
Abstract
Aim
To determine which genotypes of bovine viral diarrhoea virus (BVDV) circulate among cattle in New Zealand.
Methods
Samples comprised BVDV-1-positive sera sourced from submissions to veterinary diagnostic laboratories in 2019 (n = 25), 2020 (n = 59) and 2022 (n = 74) from both beef and dairy herds, as well as archival BVDV-1 isolates (n = 5). Fragments of the 5′ untranslated region (5′ UTR) and glycoprotein E2 coding sequence of the BVDV genome were amplified and sequenced. The sequences were aligned to each other and to international BVDV-1 sequences to determine their similarities and phylogenetic relationships. The 5′ UTR sequences were also used to create genetic haplotype networks to determine if they were correlated with selected traits (location, type of farm, and year of collection).
Results
The 5′ UTR sequences from New Zealand BVDV were closely related to each other, with pairwise identities between 89% and 100%. All clustered together and were designated as BVDV-1a (n = 144) or BVDV-1c (n = 5). There was no evidence of a correlation between the 5′ UTR sequence and the geographical origin within the country, year of collection or the type of farm. Partial E2 sequences from New Zealand BVDV (n = 76) showed 74–100% identity to each other and clustered in two main groups. The subtype assignment based on the E2 sequence was the same as based on the 5′ UTR analysis. This is the first comprehensive analysis of genomic variability of contemporary New Zealand BVDV based on the analysis of the non-coding (5′ UTR) and coding (E2) sequences.
Conclusions and clinical relevance
Knowledge of the diversity of the viruses circulating in the country is a prerequisite for the development of effective control strategies, including a selection of suitable vaccines. The data presented suggest that New Zealand BVDV are relatively homogeneous, which should facilitate eradication efforts including selection or development of the most suitable vaccines.
Introduction
Bovine viral diarrhoea virus (BVDV) is classified within the Pestivirus genus of the family Flaviviridae. Until recently, pestiviruses were classified into 11 species designated Pestivirus A through to Pestivirus K based on similarities in their genomes in combination with antigenic differences, natural host range, and pathology (Ridpath et al. Citation2006; Postel et al. Citation2021). Due to the expanding number of novel pestivirus sequences, the establishment of an additional eight species (Pestivirus L to Pestivirus S) was proposed (Postel et al. Citation2021). Cattle are considered to be the natural host for members of Pestivirus A (BVDV-1), Pestivirus B (BVDV-2), and Pestivirus H (BVDV-3 or HoBi-like pestivirus), although these viruses can also infect other cloven-hoofed animals (Nelson et al. Citation2015; de Oliveira et al. Citation2020; Evans and Reichel Citation2021). In the most recent version of the Flaviviridae taxonomy, ratified in 2023, the names of the species have been changed to Pestivirus bovis (from Pestivirus A), Pestivirus tauri (from Pestivirus B), and Pestivirus braziliense (from Pestivirus H) (Simmonds et al. Citation2017). Outside of the official classification, pestiviruses within each species are further divided into subtypes, with 21 BVDV-1 subtypes (designated a through u) currently described in the literature (Walz et al. Citation2020). Viruses from all three species have global distribution (Yesilbag et al. Citation2017), but only BVDV-1 has thus far been detected in New Zealand (Gates et al. Citation2021a, Citation2021b). Pestivirus infection is associated with economic losses in both the beef and dairy cattle industries (Lanyon et al. Citation2014; Han et al. Citation2018; Decaro Citation2020).
The genome of BVDV comprises single-stranded, positive-sense RNA that is 12.3 kb long. It encodes one open reading frame flanked by untranslated regions (UTR) (Tautz et al. Citation2015; Al-Kubati et al. Citation2021). One of the challenges in the effective control of BVDV infections is the considerable genetic variability of BVD viruses circulating in various parts of the world (Tautz et al. Citation2015; Yesilbag et al. Citation2017; Miroslaw and Polak Citation2020). Most studies of the variability of the BVDV genome used the 5′ UTR region, which is comparatively conserved (Vilcek et al. Citation2005; Deng et al. Citation2015; Stalder et al. Citation2016). Some researchers used other genomic regions for the same purpose, including those coding for N-terminal protease (Vilcek et al. Citation2004) or envelope glycoprotein E2 (Miroslaw and Polak Citation2020). The monitoring of BVDV genotypes within a given geographical region is clinically and biologically important because there are antigenic differences among BVD viruses of different subtypes (Walz et al. Citation2020).
The E2 glycoprotein plays a role in virus entry into the cell (El Omari et al. Citation2013). It is also one of the most immunodominant proteins of the virus (Deregt et al. Citation1998; Al-Kubati et al. Citation2021). As such, the high variability of E2 protein complicates the design of effective vaccines (Walz et al. Citation2020). Many currently available vaccines are offered as combination vaccines containing both BVDV-1 and BVDV-2, with BVDV-1a typically used for the BVDV-1 component.
Infection with BVDV-1 virus is common among New Zealand cattle, within both dairy and beef herds (Han et al. Citation2018; Gates et al. Citation2019, Citation2020). New Zealand comprises two main islands, with the closest neighbours (Australia and West Polynesia) located more than 1,800 km across the Pacific Ocean. The geographical isolation of the country, coupled with strict quarantine measures, assures freedom from several pathogens that are present in other parts of the world. Unlike in many other countries, beef and dairy farming systems in New Zealand are predominantly pasture-based. Altogether, this means that New Zealand provides a unique ecosystem in which pressures on viruses circulating among livestock, including BVD viruses, may differ from those in overseas herds. The only study that examined genomic variability of New Zealand BVDV was conducted 25 years ago and included only 20 viral isolates obtained between 1967 and 1997 (Vilcek et al. Citation1998). All were shown to be closely related to the international BVDV-1a NADL strain based on the analysis of the 5′ UTR fragment (Vilcek et al. Citation1998). There are no data on the genomic diversity of contemporary BVDV in New Zealand. The aim of this study was to determine which BVDV genotypes circulated among cattle in New Zealand between 2019 and 2022.
Materials and methods
Sources of samples
A total of 163 samples were included in the study. BVDV-positive sera were supplied by commercial veterinary diagnostic laboratories (Gribbles Veterinary or IDEXX Laboratories NZ) that receive submissions from throughout New Zealand. Samples from 2019 to 2020 were subjected to 5′ UTR testing as part of two separate postgraduate projects. One or two samples per farm were randomly selected from all available submissions from 2019 (beef herds (n = 25)), and 2020 (n = 59, including dairy herds (n = 56) and unknown (n = 3)). In addition, convenience samples from laboratory submissions in 2022 (n = 74, including dairy herds (n = 16), beef herds (n = 17) and unknown (n = 41)) and several archival BVDV-1 isolates stored at the Virology Laboratory at Massey University (Palmerston North, NZ) (n = 5) were included in the study.
Processing of samples
Viral RNA was extracted from the samples using commercial RNA extraction kits: either MagMAX Viral/Pathogen Nucleic Acid Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA) or QIAamp viral RNA kit (Qiagen, Venio, Netherlands). Extracted RNA was either used directly as a template for one-step reverse transcriptase (RT)-PCR to amplify 5′ UTR or for complementary DNA (cDNA) synthesis using qScript cDNA Supermix (QuantaBio, Beverly, MA, USA) according to the manufacturer’s instructions. The viral cDNA was used as a template in PCR assays to amplify 5′UTR and E2 regions of the BVDV genome.
The 5′ UTR (288 bp product) was amplified using primers 324 and 326 (Vilcek et al. Citation1994) from 2022 samples, selected 2020 samples and archival virus isolates. Amplifications were performed using HotFirePol master mix with 10 mM MgCl2 (Solis BioDyne, Tartu, Estonia) in a final reaction volume of 10 µL or 20 µL, with 0.4 μM of each primer, and 10% of the final reaction volume (1 µL or 2 µL) of template cDNA. No-template controls contained water in the place of cDNA. The PCR cycling included an initial denaturation at 95°C for 15 minutes, followed by 40 cycles of denaturation (95°C for 15 seconds), annealing (60°C for 30 seconds), elongation (72°C for 30 seconds), and a final elongation step at 72°C for 5 minutes. Sequences from 2019 and the majority of 2020 samples were amplified with primers 325 and 326 (Vilcek et al. Citation1994) in a one-step RT–PCR reaction using the SuperScript III One-Step RT–PCR with Platinum Taq enzyme mix (Thermo Fisher Scientific), as per the manufacturer’s instructions. Each PCR reaction contained 3 µL of up to 1 µg RNA template, 0.2 µM final concentration of each primer, 1 unit of SuperScript III RT/Platinum Taq enzyme mix in a supplied reaction buffer in a final volume of 50 µL. The amplification conditions were as described previously (Vilcek et al. Citation1994).
The glycoprotein E2 gene (1.1 kb) was amplified using various combinations of forward and reverse primers (). These included 810F/810F (set A), 2274F/3434R (set B), C_BVDVNZF/C_BVDVNZR (set C), and BVDV6F/BVDV5R (set D). The expected amplicon sizes were 1.2 kb (sets A and B), 1.1 kb (set C), and 0.6 kb (set D). PCR reactions were set up as described for 5′ UTR PCR, except that each 20 μL reaction contained 0.8 μM of each forward and reverse primer, and up to 4 μL of template cDNA. For some samples, nested PCR reactions were set up using forward primers from sets A, B and C and reverse primer BVDV5 R (amplicons of ∼900, 1,100, and 900 bp, respectively). Amplification was performed using a touch-down PCR cycling protocol, which consisted of an initial denaturation at 95°C for 15 minutes, followed by 10 cycles of denaturation (95°C for 10 seconds), annealing (60°C (–1°C/cycle) for 10 seconds), and elongation (72°C for 2 minutes), followed by 30 cycles of denaturation (95°C for 10 seconds), annealing (50°C for 10 seconds), and elongation (72°C for 2 minutes), and a final elongation step at 72°C for 7 minutes.
Table 1. PCR primers used to amplify viral glycoprotein E2 gene sequences from samples of bovine viral diarrhoea virus-1-positive sera collected in 2020 and 2022 from beef and dairy cattle throughout New Zealand.
The obtained PCR products were gel-purified and sent for sequencing to the Massey Genome Service (Palmerston North, NZ) and subjected to automatic dye-terminator cycle sequencing with the BigDye Terminator Version 3.1 Ready Reaction Cycle Sequencing kit and the ABI3730 Genetic Analyzer (Applied Biosystems, Waltham, MA, USA).
Phylogenetic analysis
Altogether, 149/163 New Zealand sequences were used for the 5′ UTR phylogenetic analysis including sequences from 2022 (n = 65), 2020 (n = 57), 2019 (n = 22) and older archival sequences (n = 5). The remaining 14/163 sequences were excluded due to poor quality or inadequate length. In addition, 56 international BVDV sequences representing different genotypes were obtained from GenBank for a final dataset of 205 5′ UTR sequences (see Supplementary Tables S1 and S2).
Amplification of a partial E2 coding sequence was attempted from a total of 128 samples including sera from 2022, selected sera from 2020, and archival BVDV isolates (Table S1). As not all the samples that were used for 5′ UTR sequencing in 2019/2020 were retained in storage, sera from 2019 and selected sera from 2020 were not available for E2 PCR testing. The E2 sequences (n = 76) were obtained from samples from 2022 (n = 52), 2020 (n = 20) and older archival sequences or sequences with unknown dates (n = 4). In addition, 28 international BVDV E2 sequences were obtained from GenBank for a final dataset of 104 E2 sequences.
Sequences were aligned to each other and to international sequences in Geneious version 9.1.8 (www.Geneious.com) using either Clustal or MUSCLE algorithms with default settings. The sequences in the alignments were trimmed to one length (800 nucleotides for E2 gene and 247 nucleotides for 5′ UTR) and manually edited. Sequences that were too short or of poor quality were removed from the alignment, and all remaining sequences were realigned. The final alignments were exported as fasta files and used to generate phylogenetic trees in MEGA 11 (Tamura et al. Citation2021). The trees were visualised using the Interactive Tree of Life tool (Letunic and Bork Citation2021). For the E2 phylogenetic trees, both nucleotide and deduced amino acid sequences were used. The pairwise nucleotide distances were calculated and visualised using Sequence Demarcation Tool version 1.2 (Muhire et al. Citation2014). The mean genetic distances between and within selected genotypes were calculated in MEGA 11.
Haplotype network analysis
A genetic haplotype network was generated using New Zealand 5′ UTR sequences to determine if there was any clustering of samples according to selected traits. All 149 New Zealand sequences were aligned using CLUSTAL alignment within Geneious and exported as a fasta file. Ambiguity codes were changed to “Ns” and the file was used as an input to generate a haplotype list in the DnaSP software (http://www.ub.edu/dnasp/). Sites with gaps or missing/ambiguous data were excluded. The nucleotide sequences representative of the haplotypes (n = 51) were then realigned in Geneious and exported in the nexus format with trait blocks added to represent geographical region (Northland, Auckland, Waikato, Bay of Plenty, Taranaki, Manawatū, Wellington, Canterbury, Otago, Southland, unknown), year of isolation (2022, 2020, 2018, older) and type of farm (dairy, beef, unknown). The median joining haplotype networks were drawn using default parameters in PopART version 1.7 (Leigh and Bryant Citation2015). Analysis of molecular variance (AMOVA within PopART) was used to test for correlation between population genetic structure of the 5′ UTR BVDV-1 sequences and selected traits (year of collection, farm type, geographical region). The strength of correlation was represented by a PhiPT value, with 0 indicating no correlation and 1 indicating perfect correlation. The corresponding p-values were generated by reference to 1,000 random permutations of the input data.
Results
Both 5′ UTR and E2 sequences were analysed to assess the variability of BVD viruses included in the study and to compare these with the variability of BVDV sequences worldwide. All New Zealand BVDV sequences belonged to either BVDV-1a and BVDV-1c subtypes.
5′ UTR
All samples tested with 5′ UTR primers produced the expected product, but 14/163 (8.6%) sequences were excluded from the analysis due to the poor quality of sequencing or inadequate length. All 149 New Zealand 5′ UTR sequences included in the analyses were closely related to each other ( and ). All clustered within the BVDV-1a/1c group, which was supported by a 56% bootstrap value. Within the large 1a/1c group, there was strong bootstrap support (88% and 91%, respectively) for clustering of five New Zealand sequences (numbers 26, 143, 144, 145, 149) with international BVDV-1c sequences, and for clustering of two New Zealand sequences (numbers 64 and 161) with an international subtype 1a sequence from the UK. The New Zealand sequence #27 was identical to the sequence of the subtype 1a reference strain 1-NADL. Except for a few smaller clusters of New Zealand sequences from the same geographical area (for example sequences #103–6 or #23 and #99–101), which often originated from the same farm, the clustering of the remaining New Zealand sequences within the 1a group was generally poorly supported, most likely due to close genetic distances between them. This likely led to different New Zealand sequences clustering together during the resampling process (bootstrapping).
Figure 1. Phylogenetic analysis of partial bovine viral diarrhoea virus (BVDV) 5′ UTR sequences (n = 205) identified in New Zealand (BVDV-1) and international BVDV sequences sourced from Genbank, showing the optimal tree produced by evolutionary analyses on a dataset with 239 positions conducted in MEGA11 (Tamura et al. Citation2021). The evolutionary history was inferred using the unweighted pair group method with arithmetic mean (UPGMA). The percentage of replicate trees in which associated taxa clustered together in the bootstrap test (1,000 replicates) are proportional to the width of the branches. The evolutionary distances (units = base substitutions per site) were computed using the Kimura 2-parameter method. Rate variation among sites was modelled with a gamma distribution (shape parameter = 0.27). All positions with less than 90% site coverage were eliminated, i.e. fewer than 10% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option).
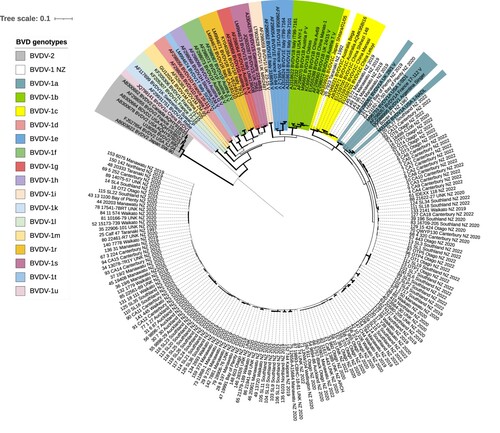
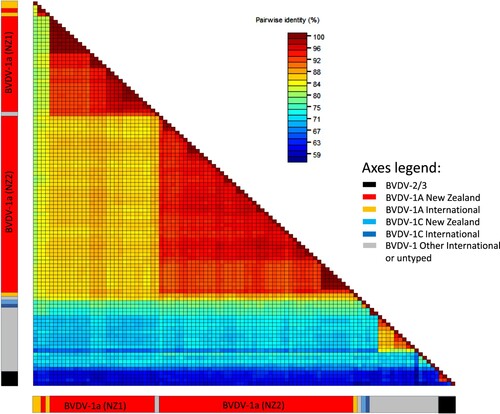
Figure 2. Heatmap showing percent identity between pairs of bovine viral diarrhoea virus (BVDV) 5′ UTR sequences. Sequences are aligned along the x and y axes and grouped into BVDV-2 and BVDV-1 subtypes 1a and 1c, of international and New Zealand origin, as shown in the bars below the map (see axis legend). Identity scores for each pairwise comparison are represented by coloured boxes with identity increasing from blue to red.
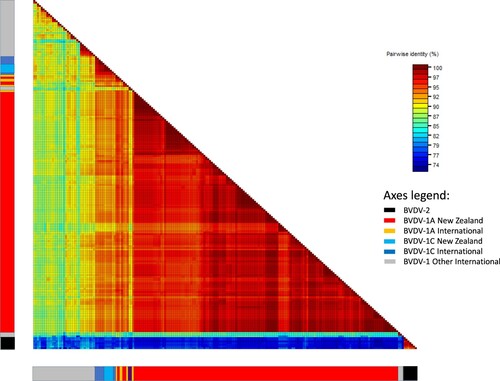
The pairwise percent identities between all New Zealand BVDV-1 5′ UTR sequences ranged from 89% to 100%. The pairwise identities between New Zealand BVDV-1a sequences ranged from 91% to 100%. New Zealand BVDV-1c sequences were 97–100% identical to each other. The pairwise identities between New Zealand sequences and international BVDV-1a and 1c sequences ranged from 89% to 100%. The percent identities between pairs of New Zealand and international sequences other than 1a and 1c ranged from 79% (BVDV-1t from China and BVDV-1 #23 from Southland) to 92% (BVDV-1d from France and BVDV-1 #27 from Manawatū) ().
The average evolutionary divergence between all pairs from the same BVDV genotype ranged from 0.009 (BVDV-1i) to 0.102 (BVDV-1 l), with NZ BVDV-1 sequences showing an average divergence of 0.01 (subtype 1c) and 0.04 (subtype 1a) (). Estimates of evolutionary divergence between New Zealand and international sequences ranged between genotypes, but were the lowest between international subtypes 1a (for NZ subtype 1a sequences) and subtype 1c (for NZ subtype 1c sequences), further supporting the classification of these sequences based on topology/clustering observed in the phylogenetic tree.
Table 2. EstimatesTable Footnotea of average evolutionary divergence over bovine viral diarrhoea virus (BVDV) 5′ untranslated region sequence pairs within BVDV genotypes and between BVDV-1 sequences identified in New Zealand (BVDV-1 NZ; n = 149) and international sequences from other genotypes (n = 56).
The current study was not designed to investigate the variability of sequences within or between farms. However, for the majority (11/19; 58%) of those farms for which more than one 5′ UTR sequence was available, all samples belonged to the same haplotype (Supplementary Table S3). Consequently, some sequences from the same geographical region were identical to each other (e.g. haplotype 27, which contained five sequences from two farms in Southland). However, haplotype network analysis did not support a correlation between the haplotype and the geographical origin of samples (, ). Samples from the same region (indicated by the same colour in ) were distributed throughout the network and some haplotypes (e.g. haplotype 5) comprised samples from several geographical locations. Similarly, there was no evidence of the correlation between the 5′ UTR sequence and the year of collection or the type of farm ().
Figure 3. International haplotype network of bovine viral diarrhoea virus 5′ untranslated region sequences from New Zealand (n = 147). The number of nucleotide substitutions between haplotypes is represented by ticks on branches. Nodes are scaled based on the number of representative sequences and coloured based on the geographical region of origin. Inferred nodes are indicated with small, closed black circles as they are not represented among sequences included in the network. Reticulation in a haplotype network indicates that the data did not contain appropriate signal to resolve a single pathway through a network, indicating that there is more than one possible sequence of mutations linking associated haplotypes.
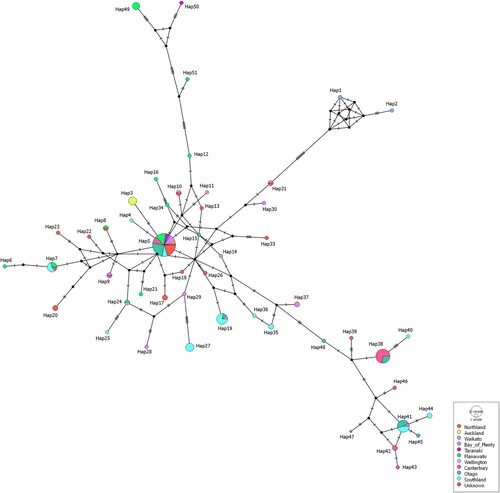
Table 3. Analysis of molecular variance (AMOVA) results indicating the strength of correlation between population genetic structure and selected traits based on partial sequence of 5′ UTR of New Zealand BVDV-1.
Viral protein E2
Of the 127 tested samples, 76 (60%) produced the expected PCR product suitable for sequencing with at least one pair of E2 primers. The remaining samples were either negative (n = 6) or produced very weak bands that could not be sequenced (n = 45) (Table S1).
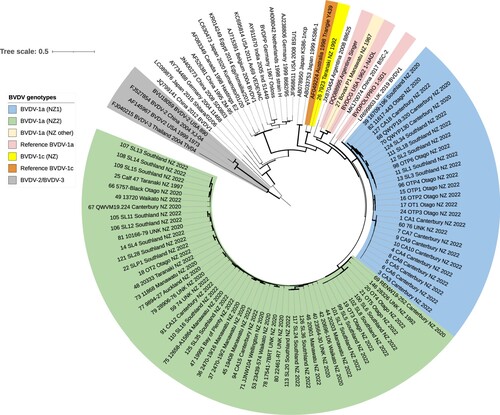
All available New Zealand BVDV-1 E2 sequences from subtype 1a viruses (based on the 5′ UTR analysis) also clustered with the international BVDV-1a sequences based on the E2 sequence analysis (). The New Zealand E2 sequences formed two main groups labelled “clade 1a (NZ1)” and “clade 1a (NZ2)” (), with strong bootstrap support (99%) for each clade. There were several smaller clades within each of the two main clades. An international sequence from UK/2019 (LR699803) clustered within the clade NZ1. Two other international BVDV-1 sequences were closely related to the New Zealand sequences: the BVDV-1a reference strain 1-SD1 and a BVDV-1 sequence from China from 2017 (MK170074), followed by a cluster of sequences containing two Argentinian sequences (including a BVDV-1a reference strain Singer), a BVDV-1a reference strain 1-NADL and a New Zealand archival isolate #27. The New Zealand archival isolate #26 clustered separately from the remaining New Zealand sequences with a BVDV-1c reference sequence from Australia (Triangie Y439). The neighbour-joining tree constructed based on the deduced amino-acid sequences showed the same topology and evolutionary relationships as the tree based on nucleotide sequences shown in (data not shown).
Figure 4. Phylogenetic analysis of partial E2 sequences (n = 104) from bovine viral diarrhoea viruses (BVDV) identified in New Zealand and international BVDV sequences sourced from Genbank, showing optimal tree produced by evolutionary analyses conducted in MEGA11 (Tamura et al. Citation2021) on a dataset with 795 positions. Evolutionary history was inferred using the neighbour joining method (Saitou and Nei Citation1987). The percentage of replicate trees in which associated taxa clustered together in the bootstrap test (1,000 replicates) are indicated by the width of the branches. The evolutionary distances (units = base substitutions per site) were computed using the Tamura-Nei method (Tamura and Nei Citation1993). The rate variation among sites was modelled with a gamma distribution (shape parameter = 0.62). All three codon positions were included. All positions with < 90% site coverage were eliminated, i.e. fewer than 10% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option).
The percent identity between pairs of New Zealand E2 nucleotide sequences ranged from 74% to 100% (). The sequences within clades 1a (NZ1) and 1a (NZ2) showed more than 91% and 90% identities, respectively. The identities between sequences from clades 1a (NZ1) and 1a (NZ2) ranged from 83% to 88%. New Zealand archival sequence #27 was identical to the reference BVDV-1a strain 1-NADL and 80–83% or 81–85% identical to sequences from clades 1a (NZ1) and 1a (NZ2), respectively. The New Zealand sequence #26 (clade 1c) was least similar to other New Zealand sequences: the pairwise identity scores between this sequence and sequences from clades 1a (NZ1) and 1a (NZ2) ranged from 74% to 80%. The identities between the New Zealand sequences from clades NZ1 and NZ2 and the international BVDV-1 sequences ranged from 65% (several New Zealand sequences and the international sequence AY911670 from India 2005) to 90% (#42 and 1-SDS).
Figure 5. Heatmap showing percent identity between pairs of bovine viral diarrhoea virus (BVDV) E2 sequences. Sequences are aligned along the x and y axes and grouped as described for with the position of New Zealand sequences from clades NZ1 and NZ2 indicated. Identity scores for each pairwise comparison are represented by coloured boxes with identity increasing from blue to red.
Discussion
The ongoing monitoring of the diversity of pestiviruses circulating in various geographical areas has been identified as important for gaining a better understanding of the epidemiology of these economically important viruses (Evans et al. Citation2019). Several European countries have implemented control strategies for BVDV, with a long-term aim of eradicating the virus from national herds (Wernike et al. Citation2017; Schweizer et al. Citation2021; Shortall Citation2022). A similar approach has been considered in New Zealand (Han et al. Citation2018; Gates et al. Citation2020, Citation2021a). Knowledge of the diversity of the viruses circulating in the country is a prerequisite for the development of effective control strategies, including a selection of suitable vaccines.
This is the first comprehensive analysis of genomic variability of contemporary New Zealand BVDV based on the analysis of the non-coding (5′ UTR) and coding (viral protein E2) sequences. The 5′ UTR has been extensively used for BVDV genotyping as it includes highly conserved regions, which facilitates amplification of a PCR product from a wide range of BVDV (Vilcek et al. Citation2004, Citation2005). As this is a non-coding region, its usefulness for typing is mostly relevant for epidemiological tracking, which can be used for the implementation and evaluation of BVDV control efforts (Vilcek et al. Citation1999; Booth et al. Citation2013). Some authors, however, questioned the usefulness of this region for genotyping purposes (Becher et al. Citation1997). The key problem identified was non-collinearity of sequences from various sources, which necessitates the introduction of gaps into the alignments. The position of gaps can influence the phylogenetic groupings, which in turn may affect the reliability of results. In addition, the clustering of sequences can differ depending on the number of sequences included and the parameters used for the construction of the phylogenetic trees. To illustrate this, viruses “190CP” and “190NCP” were classified as BVDV-1a in one study (Sakoda et al. Citation1999) and as BVDV-1c in another study (Nagai et al. Citation2001) based on the analysis of their 5′ UTR region. The same viruses were classified as BVDV-1e based on the analysis of the N-terminal E2 region (Nagai et al. Citation2004). The latter highlights the fact that variability among BVDV can arise not only due to mutation but also due to recombination, and hence clustering may differ depending on the region used for the analysis.
In our analyses, the New Zealand 5′ UTR sequences were closely related to each other, although they did not form one well-supported cluster in a phylogenetic tree. The majority were most closely related to international BVDV-1a sequences, with the remaining few clustering with international BVDV-1c sequences. None of the New Zealand BVDV-1 sequences clustered with other genotypes, including genotype 1b, which has been commonly reported in several overseas countries (Yesilbag et al. Citation2017). Thus, it appears that the predominant 5′ UTR genotype circulating among New Zealand cattle is BVDV-1a, similar to what was reported in a previous 1998 study (Vilcek et al. Citation1998). The current study represents the first report of BVDV-1c in New Zealand, which was reported to predominate in Australia (Mahony et al. Citation2005). None of the viruses belonged to BVDV-2 or BVDV-3 groups, which comprise viruses currently considered exotic to New Zealand. Our data support this classification.
The pairwise identities between New Zealand BVDV-1a 5′ UTR sequences (91–100%) were consistent with those reported for BVDV-1a viruses from other countries. Examples include BVDV-1a (n = 9) from various European countries (88–100% pairwise identity) (Vilcek et al. Citation2001) or BVDV-1a (n = 5) from Chile (92–100% pairwise identity) (Donoso et al. Citation2018).
The drivers behind the genomic diversity of pestiviruses, particularly in the non-coding regions such as 5′ UTR, are currently poorly understood. It is not known why viruses from some countries such as Australia or New Zealand appear to be relatively homogeneous, while BVDV from other countries show high heterogeneity, as illustrated by the presence of several different genotypes (Vilcek et al. Citation2005; Evans et al. Citation2019). The possible explanations include different sampling strategies, different populations sampled, different time periods for sampling or different pressures on virus evolution due to differences in the regional cattle management systems including the type of vaccines used.
The genomic diversity of BVDV is one of the factors hindering development of universally effective vaccines (Ridpath Citation2005, Citation2013; Falkenberg et al. Citation2021). Based on close similarities between New Zealand BVDV-1 viruses and international BVDV-1a viruses, including vaccine strains such as 1-SDS, Singer or 1-NADL, one would expect vaccines containing BVDV-1a strains to induce immunity that is cross-reactive with New Zealand field BVDV. However, there are considerable gaps in our understanding of the key determinants for the generation of broadly reactive and protective immune responses following BVDV-1 infection or vaccination (Falkenberg et al. Citation2021). It is well recognised that there are considerable differences between the ability of specific antisera to cross-neutralise BVDV-1 of different genotypes, although it is less clear if those differences apply to all viruses within a given genotype as opposed to the individual viruses investigated (Bachofen et al. Citation2008; Ridpath et al. Citation2010). In one study, BVDV-1a was neutralised by the homologous antiserum to a titre of 1,280, while an antiserum raised against BVDV-1f neutralised the same isolate only to a titre of 20 (Alpay and Yesilbag Citation2015). Altogether, the coefficient of antigenic similarity between six viruses from genotypes 1a, 1b, 1d, 1f, 1h and 1l used in that study ranged from 1.1% to 50%. The inconsistencies observed in the level of protection offered by the same vaccine in different cattle populations may also be influenced by host-related factors. In one study, sera from seven colostrum-deprived calves that had been vaccinated with a multicomponent vaccine containing inactivated BVDV-1 antigens were tested in a virus neutralisation assay against a selection of field and vaccine BVDV-1 and BVDV-2 isolates (Hamers et al. Citation2002). Sera from vaccinates contained antibodies that were cross-reactive against the range of BVDV tested, although there were considerable differences in the level of neutralisation observed against different viruses, as expected. In addition, there was up to a 3-log difference in the titre of sera from individual calves tested against the same virus, highlighting the influence of the host on the results obtained.
Viral protein E2 has been shown to be the main target for neutralising antibodies produced following infection with BVDV (Al-Kubati et al. Citation2021). For this reason, we have included this genetic region in the analysis. Consistent with the 5′ UTR results, the New Zealand E2 sequences were most closely related to international BVDV-1a sequences, except for sequence #26, which clustered with the Australian 1c sequence. The pairwise identity between New Zealand BVDV-1a E2 sequences ranged from 80% to 100%, with > 90% identity between sequences within the two main clusters. Comparatively few studies used E2 gene sequences for phylogenetic analysis, and the length of the E2 gene fragment used varied between studies, making direct comparison of the results difficult. Nonetheless, the pairwise percent identity between BVDV-1a sequences in Argentina was 71–77% (Pecora et al. Citation2014); between BVDV-1a subtypes in Chile 77–100% (Donoso et al. Citation2018); and the pairwise identities between BVDV-1 from 1b, 1s, and 1f subtypes in Poland were 70.4–98.5% (Miroslaw and Polak Citation2020). Thus, it appears that the New Zealand E2 gene sequences were more homogeneous than E2 gene sequences from other countries.
The grouping of New Zealand viruses with international BVDV-1a or 1c genotypes was the same for both 5′ UTR and E2 sequence analyses. Unfortunately, E2 sequence #26 represented the only New Zealand subtype 1c virus, as nucleic acids from the remaining four viruses that were subtyped as BVDV-1c based on the 5′UTR region were not available for testing. Other authors also reported similar results when BVDV-1 sequences were genotyped based on 5′ UTR and E2 sequences (Abe et al. Citation2016; Silveira et al. Citation2017; Donoso et al. Citation2018). The E2 sequence analysis in those studies produced stronger bootstrap support values for clustering of sequences within different subtypes than 5′ UTR analysis, consistent with our results.
Despite the use of several different primer pairs, E2 sequences were not obtained from 51 samples that reacted with 5′ UTR primers, either due to lack of amplification or amplification of a weak band that was not suitable for direct sequencing. This is not unexpected, as high variability in the E2 sequence has been described by others (Hertig et al. Citation1995; Miroslaw and Polak Citation2019); this is the main limitation of the use of this region for subtyping purposes. However, it raises the possibility that we have not amplified the more diverse E2 sequences and, hence, the New Zealand BVDV-1 E2 sequences may be more heterogeneous than suggested by the results presented here.
The New Zealand E2 sequences were further subdivided into two well-supported clades. Future work should include determination of the level of antigenic cross-reactivity among the viruses from the two clades. This could be done by the assessment of their reactivity with antisera raised to selected vaccine strains, as has been described by others (Mosena et al. Citation2020, Citation2022) or by in vivo trials. In one study, eight Japanese BVDV-1a from four different E2 clades were antigenically closely related, with the neutralisation titres against an antiserum raised against another BVDV-1a ranging from 1,024 to 2,048 (Abe et al. Citation2016). In contrast, the neutralisation titres of the same antiserum tested with selected BVDV-1b viruses were considerably lower and ranged from 64 to 256. Similar results were reported by Abe et al. (Citation2016), with neutralisation titres of BVDV-1a viruses from five E2 gene clusters against the same BVDV-1a antiserum ranging from 1,024 to 2,048, while the same viruses produced neutralisation titres between 16 and 128 with antisera produced against five different BVDV-1b viruses. There are no similar data available for New Zealand BVDV-1 but, by extrapolation, one would expect New Zealand viruses that showed close genetic similarity in the E2 gene to also be closely related antigenically. In agreement with this assumption, the results of an experimental infection trial using calves persistently infected with New Zealand BVDV-1a viruses showed that the fetal protection induced by vaccination with inactivated BVDV-1c vaccine was similar (72%) to the fetal protection induced by vaccination with an inactivated BVDV-1a vaccine (62%) (Packianathan et al. Citation2017). The inactivated vaccines containing either BVDV-1a (Bovilis BVD; MSD Animals Health, Rahway, NJ, USA and Hiprabovis-3; Hipra, Girona, Spain) or BVDV-1c (Ultravac BVD; Zoetis, Parsippany, NJ, USA) are the only BVDV vaccines currently registered in New Zealand. The results of the current study suggest that all three contain the relevant subtypes of BVDV-1, and that the incomplete protection anecdotally observed in the field, consistent with the results of the field trial (Packianathan et al. Citation2017), likely reflects the type of vaccines used. Although the factors affecting vaccine-induced protection are complex, live vaccines typically offer higher levels of foetal protection than killed vaccines (Ridpath Citation2013; Newcomer et al. Citation2015, Citation2017).
This study has several limitations. Relatively small numbers of samples were tested, which were sourced by convenience from submissions to the diagnostic veterinary laboratories. While the main geographical regions were represented, the number of samples available from each region was not proportional to the number of cattle in that region (Table S2). Hence, the sequences included in the study may not be representative of all viruses circulating in New Zealand. In addition, BVDV-1-positive serum samples were collected over a comparatively short period between 2019 and 2022. Although there was no indication of clustering of sequences based on the year of collection, unavailability of suitable numbers of archival sequences collected before 2019 precluded any conclusions about time as a factor for divergence of BVDV in New Zealand to be made. One of the archival New Zealand isolates (#26) from 1992 was relatively divergent from other New Zealand BVDV-1 sequences, both in the 5′ UTR and E2 regions, suggesting that New Zealand viruses may have changed since 1992. However, archival isolate #25 collected in 1997 clustered together with the contemporary New Zealand sequences, as did archival isolate #51, although the exact time of collection for the latter was not available.
The information on the identity of individual farms was coded by the laboratories and not available for all samples. With some exceptions, only one or two samples per farm were tested during the 2019 and 2020 samplings. As several different laboratories provided the samples, it is possible that the same farm could have been coded differently in 2022 and in 2019/20. For those reasons, the smallest geographical unit used for the network analyses was the region. Nonetheless, it was evident that while only a single BVDV-1 haplotype was recovered from some farms, several different haplotypes were circulating on other farms. The latter may have reflected transmission of the virus between farms, as has been shown by others (Ridpath et al. Citation2006; Booth et al. Citation2013). Data related to the movement of animals and people between farms would have been needed to assess this further. The true diversity of BVDV-1 on individual farms may have been underestimated, as the sequence data was obtained from sequencing of the single PCR product. This did not allow for the detection and differentiation of multiple genotypes that may have been present in individual samples (Dow et al. Citation2015). In addition, a very small number of samples were tested from each farm.
Notwithstanding these limitations, the data presented here suggest that New Zealand BVDV are relatively homogeneous, which should facilitate eradication efforts including selection or development of the most suitable vaccines.
Supplemental Material
Download PDF (201.9 KB)Acknowledgements
This work was supported by AgriHealth New Zealand and BVDFree New Zealand. The authors wish to thank Jessica Cheung for helping with laboratory work; staff from participating diagnostic laboratories for provision of serum samples; and Carolyn Gates for facilitating requisition of serum samples from 2019 and 2020.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
Representative New Zealand BVDV-1 sequences obtained in the study have been submitted to GenBank under accession numbers MN961220–MN961229, OR238926–OR239001 (5′UTR) and OR239002–OR239052 (protein E2 gene).
References
- Abe Y, Tamura T, Torii S, Wakamori S, Nagai M, Mitsuhashi K, Mine J, Fujimoto Y, Nagashima N, Yoshino F, et al. Genetic and antigenic characterization of bovine viral diarrhea viruses isolated from cattle in Hokkaido, Japan. Journal of Veterinary Medical Science 78, 61–70, 2016. https://doi.org/10.1292/jvms.15-0186
- Al-Kubati AAG, Hussen J, Kandeel M, Al-Mubarak AIA, Hemida MG. Recent advances on the bovine viral diarrhea virus molecular pathogenesis, immune response, and vaccines development. Frontiers in Veterinary Science 8, 665128, 2021. https://doi.org/10.3389/fvets.2021.665128
- Alpay G, Yesilbag K. Serological relationships among subgroups in bovine viral diarrhea virus genotype 1 (BVDV-1). Veterinary Microbiology 175, 1–6, 2015. https://doi.org/10.1016/j.vetmic.2014.10.034
- Bachofen C, Stalder H, Braun U, Hilbe M, Ehrensperger F, Peterhans E. Co-existence of genetically and antigenically diverse bovine viral diarrhoea viruses in an endemic situation. Veterinary Microbiology 131, 93–102, 2008. https://doi.org/10.1016/j.vetmic.2008.02.023
- Becher P, Orlich M, Shannon AD, Horner G, König M, Thiel HJ. Phylogenetic analysis of pestiviruses from domestic and wild ruminants. Journal of General Virology 78, 1357–66, 1997. https://doi.org/10.1099/0022-1317-78-6-1357
- Booth RE, Thomas CJ, El-Attar LM, Gunn G, Brownlie J. A phylogenetic analysis of bovine viral diarrhoea virus (BVDV) isolates from six different regions of the UK and links to animal movement data. Veterinary Research 44, 43, 2013. https://doi.org/10.1186/1297-9716-44-43
- de Oliveira LG, Mechler-Dreibi ML, Almeida HMS, Gatto IRH. Bovine viral diarrhea virus: recent findings about its occurrence in pigs. Viruses 12, 600, 2020. https://doi.org/10.3390/v12060600
- Decaro N. HoBi-like pestivirus and reproductive disorders. Frontiers in Veterinary Science 7, 622447, 2020. https://doi.org/10.3389/fvets.2020.622447
- Deng M, Ji S, Fei W, Raza S, He C, Chen Y, Chen H, Guo A. Prevalence study and genetic typing of bovine viral diarrhea virus (BVDV) in four bovine species in China. PLoS One 10, e0121718, 2015. https://doi.org/10.1371/journal.pone.0121718
- Deregt D, Bolin SR, van den Hurk J, Ridpath JF, Gilbert SA. Mapping of a type 1-specific and a type-common epitope on the E2 (gp53) protein of bovine viral diarrhea virus with neutralization escape mutants. Virus Research 53, 81–90, 1998. https://doi.org/10.1016/S0168-1702(97)00129-9
- Donoso A, Inostroza F, Celedon M, Pizarro-Lucero J. Genetic diversity of bovine viral diarrhea virus from cattle in Chile between 2003 and 2007. BMC Veterinary Research 14, 314, 2018. https://doi.org/10.1186/s12917-018-1641-7
- Dow N, Chernick A, Orsel K, van Marle G, van der Meer F. Genetic variability of bovine viral diarrhea virus and evidence for a possible genetic bottleneck during vertical transmission in persistently infected cattle. PLoS One 10, e0131972, 2015. https://doi.org/10.1371/journal.pone.0131972
- El Omari K, Iourin O, Harlos K, Grimes JM, Stuart DI. Structure of a pestivirus envelope glycoprotein E2 clarifies its role in cell entry. Cell Reports 3, 30–5, 2013. https://doi.org/10.1016/j.celrep.2012.12.001
- Evans CA, Reichel MP. Non-bovine species and the risk to effective control of bovine viral diarrhoea (BVD) in cattle. Pathogens 10, 1263, 2021. https://doi.org/10.3390/pathogens10101263
- Evans CA, Pinior B, Larska M, Graham D, Schweizer M, Guidarini C, Decaro N, Ridpath J, Gates MC. Global knowledge gaps in the prevention and control of bovine viral diarrhoea (BVD) virus. Transboundary and Emerging Diseases 66, 640–52, 2019. https://doi.org/10.1111/tbed.13068
- Falkenberg SM, Dassanayake RP, Terhaar B, Ridpath JF, Neill JD, Roth JA. Evaluation of antigenic comparisons among BVDV isolates as it relates to humoral and cell mediated responses. Frontiers in Veterinary Science 8, 685114, 2021. https://doi.org/10.3389/fvets.2021.685114
- Gates MC, Evans CA, Weir AM, Heuer C, Weston JF. Recommendations for the testing and control of bovine viral diarrhoea in New Zealand pastoral cattle production systems. New Zealand Veterinary Journal 67, 219–27, 2019. https://doi.org/10.1080/00480169.2019.1618745
- Gates MC, Evans CA, Han JH, Heuer C, Weston JF. Practices and opinions of New Zealand beef cattle farmers towards bovine viral diarrhoea control in relation to real and perceived herd serological status. New Zealand Veterinary Journal 68, 92–100, 2020. https://doi.org/10.1080/00480169.2019.1692735
- Gates MC, Evans CA, Weston JF. Practices and opinions of New Zealand veterinarians regarding control of bovine viral diarrhoea. New Zealand Veterinary Journal 69, 274–84, 2021a. https://doi.org/10.1080/00480169.2021.1920511
- Gates MC, Evans CA, Heuer C, Voges H, Weston JF. Temporal trends in bulk tank milk antibody ELISA and PCR test results for bovine viral diarrhoea in New Zealand pastoral dairy herds. New Zealand Veterinary Journal 69, 73–82, 2021b. https://doi.org/10.1080/00480169.2020.1806756
- Hamers C, Di Valentin E, Lecomte C, Lambot M, Joris E, Genicot B, Pastoret P. Virus neutralising antibodies against 22 bovine viral diarrhoea virus isolates in vaccinated calves. The Veterinary Journal 163, 61–7, 2002. https://doi.org/10.1053/tvjl.2001.0638
- Han JH, Weir AM, Weston JF, Heuer C, Gates MC. Elimination of bovine viral diarrhoea virus in New Zealand: a review of research progress and future directions. New Zealand Veterinary Journal 66, 273–80, 2018. https://doi.org/10.1080/00480169.2018.1509030
- Hertig C, Stalder H, Peterhans E. Genetic heterogeneity within the coding regions of E2 and NS3 in strains of bovine viral diarrhea virus. Gene 153, 191–5, 1995. https://doi.org/10.1016/0378-1119(94)00748-H
- Lanyon SR, Hill FI, Reichel MP, Brownlie J. Bovine viral diarrhoea: pathogenesis and diagnosis. The Veterinary Journal 199, 201–9, 2014. https://doi.org/10.1016/j.tvjl.2013.07.024
- Leigh J, Bryant D. Popart: full-feature software for haplotype network construction. Methods in Ecology and Evolution 6, 1110–6, 2015. https://doi.org/10.1111/2041-210X.12410
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Research 49, W293–W296, 2021. https://doi.org/10.1093/nar/gkab301
- Mahony TJ, McCarthy FM, Gravel JL, Corney B, Young PL, Vilcek S. Genetic analysis of bovine viral diarrhoea viruses from Australia. Veterinary Microbiology 106, 1–6, 2005. https://doi.org/10.1016/j.vetmic.2004.10.024
- Miroslaw P, Polak M. Increased genetic variation of bovine viral diarrhea virus in dairy cattle in Poland. BMC Veterinary Research 15, 278, 2019. https://doi.org/10.1186/s12917-019-2029-z
- Miroslaw P, Polak MP. Variability of E2 protein-coding sequences of bovine viral diarrhea virus in Polish cattle. Virus Genes 56, 515–21, 2020. https://doi.org/10.1007/s11262-020-01756-2
- Mosena ACS, Falkenberg SM, Ma H, Casas E, Dassanayake RP, Walz PH, Canal CW, Neill JD. Multivariate analysis as a method to evaluate antigenic relationships between BVDV vaccine and field strains. Vaccine 38, 5764–72, 2020. https://doi.org/10.1016/j.vaccine.2020.07.010
- Mosena ACS, Falkenberg SM, Ma H, Casas E, Dassanayake RP, Booth R, De Mia GM, Schweizer M, Canal CW, Neill JD. Use of multivariate analysis to evaluate antigenic relationships between US BVDV vaccine strains and non-US genetically divergent isolates. Journal of Virological Methods 299, 114328, 2022. https://doi.org/10.1016/j.jviromet.2021.114328
- Muhire BM, Varsani A, Martin DP. SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9, e108277, 2014. https://doi.org/10.1371/journal.pone.0108277
- Nagai M, Ito T, Sugita S, Genno A, Takeuchi K, Ozawa T, Sakoda Y, Nishimori T, Takamura K, Akashi H. Genomic and serological diversity of bovine viral diarrhea virus in Japan. Archives of Virology 146, 685–96, 2001. https://doi.org/10.1007/s007050170139
- Nagai M, Hayashi M, Sugita S, Sakoda Y, Mori M, Murakami T, Ozawa T, Yamada N, Akashi H. Phylogenetic analysis of bovine viral diarrhea viruses using five different genetic regions. Virus Research 99, 103–13, 2004. https://doi.org/10.1016/j.virusres.2003.10.006
- Nelson DD, Duprau JL, Wolff PL, Evermann JF. The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Frontiers in Microbiology 6, 1415, 2015. https://doi.org/10.3389/fmicb.2015.00713
- Newcomer BW, Walz PH, Givens MD, Wilson AE. Efficacy of bovine viral diarrhea virus vaccination to prevent reproductive disease: a meta-analysis. Theriogenology 83, 360–5.e1, 2015. https://doi.org/10.1016/j.theriogenology.2014.09.028
- Newcomer BW, Chamorro MF, Walz PH. Vaccination of cattle against bovine viral diarrhea virus. Veterinary Microbiology 206, 78–83, 2017. https://doi.org/10.1016/j.vetmic.2017.04.003
- Packianathan R, Clough WJ, Hodge A, Holz DK, Huang J, Bryant GL, Colantoni C. Prevention of fetal infection in heifers challenged with bovine viral diarrhoea virus type 1a by vaccination with a type 1c or type 1a vaccine. New Zealand Veterinary Journal 65, 134–9, 2017. https://doi.org/10.1080/00480169.2017.1291376
- Pecora A, Malacari DA, Ridpath JF, Perez Aguirreburualde MS, Combessies G, Odeón AC, Romera SA, Golemba MD, Wigdorovitz A. First finding of genetic and antigenic diversity in 1b-BVDV isolates from Argentina. Research in Veterinary Science 96, 204–12, 2014. https://doi.org/10.1016/j.rvsc.2013.11.004
- Postel A, Smith DB, Becher P. Proposed update to the taxonomy of pestiviruses: eight additional species within the genus Pestivirus, family Flaviviridae. Viruses 13, 1542, 2021 https://doi.org/10.3390/v13081542
- Ridpath JF. Practical significance of heterogeneity among BVDV strains: impact of biotype and genotype on U.S. control programs. Preventive Veterinary Medicine 72, 17–30, 2005. https://doi.org/10.1016/j.prevetmed.2005.08.003
- Ridpath JF. Immunology of BVDV vaccines. Biologicals 41, 14–9, 2013. https://doi.org/10.1016/j.biologicals.2012.07.003
- Ridpath JF, Neill JD, Vilcek S, Dubovi EJ, Carman S. Multiple outbreaks of severe acute BVDV in North America occurring between 1993 and 1995 linked to the same BVDV2 strain. Veterinary Microbiology 114, 196–204, 2006. https://doi.org/10.1016/j.vetmic.2005.11.059
- Ridpath JF, Fulton RW, Kirkland PD, Neill JD. Prevalence and antigenic differences observed between bovine viral diarrhea virus subgenotypes isolated from cattle in Australia and feedlots in the southwestern United States. Journal of Veterinary Diagnostic Investigation 22, 184–91, 2010. https://doi.org/10.1177/104063871002200203
- Russell GC, Zadoks RN, Willoughby K, Bachofen C. Bovine viral diarrhoea virus loses quasispecies diversity rapidly in culture. Microbial Genomics 6, 2020. https://doi.org/10.1099/mgen.0.000343
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4, 406–25, 1987. https://doi.org/10.1093/oxfordjournals.molbev.a040454
- Sakoda Y, Ozawa S, Damrongwatanapokin S, Sato M, Ishikawa K, Fukusho A. Genetic heterogeneity of porcine and ruminant pestiviruses mainly isolated in Japan. Veterinary Microbiology 65, 75–86, 1999. https://doi.org/10.1016/S0378-1135(98)00284-3
- Schweizer M, Stalder H, Haslebacher A, Grisiger M, Schwermer H, Di Labio E. Eradication of bovine viral diarrhoea (BVD) in cattle in Switzerland: lessons taught by the complex biology of the virus. Frontiers in Veterinary Science 8, 702730, 2021. https://doi.org/10.3389/fvets.2021.702730
- Shortall O. Lessons learned for animal health governance from bovine viral diarrhea eradication schemes in Scotland and Ireland. Frontiers in Veterinary Science 9, 956635, 2022. https://doi.org/10.3389/fvets.2022.956635
- Silveira S, Weber MN, Mosena AC, da Silva MS, Streck AF, Pescador CA, Flores EF, Weiblen R, Driemeier D, Ridpath JF, et al. Genetic diversity of Brazilian bovine pestiviruses detected between 1995 and 2014. Transboundary and Emerging Diseases 64, 613–23, 2017. https://doi.org/10.1111/tbed.12427
- Simmonds P, Becher P, Bukh J, Gould EA, Meyers G, Monath T, Muerhoff S, Pletnev A, Rico-Hesse R, Smith DB, et al. ICTV virus taxonomy profile: Flaviviridae. Journal of General Virology 98, 2, 2017. https://doi.org/10.1099/jgv.0.000672
- Stalder H, Hug C, Zanoni R, Vogt HR, Peterhans E, Schweizer M, Bachofen C. A nationwide database linking information on the hosts with sequence data of their virus strains: a useful tool for the eradication of bovine viral diarrhea (BVD) in Switzerland. Virus Research 218, 49–56, 2016. https://doi.org/10.1016/j.virusres.2015.09.012
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10, 512–26, 1993. https://doi.org/10.1093/oxfordjournals.molbev.a040023
- Tamura K, Stecher G, Kumar S. Mega11: molecular evolutionary genetics analysis version 11. Molecular Biology and Evolution 38, 3022–7, 2021. https://doi.org/10.1093/molbev/msab120
- Tautz N, Tews BA, Meyers G. The molecular biology of pestiviruses. Advances in Virus Research 93, 47–160, 2015. https://doi.org/10.1016/bs.aivir.2015.03.002
- Vilcek S, Herring AJ, Herring JA, Nettleton PF, Lowings JP, Paton DJ. Pestiviruses isolated from pigs, cattle and sheep can be allocated into at least three genogroups using polymerase chain reaction and restriction endonuclease analysis. Archives of Virology 136, 309–23, 1994. https://doi.org/10.1007/BF01321060
- Vilcek S, Bjorklund HV, Horner GW, Meers J, Belak S. Genetic typing of pestiviruses from New Zealand. New Zealand Veterinary Journal 46, 35–7, 1998. https://doi.org/10.1080/00480169.1998.36048
- Vilcek S, Alenius S, Paton DJ, Mittelholzer C, Belak S. Genetic clustering of bovine viral diarrhoea viruses in cattle farms: genetic identification and analysis of viruses directly from cattle sera. The Veterinary Journal 158, 33–8, 1999. https://doi.org/10.1053/tvjl.1999.0363
- Vilcek S, Paton DJ, Durkovic B, Strojny L, Ibata G, Moussa A, Loitsch A, Rossmanith W, Vega S, Scicluna MT, et al. Bovine viral diarrhoea virus genotype 1 can be separated into at least eleven genetic groups. Archives of Virology 146, 99–115, 2001. https://doi.org/10.1007/s007050170194
- Vilcek S, Durkovic B, Kolesarova M, Greiser-Wilke I, Paton D. Genetic diversity of international bovine viral diarrhoea virus (BVDV) isolates: identification of a new BVDV-1 genetic group. Veterinary Research 35, 609–15, 2004. https://doi.org/10.1051/vetres:2004036
- Vilcek S, Durkovic B, Kolesarova M, Paton DJ. Genetic diversity of BVDV: consequences for classification and molecular epidemiology. Preventive Veterinary Medicine 72, 31–5, 2005. https://doi.org/10.1016/j.prevetmed.2005.08.004
- Walz PH, Chamorro MF, Falkenberg SM, Passler T, van der Meer F, Woolums AR. Bovine viral diarrhea virus: an updated American College of Veterinary Internal Medicine consensus statement with focus on virus biology, hosts, immunosuppression, and vaccination. Journal of Veterinary Internal Medicine 34, 1690–706, 2020. https://doi.org/10.1111/jvim.15816
- Wernike K, Schirrmeier H, Strebelow HG, Beer M. Eradication of bovine viral diarrhea virus in Germany—diversity of subtypes and detection of live-vaccine viruses. Veterinary Microbiology 208, 25–9, 2017. https://doi.org/10.1016/j.vetmic.2017.07.009
- Yesilbag K, Alpay G, Becher P. Variability and global distribution of subgenotypes of bovine viral diarrhea virus. Viruses 9, 128, 2017. https://doi.org/10.3390/v9060128