
ABSTRACT
Aims
To investigate the pathogenesis of a disease in takahē (Porphyrio hochstetteri) with intracytoplasmic inclusion bodies in lower motor neurons.
Methods
Four birds aged between 5 and 12 years, from three different wildlife sanctuaries in New Zealand were examined. Of these, only one had signs of spinal dysfunction in the form of paresis. Stained paraffin sections of tissues were examined by light microscopy and immunostained sections of the ventral horn of the spinal cord by confocal microscopy. Epoxy resin sections of the spinal cord from the bird with spinal dysfunction were examined by electron microscopy.
Results
Two types of inclusion bodies were noted, but only in motor neurons of the ventral spinal cord and brain stem. These were large globoid eosinophilic bodies up to 5 µm in diameter, and yellow/brown granular inclusions mostly at the pole of the cell. The globoid bodies stained with Luxol fast blue but not with periodic acid Schiff (PAS), or Sudan black. The granular inclusions stained with Luxol fast blue, PAS and Sudan black. Both bodies were slightly autofluorescent. On electron microscopy the globoid bodies had an even electron-dense texture and were bound by a membrane. Beneath the membrane were large numbers of small intraluminal vesicles. The smaller granular bodies were more heterogeneous, irregularly rounded and membrane-bound accumulations of granular electron-dense material, often with electron-lucent vacuoles. Others were more vesicular but contained varying amounts of electron-dense material. The large globoid bodies did not immunostain for lysosomal markers lysosomal associated protein 1 (LAMP1) or cathepsin D, so were not lysosomal. The small granular bodies stained for cathepsin D by a chromogenic method.
A kindred matrix analysis showed two cases to be as closely related as first cousins, and another case was almost as closely related to one of them, but the fourth bird was unrelated to any other.
Conclusions
It was concluded that this was an endoplasmic reticulum storage disease due to a specific protein misfolding within endoplasmic reticulum. It was rationalised that the two types of inclusions reflected the same aetiology, but that misfolded protein in the smaller granular bodies had entered the lysosomal system via endoplasmic reticulum autophagy. Although the cause was unclear, it most likely had a genetic aetiology or predisposition and, as such, has clinical relevance.
Introduction
This report describes a novel disease in four adult takahē (Porphyrio hochstetteri) characterised by cytoplasmic inclusions in motor neurons of the brain stem and spinal cord. These cases were not recorded in a previous retrospective study of post-mortem findings in takahē (McClelland et al. Citation2011).
This species was once thought to be extinct until a small, isolated population was discovered in 1948 in remote alpine grasslands of the South Island of New Zealand. They are the largest living member species of the rail family and measure up to 60 cm in length and 3.8 kg in weight. Over many years, a captive breeding programme has resulted in a current population of approximately 440 birds, evenly split between monitored sanctuaries and their original wild habitat. A controlled population of 220 banded pedigree birds is maintained in 17 sanctuaries located in diverse environments ranging from coastal, island, mountain forest, inland grassland to South Island tussock. Individuals are regularly exchanged between sanctuaries to help maintain genetic diversity.
Intraneuronal inclusions are a hallmark of disease and in some cases of ageing. They may be associated with viral infections, lysosomal storage diseases (LSD), some toxicoses and proteinopathies associated with protein conformational changes. Of the approximately 70 LSD identified in humans (Mehta and Winchester Citation2012; Platt et al. Citation2018), many are also found in domestic animals (Jolly and Walkley Citation1997; Hopwood et al. Citation2004; Alroy and Lyons Citation2014) and in avian species (Jolly et al. Citation2021).
Proteinopathies are characterised by accumulation of a specific wild-type, or a mutant protein, with altered conformation that facilitates aggregation into amorphous or fibrillar formations. Among the many such diseases, neurodegenerative forms associated with ageing are common, but they may also have disease-related gene mutations, or be affected by cellular stressors. In the central nervous system, the type and distribution of inclusion, and age of the individual may be informative as to diagnosis of a particular disease group; for example, the microtubule associated protein (Tau) in neurofibrillary tangles and amyloid-β in Alzheimer’s disease (Busche and Hyman Citation2020), synuclein in Lewy bodies of Parkinson’s disease (Stefanis Citation2012) and TAR DNA-binding protein 43 (TDP-43) in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (Hardiman et al. Citation2017). Neurons are vulnerable to such aggregations as, being post-mitotic cells, the misfolded proteins are not diluted by cell division. Moreover, once formed, the misfolded proteins may undergo self-propagation and even spread to other cells in a prion-like manner, but unlike prions, they are not transmissible between individuals (Walker and LeVine Citation2000a; Marsh Citation2019, Padilla-Godínez et al. Citation2021).
Peptides, during synthesis in rough endoplasmic reticulum (RER), undergo complex folding and maturation which is mediated by numerous chaperones, enzymes, and their own intrinsic amino acid structure. They are then sorted in smooth endoplasmic reticulum (SER) and directed according to their functional destiny, whether it be secretory, or within the cell. Misfolding is common and normally dealt with by a series of steps in SER, with segregation into the cytosol and digestion via proteasomes or lysosomes. These processes are known as “endoplasmic reticulum (ER) associated degradation.” Ubiquitination is a primary step in initiating degradation of misfolded protein by the proteasome apparatus. Other misfolded proteins or small aggregates, including those associated with membranes, are dealt with through endoplasmic reticulum-associated autophagy (ER-phagy) which is an autophagy/lysosome-mediated selective degradation of ER domains or fragments (Tepedelen and Kirmizibayrak Citation2019; Johnston and Samant Citation2021; Li and Sun Citation2021). When these mechanisms fail or are over-loaded, misfolded proteins may aggregate leading to loss of function or cell toxicity. The most common, but not the only cause of disease resulting from protein misfolding, is a genetic mutation of the protein in question. There are many such diseases (Needham et al. Citation2019), but relatively few have been described with massive accumulation of aggregated protein in distended SER or RER to give a morphological meaning to the term “endoplasmic reticulum storage disease.” Aggregation of protein may also occur in RER when produced in excess of a cell’s ability to secrete it; for example, immunoglobulin in Russell bodies of plasma cells (Gray and Doniach Citation1970; Mossuto et al. Citation2015).
The aim of this study was to identify the nature and pathogenesis of intraneuronal inclusions that characterised a novel disease in takahē.
Materials and methods
Birds
The four takahē involved were from a population of sick or dead birds routinely submitted by the Department of Conservation, or others, to Wildbase (Massey University, Palmerston North, NZ) for post-mortem examination. A 5-year-old male takahē (case 1), a resident of Tāwharanui Regional Park (a coastal area near Auckland), was observed showing neurological signs of paresis in February 2019. On clinical examination at the New Zealand Centre for Conservation Medicine (Auckland Zoo, Auckland, NZ) the bird was emaciated, and showed hyperextension of both legs with little ability to move them. There was no proprioception and a reduced pain response. No skeletal abnormalities were observed in radiographs. Blood chemistry showed mild increases (based on previous records of tahakē held on the Zoo’s database) in serum creatine kinase and aspartate aminotransferase activities, indicative of muscle and liver degeneration respectively. Serum lead and zinc concentrations were within normal limits (reference ranges for pet birds from Gribbles Veterinary, Auckland, NZ). The bird was treated symptomatically using fluid therapy, tube-feeding and antibiotics (amoxicillin/clavulanate). After 4 weeks’ hospitalisation it showed little improvement and was euthanised on welfare grounds. Formalised tissues were submitted to Wildbase.
Two further takahē (cases 2 and 3) submitted to Wildbase from the Burwood Takahē Breeding Centre (Te Anau, NZ) had similar histological lesions to those of case 1 (see below), but without a history of neurological dysfunction. Case 2 was a 5-year-old female found dead in a breeding pen with a male in April 2019. At necropsy, the cause of death was uncertain but there was pulmonary congestion and oedema. Case 3, a 2-year-old male, was also found dead in a breeding pen with a female in May 2021 and, although he had a verminous entero-typhlitis, the cause of death was not ascertained. Case 4, a 12-year-old female, was euthanised in June 2021 due to blindness associated with a mature cataract in one eye and a severe bacterial endophthalmitis of the other. This case was from Wairakei Golf and Sanctuary (Taupo, NZ), an inland grassy sanctuary in the North Island. Cases 2–4 were submitted fresh for necropsy at Wildbase.
Light and electron microscopy
Tissues from all four birds were fixed in 10% neutral buffered formalin and embedded in paraffin wax for histopathology. Brain, spinal cord, peripheral nerve, liver, kidney and muscle sections were stained with H&E. Selected brain and spinal cord sections were also stained by periodic acid Schiff (PAS), Sudan black, Alcian blue, Luxol fast blue and Perls (Prussian blue) iron staining methods. Liver sections were also stained by Perls stain. Spinal cord from case 1 was post-fixed in 2% glutaraldehyde, 1% osmium tetroxide and embedded in epoxy resin. Semi-thin sections were stained with toluidine blue, and ultra-thin sections were stained with uranyl acetate and lead citrate for electron microscopy.
Immunocytochemistry
Paraffin sections were immuno-stained using standard immunofluorescence or chromogenic methods following heat-mediated pre-treatment in citrate buffer, pH 6. Primary antibodies to cathepsin D (1:500 dilution; Abcam, Cambridge, UK), or lysosomal protein 1 (LAMP1) (#4170, 1:200 dilution; Abcam) were applied to sections overnight and detected with either species-specific fluorophore conjugated secondary antibodies by confocal microscopy (excitation 550 nm; emission 570 nm), or species-specific biotinylated secondary antibodies, avidin–biotin-peroxidase complex and diaminobenzidine (DAB) for chromogenic detection by standard light microscopy. For each stain system, controls were run simultaneously, i.e. positive control with known antigen and negative controls omitting primary antibody or secondary antibody.
Pedigree analysis
Pedigrees of affected birds were accessed and compared directly and through constructing a kindred matrix (Ballou et al. Citation2020).
Results
Pathology
There were no significant gross lesions directly attributable to the disorder in any of the four cases other than emaciation associated with case 1. Significant histological lesions were noted only in large motor neurons of the ventral horn of the spinal cord and the brain stem in all four birds. The most obvious lesions in H&E-stained sections were large globoid, colloid-like bodies up to 5 µm in diameter in ventral horns and brain stem motor neurons. In some sections, 60–70% of neurons were affected, with several to many of these inclusions filling much of the cytoplasm ((a)). Large inclusions tended to obscure many smaller, but similar ones. These globoid bodies stained with eosin, which was more intense at their rim ((a)), faintly with Luxol fast blue ((b)), but not with the PAS ((e)), Alcian blue, or Sudan black stains. In semi-thin epoxy resin sections, there was strong affinity for toluidine blue, which stained them a dark blue. On fluorescent microscopy the globoid bodies in unstained sections were faintly auto-fluorescent (excitation 550 nm; emission 570 nm).
Figure 1. Photomicrographs of neuronal lesions from affected takahē (Porphyrio hochstetteri). (a) A motor neuron of the spinal cord ventral horn has numerous globoid eosinophilic inclusions (H&E; bar = 6 µm). (b) The neuronal globoid inclusions stain with Luxol fast blue (bar = 12 µm). (c) A motor neuron in the brain stem shows a large granular light brown pigment formation (H&E; bar = 12 µm). (d) A ventral horn neuron with granular material at the pole that stains with Luxol fast blue (bar = 10 µm). (e) A ventral horn neuron contains PAS-positive granular material; in contrast the round unstained vacuolar areas are interpreted as globoid bodies as are the empty vacuoles in the neighbouring neuron which do not stain with PAS (bar = 12 µm). (f) A dead ventral horn neuron with eosinophilic globoid bodies and their ghosts (H&E; bar = 5 µm); compare to (a) from the same section. (g) A fluoresent immunostain for lysosomal associated protein 1 (LAMP1) did not stain the large globoid bodies but stained a myriad small bodies in the cytosol indicating their lysosomal nature (bar = 3 µm). (h) The fluorescent immunostain for cathepsin D did not stain the large globoid bodies (arrow) but stained many small bodies in the cytosol (bar = 3 µm). (i) The colourimetric stain (diaminobenzidine; DAB) for LAMP1 stained many small bodies in this ventral horn neuron. Note that in the area of the arrow it stained the periphery of the bodies as expected with LAMP1 primary antibody. The large globoid inclusion did not immunostain (bar = 4 µm). (j) The colourimetric immunostain (DAB) for cathepsin D stained a large number of concentrated small lysosomal bodies at the pole of this ventral horn neuron, which are interpreted as the small granular bodies in other neurons (bar = 4 µm).
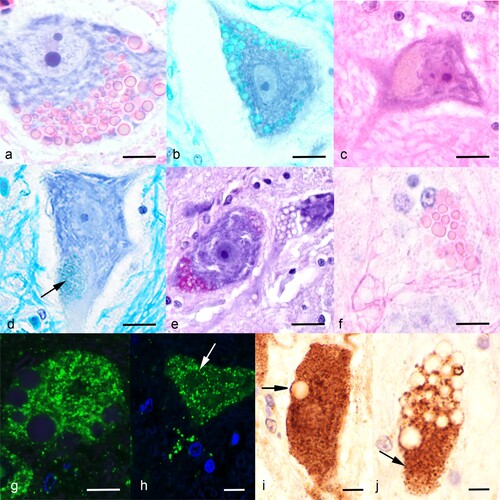
The second type of inclusion was more ubiquitous, and in H&E-stained sections appeared as indistinct light grey/brown granular material ((c)), often at the poles of the cell. It stained moderately with Luxol fast blue ((d)), was strongly PAS-positive ((e)), and stained with Sudan black. PAS-positive granules were also seen between the larger unstained globoid bodies. This granular material was also mildly auto-fluorescent.
In case 4 there was evidence of necrosis of occasional ventral horn neurons as shown by many globoid bodies and their ghosts within remnant dead neurons ((f)).
There were no storage-type lesions in liver, spleen or kidney, except for a brown pigment in the Küpffer cells of the liver, which stained with Perls (Prussian blue) iron stain. There were also large amounts of iron-staining granules in hepatocytes. The spinal cord neuronal lesions did not stain for iron.
Immunocytochemistry (case 1)
There was no immunostaining for the lysosomal markers LAMP1 and cathepsin D of the large globoid bodies or their periphery when viewed by either the fluorescent ((g,h)) or chromogenic methods ((i,j)). In contrast, there were a multitude of fluorescent small bodies in the cytosol indicative of lysosomes, but it was uncertain as to whether these were disease-related or part of the normal complement of lysosomes. Chromogenic staining by both LAMP1 and cathepsin D of concentrated lysosomes in ventral horn neurons was interpreted as disease-related ((i,j)).
Ultrastructure (case 1)
There was very severe autolysis of the cytosol, but the inclusions were robust and intact. Ultra-structurally, the large globoid bodies were membrane-bound, electron-dense spherical cytosomes ranging up to 5 µm in diameter and with an even, fine, granular texture ((a,b)). There were often many small intraluminal vesicles between the unit-membrane and the electron-dense material ((b,c)).
Figure 2. Transmission electron micrographs of the neuronal lesions in a takahē (Porphyrio hochstetteri) (Case 1) with neurological signs. (a) Low power micrograph of large dense globoid bodies of various sizes in the cytosol of a ventral horn neuron (bar = 10 µm). (b) A large electron dense, membrane-bound globoid body is contrasted with numerous small irregular granular bodies in the cytosol (bar = 1 µm). The large body is surrounded by small intraluminal vesicles. (c) High power electron micrograph shows intraluminal vesicles beneath the surrounding membrane (arrow). (d) The electron dense granular bodies are surrounded by a membrane (arrow) and contain electron lucent areas (bar = 500 nm). (e) A vesicle contains some electron dense granular material (bar = 200 nm). (f) A small round granular body within a limiting membrane contains an electron lucent area (bar = 200 nm).
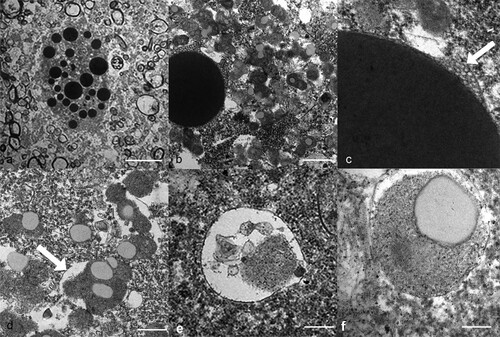
The granular material noted using light microscopy was made up of many irregularly rounded smaller electron-dense bodies with a granular texture, which sometimes contained electron-lucent round areas ((b,d)). Although a surrounding membrane was often difficult to see, it was clearly present in some bodies ((d)). The irregularity of the granular bodies implied that smaller bodies were fusing together and with the lucent areas. A small minority of these contained membranous laminar material, but this form was relatively uncommon. Others were essentially vesicular with varying amounts of electron-dense or lucent material ((e,f)). The granular bodies were more ubiquitous than indicated by light microscopy and were found throughout the cytosol and between the larger globoid bodies.
Pedigree analysis
A kindred matrix table, in which a figure of 0.1250 or above denotes first cousins, showed that cases 1 and 4 had a first cousin relationship (0.1730), and that cases 2 and 4 were slightly less related (0.1200). Cases 1 and 2 had a more distant relationship (0.0294), while case 3 had no recorded relationship with the others.
Discussion
The four cases of this novel neurological storage disease, with conflicting clinical histories, occurred in a population of approximately 440 individuals bred up from 44 founders from the original wild habitat. The disease was characterised by two distinct histological inclusions in motor neurons of the brain stem and spinal cord, i.e. large, spherical eosinophilic globoid bodies and smaller, more ubiquitous, heterogeneous granular inclusions. The globoid bodies were considered the signature lesion for this disease. They were membrane-bound, which qualifies them as cellular organelles, most logically of the phagosome/endosomal/lysosomal pathway, and possibly indicative of an LSD. Their histochemistry was moderately informative. The lack of sudanophilia, or staining with PAS or Alcian blue rules out neutral lipids, complex sugars of mannose-containing oligosaccharides, and mucopolysaccharides respectively, found in known LSD. The eosinophilia is probably indicative of protein. There was moderate staining with Luxol fast blue, a stain usually associated with myelin. The strong toluidine blue staining in epoxy resin sections is indicative of acidic material such as sulphates, carboxylates, phosphate radicals or nucleic acids (Sridharan and Shankar Citation2012).
The spherical shape and even electron-dense appearance of the globoid bodies is dissimilar to that of any secondary lysosome in the known LSD. The inclusion of many peripheral intraluminal vesicles suggests that they might be a form of multivesicular body with material entering by micro-autophagy. If so, then the globoid bodies could be interpreted as late endosomes. However, they did not immuno-stain with antibodies to lysosomal marker proteins LAMP1 and cathepsin D as would be expected, so this is not a lysosomal disease of either genetic or toxic origin. In addition, the limited distribution to neurons of the brain stem and spinal cord is not mirrored by any known LSD. They are also dissimilar to inclusions formed from non-digestible xenobiotics such as those associated with Phalaris spp. (Gallagher et al. Citation1966; Jolly and Hartley Citation1977, Alden et al. Citation2014) or Trachyandra spp. (Huxtable et al. Citation1987), in which there are numerous small intracytoplasmic brown inclusions. The limited distribution to motor neurons, and the finding of cases in different habitats with different environments and flora, at different times of the year, did not support a toxic plant aetiology.
The distribution of lesions in these takahē was that of a lower motor neuron disease. A heterogeneous group of human motor neuron diseases are known as “amyotrophic lateral sclerosis” affecting both upper and lower motor neurons. Although most cases (90%) appear to be sporadic, others have a variety of mutant genes as causative factors (Hardiman et al. Citation2017). Epidemiology studies indicate that environmental stressors are also involved, although most are not defined (Hardiman et al. Citation2017). However, an epidemic of ALS and Parkinson’s disease on the island of Guam has long been linked with exposure to β-N-metylamino-L-alanine found in seeds of cycad plants. A possible mechanism in that epidemic involves the incorporation of this molecule, as an abnormal amino acid, into protein, leading to protein misfolding (Dunlop et al. Citation2021). This abnormal amino acid also occurs in cyanotoxins produced by cyanobacteria. Cyanobacterial algal blooms in lakes in the USA have been identified as a risk factor for ALS (Torbick et al. Citation2018). In most cases of ALS, the aggregating protein is phosphorylated TDP-43, regardless of the cause or mutant gene (Brettschneider et al. Citation2013). This accumulates as skeins of fibrillar material in the cytoplasm. ALS may also be characterised by more solid round inclusions known as Bunina bodies, which are eosinophilic and stain with Luxol fast blue. They are far less numerous than those in the takahē neurons under study, are ultrastructurally different and are not surrounded by a unit-membrane (Wada et al. Citation1999). A lower motor neuron disease occurs in horses that has a strong association with vitamin E deficiency (Cummings et al. Citation1993; Mohammed et al. Citation2007). In that disorder there are large oval granular moderately electron dense inclusions surrounded by a membrane studded with ribosomes.
Amyotrophic lateral sclerosis is classified as a proteinopathy due to aggregation of misfolded protein. Despite the differences in the two diseases, it is probable that the takahē disease is also a proteinopathy. There are many proteinopathies affecting different organs and proteins that are characterised by a variety of non-membrane-bound cellular inclusions, and sometimes extracellular deposits. In contrast, there are membrane-bound inclusion bodies which are within RER. These were first described in the liver of human patients with a form of hereditary hypofibrinogenaemia caused by the Z mutation in α-1 antitrypsin, and gave rise to the name “endoplasmic reticulum storage disease” (Lomas et al. Citation1992; Callea and Desmet Citation2021, Padilla-Godínez et al. Citation2021). A similar disease has been described in an elephant (Yamada et al. Citation2003). Other disorders with similar RER inclusions are Russell bodies in plasma cells (Gray and Doniach Citation1970; Mossuto et al. Citation2015); Collins bodies in the nervous system in a familial encephalopathy in which neuroserpin aggregates accumulate (Davis et al. Citation1999; Walker and LeVine, Citation2000b); and inclusions in equine motor neuron disease as noted above (Cummings et al. Citation1993). The morphology of Russell bodies, as observed in H&E sections and electron micrographs (Gray and Doniach Citation1970), and those of Collins bodies are similar, although not identical, to those in these takahē. However, an association with ribosomes of RER was not noted in the takahē disease, although this may have been obscured in electron micrographs through very poor cellular detail resulting from marked autolysis and sub-optimal fixation. The takahē bodies also show a similarity to secretory vesicles such as zymogen granules in exocrine pancreas (Longnecker Citation2014). The secretory products of ventral cord motor neurons are mainly acetylcholine from clear-core secretory vesicles, and neuropeptide neurotransmitters from dense-core vesicles. These latter organelles contain the precursor protein of the neuropeptides, proteases to process them, other proteins, biogenic amines, and microRNA (Merigihi Citation2018).
The granular material was also a significant lesion and, in some neurons, the dominant one. Its nature was heterogeneous and only superficially similar to lipofuscin. Sudanophilia indicates neutral lipid and the strong PAS staining indicates the likely presence of complex sugars. This granular material stained with the lysosomal marker cathepsin D ((i,j)) so does have a lysosomal association. It is concluded that it is related to that of the globoid bodies and reflects the same origins, but that misfolded aggregated protein has entered the lysosomal pathway by ER-phagy and become complexed with other lysosomal catabolites. This aetiological relationship is supported by the observation that staining by Luxol fast blue was present in both types of bodies. As the larger globoid bodies clearly did not contain complex lipids such as those in myelin, Luxol fast blue staining in both types of storage material may indicate hydrophobic amino acids exposed on peptide misfolding and aggregation.
Iron was not stained in either of the two types of neuronal storage bodies. The large amount of iron in Küpffer cells and hepatocytes is probably not related to the storage material.
The lesions in takahē occurred in birds aged between 2 and 12 years, but only case 1 was recorded to have clinical signs commensurate with spinal cord dysfunction. The florid nature of spinal cord inclusions suggests that they were not insignificant. The cause of the disease is uncertain. Despite the disparate age of birds when lesions were observed, a genetic aetiology or predisposition to disease is likely and supported by the small gene pool of the species and close breeding of three of the cases. Some proteinopathies have complex, ill-defined causes with environmental factors involved that trigger cellular stress initiating protein aggregation. A small aggregate may act as a nidus and accelerate further self-aggregation (Marsh Citation2019; Walker and LeVine Citation2000a; Padilla-Godínez et al. Citation2021).
The precise cause and clinical significance of this disorder to the breeding programme for this endangered species are unknown. The nature of the lesions, clinical disease in one of the cases, and the likelihood of a genetic component to its pathogenesis, are causes for concern for the recovery programme.
Acknowledgements
The paraffin sections for light microscopy were prepared by Evelyn Lupton and Petru Daniels, Histology Laboratory, School of Veterinary Science, Massey University (Palmerston North, NZ). Electron microscopy was conducted with the help of Yanyu He and Raoul Solomon of the Manawatū Microscopy and Imaging Centre, Massey University. The background epidemiological data and information was supplied by members of the takahē recovery team, Department of Conservation. The research was supported by the Department of Conservation “Wildlife Institutions Relief Fund, NZ.”
References
- Alden R, Hockney B, Weston LA, Quin JC. Phalaris toxicoses in Australian livestock production systems: prevalence, aetiology, and toxicology. Journal of Toxins 1, 7, 2014. https://doi.org/10.13188/2328-1723.1000003
- Alroy J, Lyons JA. Lysosomal storage diseases. Journal of Inborn Errors of Metabolism and Screening 1, 20, 2014. https://doi.org/10.1177/2326409813517663
- *Ballou JD, Lacy RC, Poliak JP. PMx Software for Demographic and Genetic Analysis and Management of Pedigreed Populations (Version.1.62.20200110). http://scti.tools (accessed 2 March 2023). Chicago Zoological Society, Brookfield, IL, USA, 2020
- Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Annals of Neurology 74, 20–38, 2013. https://doi.org/10.1002/ana.23937
- Busche MA, Hyman BT. Synergy between amyloid-β and Tau in Alzheimer’s disease. Nature Neuroscience 23, 1183–93, 2020. https://doi.org/10.1038/s41593-020-0687-6
- Callea F, Desmet V. The discovery of endoplasmic reticulum storage disease. The connection between an H&E slide and the brain. International Journal of Molecular Sciences 22, 2899, 2021. https://doi.org/10.3390/ijms22062899
- Cummings JF, de Lahunta A, Summers BA, Mohammed HO, Divers TJ, Valentine BA, Trembicki-Graves K. Eosinophilic cytoplasmic inclusions in sporadic equine motor neuron disease: an electron microscopic study. Acta Neuropatholica 85, 201–97, 1993. https://doi.org/10.1007/BF00227725
- Davis RL, Holohan PD, Shrimpton AE, Tatum AH, Daucher J, Collins GH, Todd R, Bradshaw C, Kent P, Feiglin D, et al. Familial encephalopathy with neuroserpin inclusion bodies. American Journal of Pathology 155, 1901–13, 1999. https://doi.org/10.1016/S0002-9440(10)65510-1
- Dunlop RA, Banack SA, Bishop SL, Metcalf JS, Murch SJ, Davis DA, Stommel KW, Karlson O, Brittebo EB, Chatziefthimiou AD, et al. Is exposure to BMMA a risk factor for neurodegenerative diseases? A response to a critical review of the BMMA hypothesis. Neurotoxicity Research 39, 81–106, 2021. https://doi.org/10.1007/s12640-020-00302-0
- Gallagher CH, Koch JH, Hoffman H. Diseases of sheep due to ingestion of Phalaris tuberosa. Australian Veterinary Journal 42, 279–84, 1966. https://doi.org/10.1111/j.1751-0813.1966.tb08836.x
- Gray A, Doniach I. Ultrastructure of plasma cells containing Russell bodies in human stomach and thyroid. Journal of Clinical Pathology 23, 608–12, 1970. https://doi.org/10.1136/jcp.23.7.608
- Hardiman O, Al-Chalabi A, Chio A, Carr EM, Lagroscino G, Robbercht W, Shaw PM, Simmons Z, van den Berg LH. Amyotrophic lateral sclerosis. Nature Reviews Disease Primers 3, 17071, 2017. https://doi.org/10.1038/nrdp.2017.71
- Hopwood JJ, Crawley AC, Taylor FM. Spontaneous and engineered disease models. In: Platt FM, Walkely SU (eds). Lysosomal Diseases of the Brain. Pp 257–89. Oxford University Press, Oxford, UK, 2004. https://doi.org/10.1093/acprof:oso/9780198508786.003.0011
- Huxtable CR, Chapman HM, Main DC, Vass D, Pearse BHG, Hilbert BJ. Neurological disease and lipofuscinosis in horses and sheep grazing Trachyandra divaricate (branched onion weed) in south Western Australia. Australian Veterinary Journal 64, 105–8, 1987. https://doi.org/10.1111/j.1751-0813.1987.tb09639.x
- Johnston HE, Samant RS. Alternative systems for misfolded protein clearance: life beyond the proteosome. FEBS Journal 288, 4464–87, 2021. https://doi.org/10.1111/febs.15617
- Jolly RD, Hartley WJ. Storage diseases of domestic animals. Australian Veterinary Journal 53, 1–8, 1977. https://doi.org/10.1111/j.1751-0813.1977.tb15809.x
- Jolly RD, Walkley SU. Lysosomal storage diseases of domestic animals: an essay in comparative pathology. Veterinary Pathology 34, 527–48, 1997. https://doi.org/10.1177/030098589703400601
- Jolly RD, Hunter SA, Alley MR, King BM, Lau AA, Trim PJ, Snel MF, Hemsley KM. Mucopolysaccharidosis II (MPS II) in a free-living kaka (Nestor meridionalis) in New Zealand. Journal of Wildlife Diseases 57, 884–90, 2021. https://doi.org/10.7589/JWD-D-20-00173
- Li H, Sun S. Protein aggregation in the ER: calm behind the storm. Cells 10, 3337, 2021. https://doi.org/10.3390/cells10123337
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z α1-antitrypsin accumulation in the liver. Nature 357, 605–7, 1992. https://doi.org/10.1038/357605a0
- Longnecker DS. Anatomy and Histology of the Pancreas. https://doi.org/10.3998/panc.2014.3 (accessed 2 March 2023). Pancreapedia, Ann Arbor, MI, USA, 2014
- Marsh AP. Molecular mechanisms of proteinopathies across neurodegenerative disease: a review. Neurological Research and Practice 1, 35, 2019. https://doi.org/10.1186/s42466-019-0039-8
- McClelland JM, Gartrell BD, Roe WD. A retrospective study of post-mortem examination findings in takahē (Porphyria hochstetteri). New Zealand Veterinary Journal 59, 160–5, 2011. https://doi.org/10.1080/00480169.2011.579243
- Mehta A, Winchester B (eds). Lysosomal Storage Disorders: A Practical Guide. Wiley-Blackwell, Chichester, UK, 2012. https://doi.org/10.1002/9781118514672
- Merigihi A. Co-storage of high molecular weight neurotransmitters in large dense core vesicles of mammalian neurons. Frontiers of Cellular Neuroscience 12, 2018. https://doi.org/10.3389/fncel.2018.00272
- Mohammed HO, Divers TJ, Summers BA, de Lahunta A. Vitamin E deficiency and risk of equine motor neuron disease. Acta Veterinaria Scandinavica 49, 17–25, 2007. https://doi.org/10.1186/1751-0147-49-17
- Mossuto MF, Ami D, Anelli T, Fagioli C, Doglia SM, Sitia R. Biochemical nature of Russell bodies. Scientific Reports 5, 12585, 2015. https://doi.org/10.1038/srep12585
- Needham PG, Guerriero CJ, Brodsky JL. Chaperoning endoplasmic reiticulum-associated degradation and protein conformational diseases. Cold Spring Harbor Perspectives in Biology 11, a033928, 2019. https://doi.org/10.1101/cshperspect.a033928
- Padilla-Godínez FJ, Ramos-Acevedo R, Martínez-Becerril HA, Bernal-Conde LD, Garrido-Figueroa JF, Hiriart M, Hernández-López A, Argüero-Sánchez R, Callea F, Guerra-Crespo M. Protein misfolding and aggregation: the relatedness between Parkinson’s disease and endoplasmic reticulum storage disorders. International Journal of Molecular Sciences 22, 1–46, 2021. https://doi.org/10.3390/ijms222212467
- Platt FM, d’Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nature Reviews Disease Primers 4, article 27, 2018. https://doi.org/10.1038/s41572-018-0025-4
- Sridharan G, Shankar AA. Toluidine blue: a review of its chemistry and clinical utility. Journal of Oral Maxillofacial Pathology 16, 251–5, 2012. https://doi.org/10.4103/0973-029X.99081
- Stefanis L. α-Synuclein in Parkinson’s disease. Cold Spring Harbor Perspectives in Medicine 2, a009399, 2012. https://doi.org/10.1101/cshperspect.a009399
- Tepedelen BE, Kirmizibayrak PB. Endoplasmic-associated degradation (ERAD). In: Català A (ed). Endoplasmic Reticulum. InTech, London, UK, 2019. https://doi.org/10.5772/intechopen.82043
- Torbick N, Ziniti B, Stommel E, Linder E, Andrew A, Caller T, Haney T, Bradley W, Henegan PL, Xun S. Assessing cyanobacterial harmful algal blooms as risk factors for amyotrophic lateral sclerosis. Neurotoxicity Research 33, 199–212, 2018. https://doi.org/10.1007/s12640-017-9740-y
- Wada N, Uchiara T, Nakamura A, Oyanagi K. Bunina bodies in amyotrophic lateral sclerosis on Guam: a histological, immunohistochemical and ultrastructural investigation. Acta Neuropatholica 98, 150–6, 1999. https://doi.org/10.1007/s004010051063
- Walker LC, LeVine H. The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly. Molecular Neurobiology 21, 83–96, 2000a. https://doi.org/10.1385/MN:21:1-2:083
- Walker LC, LeVine H. The cerebral proteopathies. Neurobiology of Aging 21, 559–61, 2000b. https://doi.org/10.1016/S0197-4580(00)00160-3
- Yamada M, Nakamura K, Nazaki H, Tanaka H. Hepatocelluar endoplasmic reticulum storage disease in an African elephant (Loxodanta africana). Journal of Comparative Pathology 128, 192–4, 2003. https://doi.org/10.1053/jcpa.2002.0608
- * Non-peer-reviewed