (E1/2 = +0.70 V vs. SCE)[Citation16] with N-tosylhydrazone 34 was also studied, and a hydrazide product 53 was isolated in 12% yield and >99:1 dr (though it is possible that a minor diastereomer was removed during chromatographic purification) (Scheme 5b). In this case, a new hydrazide species 54 (a reduction product of 34) was detected, and mass spectroscopic evidence consistent with enamine 55 was also found (i.e., m/z peaks of 146 for [M + H]+ and 289 for [2M + H]+).[Citation18]
Scheme 5. (a) Photoredox coupling of N,N-dimethylaniline 49 with N-Ts hydrazone 34. (b) Photoredox coupling of N-Ph pyrrolidine 52 with N-Ts hydrazone 34.
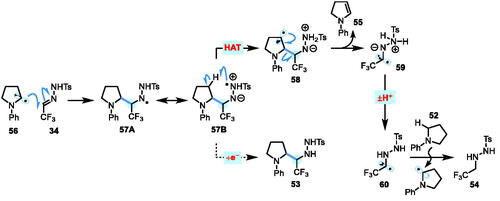
Given that none of the reduced hydrazone by-product 54 was detected in the reaction of N,N-dimethylaniline 49, this suggests that cyclic amine substrate 52 is prone to a side-reaction to afford 54 that is not possible with compound 49. On the basis that N-Ph enamine 55 was also detected by MS analysis from 52 (see Scheme 5b), a speculative mechanistic rationalization to account for these findings is presented in Scheme 6. Assuming that the reaction proceeds via α-amino radical attack onto the C=N bond of the CF3 hydrazone 34, the resultant hydrazinyl radical 57 may be able to engage in a 1,5-HAT with a C–H bond on the ring of the cyclic amine. β-Scission of the alkyl radical would then generate enamine by-product 55, alongside the α-CF3 radical species 59. Proton transfer (to give 60) could potentially be followed by an intermolecular HAT from the amine substrate, delivering the reduced hydrazone species 54. Concomitant generation of an α-amino radical from 52 could then feasibly lead to a chain process.
Scheme 6. Proposed mechanistic rationale for the generation of reduced hydrazone by-product 54 when using cyclic amine 52.
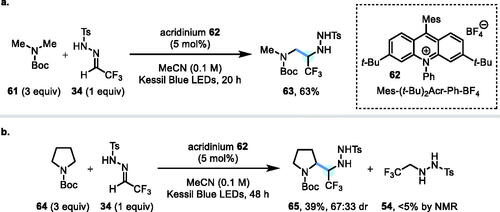
In parallel to investigations with N-Ph amines as radical precursors, N-Boc amines were also tested as substrates for a photoredox-catalyzed α-C–H functionalization with α-CF3 hydrazone 34. Given that carbamates possess much higher oxidation potentials than N-Ph amines (i.e., E1/2 = +1.96 V vs. SCE for N-Boc piperidine),[Citation19] we opted to use the strongly oxidizing acridinium photocatalyst 62 [E½ (PC*/PC•–) = +2.15 V vs. SCE in MeCN] recently developed by Nicewicz and coworkers.[Citation19] Thus, N-Boc dimethylamine 61 in MeCN was reacted with α-CF3 hydrazone 34 in the presence of acridinium catalyst 62 (5 mol%) with irradiation by Kessil blue LEDs for 20 h (Scheme 7a). Under these conditions, the α-CF3 hydrazide 63 was isolated in a 63% yield. Unfortunately, attempts to reduce the stoichiometry of N-Boc dimethylamine 61 to 1.0 equiv. led to extensive by-product formation and a complex mixture. A similar reaction using N-Boc pyrrolidine 64 gave the corresponding α-CF3 hydrazide 65 in only 39% isolated yield (67:33 dr). The reduced hydrazone by-product 54 was only observed at a trace level (<5% by NMR), suggesting that the side reaction operative for N-Ph pyrrolidine 52 does not make a significant contribution in this case (Scheme 7b).
Scheme 7. (a) Photoredox coupling of N-Boc dimethylamine 61 with N-Ts hydrazone 34. (b) Photoredox coupling of N-Boc pyrrolidine 64 with N-Ts hydrazone 34.
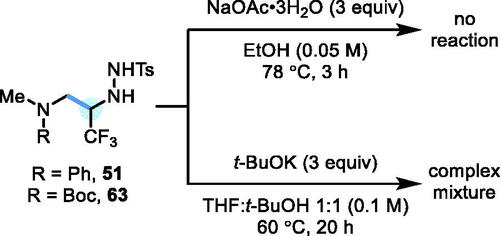
At this stage, we attempted a base-induced denitrogenative decomposition of N-Ts hydrazide products 51 and 63, to gauge the possibility of developing a one-pot protocol for “traceless” α-C–H alkylation (or, more specifically, trifluoroethylation if using 34). Using literature conditions for N-sulfonyl hydrazide decomposition with NaOAc (via a radical pathway from an intermediate N-H diazene),[Citation9b,Citation20] no reaction was observed. The complete lack of reactivity is surprising, and we postulate that the strongly inductively-withdrawing CF3 group may render the transient N-centered anion too thermodynamically stable to eliminate the β-sulfinyl group. Further attempts to induce denitrogenative decomposition of 51 and 63 using the stronger base t-BuOK[Citation21] were also unsuccessful, with both compounds suffering extensive degradation to afford complex mixtures of unidentifiable products (Scheme 8). A β-elimination of fluoride ion from a putative α-CF3 carbanion in the latter case may be to blame.[Citation22]
Scheme 8. Attempted denitrogenative decomposition of N-Ts hydrazides 51 and 63.
Conclusions
The reactivity of N-tosylhydrazones as acceptors for nucleophilic radicals generated by photoredox catalysis has been briefly explored, as part of studies directed toward a “traceless,” metal-free α-C–H alkylation of aliphatic amine derivatives. The introduction of an electron-withdrawing trifluoromethyl group on the hydrazone carbon proved necessary to switch on reactivity for intermolecular capture with nucleophilic radicals, presumably by increasing the electrophilicity of what is otherwise a relatively nucleophilic site. The intended β-scission step from the hydrazinyl radical intermediates—to expel a sulfonyl radical and generate N-H diazenes—was outcompeted by SET reduction by the reduced form of the photocatalyst, leading to N-Ts hydrazide products. A side reaction involving a suspected 1,5-HAT process and subsequent fragmentation appears to operate with N-Ph pyrrolidine as the substrate, eroding the yield of N-Ts hydrazide product. Attempted denitrogenative decomposition of N-Ts hydrazide products was not fruitful and gave either no reaction or complex mixtures of unidentified species. Although unsuccessful, these studies do serve to identify challenges for future researchers attempting to develop similar radical-based transformations with N-tosylhydrazones.
Experimental
Synthesis of N-(1-cyclohexyl-2,2,2-trifluoroethyl)-4-methylbenzenesulfonylhydrazide (35)
An oven-dried, 20-mL scintillation vial equipped with a stirrer bar was charged with silicate 32 (96 mg, 0.22 mmol, 1.0 equiv.), hydrazone 34 (238 mg, 0.89 mmol, 4.0 equiv.) and 4CzIPN 33 (1.8 mg, 2.3 µmol, 1 mol%). The vial was sealed with an upturned septa and evacuated and backfilled with nitrogen three times. DMF (anhydrous, 2.5 mL) was added and the mixture was sparged with nitrogen for 15 min. The resultant solution was stirred under irradiation (Kessil H150-Blue LED lamp) for 18 h. The reaction mixture was diluted with EtOAc (15 mL) and washed with H2O (5 × 15 mL). The organic layer was dried with a phase separating cartridge and then concentrated in vacuo. Purification via preparative HPLC (Waters CSH C18 OBD column, 5 µ silica, 30 mm diameter, 100 mm length) using decreasingly polar mixtures of H2O [containing 1% by volume of NH4OH (28–30% in H2O)] and MeCN as eluents gave 35 as a white, crystalline solid (32 mg, 41%). mp 161 °C. 1H NMR (500 MHz, chloroform-d) 7.71–7.91 (m, 2H, ArH), 7.34 (d, J = 7.9 Hz, 2H, ArH), 6.10 (s, 1H, NH), 3.71 (d, J = 8.3 Hz, 1H, NH), 2.94–3.11 (m, 1H, C(1′′)H), 2.45 (s, 3H, C(9)H3), 1.57–1.78 (m, 5H, Cy), 1.49 (s, 1H, Cy), 1.04–1.25 (m, 4H, Cy), 0.83–1.01 (m, 1H, Cy). 13C NMR (126 MHz, DMSO-d6) δ 143.7 (Ar), 137.0 (Ar), 130.2 (Ar), 128.7 (Ar), 127.4 (q, J = 287 Hz, C(2′′)), 67.2 (q, J = 24.4 Hz, C(1′′), 37.3 (C(1)), 29.9 (Cy), 29.2 (Cy), 26.7 (Cy), 26.7 (Cy), 26.6 (Cy), 21.9 (C(9)). 19F NMR (376 MHz, chloroform-d) δ −73.3 (d, J = 8.0 Hz). IR (neat, cm−1) 3297 (w), 3249 (w), 2923 (w), 2853 (w), 1601 (w), 1509 (w), 1497 (w), 1455 (w), 1354 (w), 1320 (w), 1258 (w), 1228 (w), 1189 (w), 1156 (m), 1145 (m), 1086 (w), 971 (w), 842 (w), 817 (w), 801 (w), 748 (w), 707 (w), 689 (w), 666 (w). HRMS (ESI+) calculated for C15H21F3N2O2S: [M + H]+ 351.1354; found: [M + H]+ 351.1363.
Supplemental Material
Download PDF (2 MB)Acknowledgments
The authors gratefully acknowledge the technical staff within Chemistry at the University of Bath for technical support and assistance in this work, including the Material and Chemical Characterization Facility (MC2) (https://doi.org/10.15125/mx6j-3r54).
Additional information
Funding
References
- Blakemore, D. C.; Castro, L.; Churcher, I.; Rees, D. C.; Thomas, A. W.; Wilson, D. M.; Wood, A. Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem. 2018, 10, 383–394. DOI: 10.1038/s41557-018-0021-z.
- Nugent, T. C. Chiral Amine Synthesis: Methods, Developments and Applications; Wiley-VCH: Weinheim, 2010.
- Selected reviews: (a) Trowbridge, A.; Walton, S. M.; Gaunt, M. J. New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev. 2020, 120, 2613–2692. DOI: 10.1021/acs.chemrev.9b00462. (b) Gonnard, L.; Guérinot, A.; Cossy, J. Transition Metal Catalyzed α-Alkylation of Amines by Csp3–H Bond Activation. Tetrahedron 2019, 75, 145–163. DOI: 10.1016/j.tet.2018.11.034. (c) Mitchell, E. A.; Peschiulli, A.; Lefevre, N.; Meerpoel, L.; Maes, B. U. W. Direct α-Functionalization of Saturated Cyclic Amines. Chemistry 2012, 18, 10092–10142. DOI: 10.1002/chem.201201539. (d) Shi, L.; Xia, W. Photoredox Functionalization of C–H Bonds Adjacent to a Nitrogen Atom. Chem. Soc. Rev. 2012, 41, 7687–7697. DOI: 10.1039/c2cs35203f. (e) Campos, K. R. Direct sp3 C–H Bond Activation Adjacent to Nitrogen in Heterocycles. Chem. Soc. Rev. 2007, 36, 1069–1084. DOI: 10.1039/B607547A.
- (a) Grayson, J. D.; Cresswell, A. J. γ-Amino Phosphonates via the Photocatalytic α-C–H Alkylation of Primary Amines. Tetrahedron 2021, 81, 131896. DOI: 10.1016/j.tet.2020.131896. (b) Ryder, A. S. H.; Cunningham, W. B.; Ballantyne, G.; Mules, T.; Kinsella, A. G.; Turner‐Dore, J.; Alder, C. M.; Edwards, L. J.; McKay, B. S. J.; Grayson, M. N.; Cresswell, A. J. Photocatalytic α-Tertiary Amine Synthesis via C − H Alkylation of Unmasked Primary Amines. Angew. Chem. Int. Ed. Engl. 2020, 59, 14986–14991. DOI: 10.1002/anie.202005294.
- Reviews of visible-light photoredox catalysis: (a) Zilate, B.; Fischer, C.; Sparr, C. Design and Application of Aminoacridinium Organophotoredox Catalysts. Chem. Commun. 2020, 56, 1767–1775. DOI: 10.1039/C9CC08524F. (b) McAtee, R. C.; McClain, E. J.; Stephenson, C. R. J. Illuminating Photoredox Catalysis. Trends Chem. 2019, 1, 111–125. DOI: 10.1016/j.trechm.2019.01.008. (c) Shang, T.-Y.; Lu, L.-H.; Cao, Z.; Liu, Y.; He, W.-M.; Yu, B. Recent Advances of 1,2,3,5-Tetrakis(Carbazol-9-yl)-4,6-Dicyanobenzene (4CzIPN) in Photocatalytic Transformation. Chem. Commun. 2019, 55, 5408–5419. DOI: 10.1039/C9CC01047E. (d) Stephenson, C.R.J.; Yoon, T.; MacMillan, D.W.C., Eds. Visible Light Photocatalysis in Organic Chemistry; Wiley-VCH, Berlin, 2018. (e) Marzo, L.; Pagire, S. K.; Reiser, O.; König, B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. Engl. 2018, 57, 10034–10041. DOI: 10.1002/anie.201709766. (f) Srivastava, V.; Singh, P. Eosin Y Catalysed Photoredox Synthesis: A Review. RSC Adv. 2017, 7, 31377–31392. DOI: 10.1039/C7RA05444K. (g) Romero, N. A.; Nicewicz, D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. DOI: 10.1021/acs.chemrev.6b00057. (h) Ravelli, D.; Protti, S.; Fagnoni, M. Carbon-Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. DOI: 10.1021/acs.chemrev.5b00662. (i) Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. DOI: 10.1021/acs.joc.6b01449. (j) Schultz, D. M.; Yoon, T. P. Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343, 1239176. DOI: 10.1126/science.1239176. (k) Prier, C. K.; Rankic, D. A.; Macmillan, D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. DOI: 10.1021/cr300503r.
- Selected reviews on HAT catalysis, see: (a) Chu, J. C. K.; Rovis, T. Complementary Strategies for Directed C(sp3)–H Functionalization: A Comparison of Transition‐Metal‐Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed. 2018, 57, 62–101. DOI: 10.1002/anie.201703743. (b) Capaldo, L.; Ravelli, D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071. DOI: 10.1002/ejoc.201601485.
- (a) Aynetdinova, D.; Callens, M. C.; Hicks, H. B.; Poh, C. Y. X.; Shennan, B. D. A.; Boyd, A. M.; Lim, Z. H.; Leitch, J. A.; Dixon, D. J. Installing the “Magic Methyl” – C–H Methylation in Synthesis. Chem. Soc. Rev. 2021, 50, 5517–5563. DOI: 10.1039/D0CS00973C. (b) Schonherr, H.; Cernak, T. Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem. Int. Ed. 2013, 52, 12256–12267. DOI: 10.1002/anie.201303207.
- For reactions of N-silylated/metalated N-Ts hydrazones with organometallic nucleophiles, followed by denitrogenative decomposition see: (a) Myers, A. G.; Movassaghi, M. Highly Efficient Methodology for the Reductive Coupling of Aldehyde Tosylhydrazones with Alkyllithium Reagents. J. Am. Chem. Soc. 1998, 120, 8891–8892. DOI: 10.1021/ja981918h. (b) Myers, A. G.; Kukkola, P. J. Stereoselective Synthesis of Olefins from Silylated Sulfonylhydrazones.J. Am. Chem. Soc. 1990, 112, 8208–8210. DOI: 10.1021/ja00178a078. (c) Vedejs, E.; Dolphin, J.; Stolle, W. A New Olefin Synthesis: Condensation of Aldehyde Tosylhydrazones with Stabilized Carbanions. J. Am. Chem. Soc. 1979, 101, 249–251. DOI: 10.1021/ja00495a057. (d) Vedejs, E.; Stolle, W. T. Reductive Alkylation of Aldehyde Tosylhydrazones with Organolithium Reagents. Tetrahedron Lett. 1977, 18, 135–138. DOI: 10.1016/S0040-4039(01)92569-9.
- (a) Wang, S.; Cheng, B.-Y.; Sršen, M.; König, B. Umpolung Difunctionalization of Carbonyls via Visible-Light Photoredox Catalytic Radical-Carbanion Relay. J. Am. Chem. Soc. 2020, 142, 7524–7531. DOI: 10.1021/jacs.0c00629. (b) Dao, H. T.; Li, C.; Michaudel, Q.; Maxwell, B. D.; Baran, P. S. Hydromethylation of Unactivated Olefins. J. Am. Chem. Soc. 2015, 137, 8046–8049. DOI: 10.1021/jacs.5b05144. (c) Campbell, N. E.; Sammis, G. M. Single-Electron/Pericyclic Cascade for the Synthesis of Dienes. Angew. Chem. Int. Ed. Engl. 2014, 53, 6228–6231. DOI: 10.1002/anie.201403234. (d) Kim, S.; Cho, J. R. Radical Cyclization of Mesitylsulfonylhydrazones. Synlett 1992, 1992, 629–630. DOI: 10.1055/s-1992-21435.
- For umpolung reactions of N-deprotonated N-Ts hydrazones with electrophiles, followed by denitrogenative decomposition see: (a) Reyes, J. R.; Rawal, V. H. Reductive Chlorination and Bromination of Ketones via Trityl Hydrazones. Angew. Chem. 2016, 128, 3129–3132. DOI: 10.1002/ange.201510909. (b) Baldwin, J. E.; Adlington, R. M.; Bottaro, J. C.; Kolhe, J. N.; Newington, I. M.; Perry, M. W. D. Azo Anions in Synthesis: Use of Trityl- and Diphenyl-4-Pyridylmethylhydrazones for Reductive C–C Bond Formation. Tetrahedron 1986, 42, 4235–4246. DOI: 10.1016/S0040-4020(01)87648-1. (c) Baldwin, J. E.; Bottaro, J. C.; Kolhe, J. N.; Adlington, R. M. Azo Anions in Synthesis. Use of Trityl- and Diphenyl-4-Pyridylmethyl-Hydrazones for Reductive C–C Bond Formation from Aldehydes and Ketones. J. Chem. Soc. Chem. Commun. 1984, 22–23. DOI: 10.1039/C39840000022.
- For reviews, see: (a) Trowbridge, A.; Walton, S. M.; Gaunt, M. J. New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev. 2020, 120, 2613–2692. DOI: 10.1021/acs.chemrev.9b00462. (b) Leitch, J. A.; Rossolini, T.; Rogova, T.; Maitland, J. A. P.; Dixon, D. J. α-Amino Radicals via Photocatalytic Single-Electron Reduction of Imine Derivatives. ACS Catal. 2020, 10, 2009–2025. DOI: 10.1021/acscatal.9b05011. (c) Zou, Y.-Q.; Xiao, W.-J. In Visible Light Photocatalysis in Organic Chemistry; Stephenson, C. R. J., Yoon, T., MacMillan, D. W. C., Eds.; Wiley-VCH, Berlin, 2018; Chapter 4, pp 93–128. (d) Nakajima, K.; Miyake, Y.; Nishibayashi, Y. Synthetic Utilization of α-Aminoalkyl Radicals and Related Species in Visible Light Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1946–1956. DOI: 10.1021/acs.accounts.6b00251.
- Zhang, H.; Hay, E. B.; Geib, S. J.; Curran, D. P. Fates of Imine Intermediates in Radical Cyclizations of N-Sulfonylindoles and Ene-Sulfonamides. Beilstein J. Org. Chem. 2015, 11, 1649–1655. DOI: 10.3762/bjoc.11.181.
- Corcé, V.; Chamoreau, L. M.; Derat, E.; Goddard, J. P.; Ollivier, C.; Fensterbank, L. Silicates as Latent Alkyl Radical Precursors: Visible-Light Photocatalytic Oxidation of Hypervalent Bis-Catecholato Silicon Compounds. Angew. Chem. Int. Ed. Engl. 2015, 54, 11414–11418. DOI: 10.1002/anie.201504963.
- Luo, J.; Zhang, J. Donor–Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)–C(sp2) Cross Coupling. ACS Catal. 2016, 6, 873–877. DOI: 10.1021/acscatal.5b02204.
- (a) Pratsch, G.; Lackner, G. L.; Overman, L. E. Constructing Quaternary Carbons from N-(Acyloxy)Phthalimide Precursors of Tertiary Radicals Using Visible-Light Photocatalysis. J. Org. Chem. 2015, 80, 6025–6036. DOI: 10.1021/acs.joc.5b00795. (b) Fan, X.-Z.; Rong, J.-W.; Wu, H.-L.; Zhou, Q.; Deng, H.-P. D.; Tan, J.; Xue, C.-W.; Wu, L.-Z.; Tao, H.-R.; Wu, J. Eosin Y as a Direct Hydrogen-Atom Transfer Photocatalyst for the Functionalization of C–H Bonds. Angew. Chem. Int. Ed. Engl. 2018, 57, 8514–8518. DOI: 10.1002/anie.201803220.
- Ahneman, D. T.; Doyle, A. G. C–H Functionalization of Amines with Aryl Halides by Nickel-Photoredox Catalysis. Chem. Sci. 2016, 7, 7002–7006. DOI: 10.1039/C6SC02815B.
- Das, P.; Begam, H. M.; Bhunia, S.; Jana, R. Photoredox-Catalyzed Tandem Demethylation of N,N-Dimethyl Anilines Followed by Amidation with α-Keto or Alkynyl Carboxylic Acids. Adv. Synth. Catal. 2019, 361, 4048–4054. DOI: 10.1002/adsc.201900525.
- Analysis of the 1H NMR spectrum was not fruitful, given the complexity of the mixture and signal overlap.
- McManus, J. B.; Onuska, N. P. R.; Nicewicz, D. A. Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 9056–9060. DOI: 10.1021/jacs.8b04890.
- Zhang, J.; Eisink, N. N. H. M.; Witte, M. D.; Minnaard, A. J. Regioselective Manipulation of GlcNAc Provides Allosamine, Lividosamine, and Related Compounds. J. Org. Chem. 2019, 84, 516–525. DOI: 10.1021/acs.joc.8b01949.
- Kedrowski, S. M. A.; Dougherty, D. A. Room-Temperature Alternative to the Arbuzov Reaction: The Reductive Deoxygenation of Acyl Phosphonates. Org. Lett. 2010, 12, 3990–3993. DOI: 10.1021/ol1015493.
- Ramanathan, S.; Lemal, D. M. Synthesis, Mechanism of Formation, and Dynamics of a Highly Fluorinated Methylenecyclobutene. J. Org. Chem. 2007, 72, 1570–1574. DOI: 10.1021/jo061946f.