
Abstract
1. Among the different in vitro studies recommended by the regulatory agencies, no gold-standard model can easily and directly measure the quantitative CYP450 contributions to drug biotransformation. In this article, we propose an original strategy, called SilensomesTM, to produce human liver microsomes silenced for one specific CYP450, thanks to specific mechanism-based inhibitors (MBI).
2. Using azamulin as a specific CYP3A4 MBI, we demonstrated the proof of concept that CYP3A4 can be totally, specifically (even against 3A5) and permanently (at least for six years) inhibited by our process. Thus, comparing clearance in control and CYP3A4-SilensomesTM, CYP3A4 contributions were determined for 11 CYP3A4 substrates which correlated with known in vivo contributions and revealed accuracy with less than 10% error. In comparison, contributions determined using recombinant human CYP450 (rhCYP450s) were less accurate (more than 10% error for 30% of the tested CYP3A4 substrates).
3. This easy and ready-to-use in vitro method combines the advantages of existing models (specificity of rhCYP450s and representativeness of HLM) without their drawbacks. The same strategy could be used to silence other major CYP450s one-by-one to provide a complete direct CYP450 quantitative phenotyping kit.
Introduction
Pharmacokinetic drug–drug interaction can significantly impact drug safety and efficacy. The prediction of drug–drug interaction risk is now a requisite in the development plan, from the selection of a new drug candidate to the submission of the registration dossier (FDA, Guidance for Industry, 2012, http://www.fda.gov/cder/guidance/index.htm; EMA, Guideline on the investigation of drug interactions, 2012, http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf).
In vitro identification and measurement of the contribution of the major cytochrome P450 enzymes involved in the metabolism of a new drug candidate, a test also called the “CYP450 phenotyping assay,” allows the prediction of the impact of other co-administered drugs (perpetrators) on the pharmacokinetics of the new chemical entity (NCE = victim). Today, a battery of in vitro tests (recommended by the regulatory agencies) is required for this CYP450 phenotyping assay, with each of these tests suffering from numerous limitations (Ogilvie et al., Citation2008; Wienkers & Stevens, Citation2003). For instance, human recombinant CYP450 enzymes (rhCYP450), as an overexpressing system, cannot allow a direct quantitative measurement of the contribution of each CYP450 to the metabolism of a drug, necessitating the use of a relative activity factor (RAF) to extrapolate the true in vitro situation (Proctor et al., Citation2004). Moreover, this in vitro system is not fully representative of the situation in the liver (different electrophysiological environment, host sometimes non-human, truncated protein, expression ratio with human NADPH-cytochrome P450 reductase and cytochrome b5, no heterologous oligomers, etc.) (Parmentier et al., Citation2007). Other in vitro techniques, like human liver microsomes (HLM), require the use of inhibitors, which suffer from a lack of specificity (e.g. antibody anti-CYP450) or have too specific in vitro conditions of use (e.g. chemical competitive CYP450 specific inhibitors) that are not always adapted for the substrate under investigation, especially for low-turnover substrate.
An in vitro model, called SilensomesTM, has been developed to encounter the disadvantages of the current methodologies and should provide advantages over the currently used approaches . Indeed, they are as physiologic and as HLM and as specific as rhCYP450. SilensomesTM correspond to batches of cryopreserved pooled HLM chemically silenced for one specific CYP450 using a mechanism-based inhibitor (MBI). SilensomesTM can be handled like conventional HLM and are ready-to-use for the phenotyping assay of a NCE.
CYP3A4 is the main CYP450 involved in drug metabolism; approximately half the drugs in the market today are CYP3A4 substrates, and co-administration of CYP3A4 inhibitors can lead to serious and even lethal drug–drug interactions (Wienkers & Heath, Citation2005). Consequently, CYP3A4 drug–drug and drug–food interactions are considered as an integral part of drug research from screening to post-marketing stages. For this reason, CYP3A4 was selected for the proof of concept for SilensomesTM.
The objective of this article is to demonstrate that, through insuring complete, long-lasting and specific CYP3A4 inhibition, SilensomesTM constitutes a reliable, easy- and ready-to-use system that allows accurate prediction of the in vivo human CYP3A4 contribution to the metabolism of a specific drug or NCE.
First, in vitro experimental conditions were adjusted to prepare batches of SilensomesTM directed against CYP3A4. Second, the CYP3A4 inhibition potency of these CYP3A4-SilensomesTM and their specificity against the major human CYP450s, i.e. CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP2E1, were evaluated together with the maintenance of this CYP450-quenching property after several months of storage at −80 °C. Finally, CYP3A4-SilensomesTM were then validated using molecules known to be metabolized in humans by CYP3A4 to various degrees.
Materials and methods
Chemicals
Testosterone, 6-β-hydroxytestosterone, midazolam, 1′-hydroxymidazolam, nifedipine, oxidized nifedipine, phenacetin, paracetamol, bupropion, omeprazole, paclitaxel, diclofenac, 4′-hydroxydiclofenac, dextromethorphan, dextrorphan, chlorozoxazone, 6-hydroxychlorzoxazone, amodiaquine, desethylamodiaquine, S-mephenytoin, 4′-hydroxymephenytoin, loperamide, mirtazapine, ketoconazole, buspirone, domperidone, indinavir, ranolazine, simvastatine, tamsulosin, ammonium formiate, NADPH, MgCl2, trizma base, bovine serum albumin were purchased from Sigma Aldrich (St Louis, MO); Bortezomib from Toronto Research Chemicals (North York, Canada); azamulin, hydroxybupropion, 6α-hydroxypaclitaxel from Corning (New York, USA); 5-hydroxyomeprazole from Synfine Research (Richmond Hill, Canada); formic acid, dimethyl sulfoxide and acetonitrile from MERCK (Darmstadt, Germany) and methanol from PROLABO (Fontenay-sous-Bois, France).
Source of HLM and rhCYP450
HLM prepared from different donors were provided from BD Gentest, (Woburn, MD) and Biopredic International (Saint-Grégoire, France). CYP3A4-SilensomesTM and control-SilensomesTM were prepared from BD Gentest HLM or Biopredic HLM. Bactosomes heterologously expressing individual human cytochrome P450 enzymes (rhCYP450) were purchased from Cypex (Dundee, UK).
Determination of azamulin KI and kinact toward CYP3A4
HLM were pre-incubated at 37 °C in a Tris-HCl buffer solution (0.1 M, pH 7.4) supplemented with MgCl2 (5 mM) and azamulin at 0, 0.075, 0.2, 0.4, 1.5, 5 and 10 μM. The inactivation reaction was initiated by the addition of NADPH 1 mM and performed during 0, 1, 2, 5, 7.5, 10 and 15 min. The incubate was then diluted at 1/20 or 1/200 to reach an HLM protein concentration of respectively 0.1 mg/mL or 0.01 mg/mL in Tris-HCl (0.1 M, pH 7.4), MgCl2 (5 mM), and NADPH (1 mM). Remaining CYP3A4 activity was measured on midazolam (50 μM) after 7 min of incubation.
Preparation of CYP3A4-SilensomesTM and homologous controls
HLMs were first pre-incubated with azamulin at 5 μM for 5 min in Tris-HCl (0.1 M, pH 7.4), MgCl2 (5 mM) at 37 °C. NADPH 1 mM was added for a 15 min inactivation reaction. After cooling on ice, the incubate was concentrated using Centricon systems® (Millipore) according to the manufacturer recommendations and then ultracentrifuged. The pellet was suspended in Tris-HCl buffer (0.1 M, pH 7.4) at approximately 20 mg/mL of microsomal proteins. Microsomal proteins were quantified with the BC ASSAY kit (Interchim, Montluçon, France). CYP3A4-silenced HLM generated according to this protocol, namely CYP3A4-SilensomesTM, were stored at −80 °C. At the same time, the homologous control counterparts, namely control-SilensomesTM, were prepared under the same conditions, except that azamulin was replaced by an equivalent volume of solvent. Characterizations (specificity and inhibition potency) were performed on SilensomesTM batches after at least one night of freezing.
Characterization of non-CYP3A4 CYP450 activity maintenance, CYP3A4 inhibition potency and specificity of CYP3A4-SilensomesTM and control-SilensomesTM
Specific CYP450 (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 2E1) substrates (Parmentier et al., Citation2007) were incubated in the presence of native HLM, control-SilensomesTM or CYP3A4-SilensomesTM. CYP450-specific activities were measured under initial rate and saturation conditions (see details in respectively).
Table 1. Initial rate incubation conditions.
Table 2. Saturating incubation conditions.
Microsomal incubations were carried out in a buffered medium (Tris 0.1 M, pH 7.4) with CYP3A4- and control-SilensomesTM supplemented with 5 mM MgCl2 with the specific substrate at 37 °C in a shaking water bath. Reactions were started after a 5 min pre-incubation period by adding 1 mM NADPH, and were terminated (time depending on tested substrate) by protein precipitation; each sample was then vortex mixed and allowed to stand on ice for 10 min after which it was centrifuged. The supernatants were analyzed using liquid chromatography with tandem mass spectrometry detection (LC-MS/MS).
LC/MS/MS analysis of samples
Supernatants were analyzed using liquid chromatography UPLC Acquity system (Waters, Milford, MA) or Agilent HP1100 (Agilent Technologie, Santa Clara, CA) with tandem mass spectrometry detection API4000 (Sciex, Framingham, MA) or Ultima Mass Spectrometer (Waters, Milford, OR) to determine specific metabolite concentrations. Identities were confirmed using authentic standards of parent compounds and metabolites.
Specificity of azamulin for CYP3A4 vs. 3A5
RhCYP3A4 and rhCYP3A5 were incubated with azamulin (5 μM) for 15 min in Tris-HCl (0.1 M, pH 7.4), MgCl2 5 mM, and NADPH 1 mM at 0.35 mg/mL of proteins. The pre-incubate was then diluted 1/10 in the same buffer. Remaining CYP3A4 activity was measured by incubating the diluted aliquot with midazolam (50 μM) or nifedipine (50 μM) for 5 min.
Intrinsic clearance of midazolam and nifedipine was also measured in CYP3A4-SilensomesTM and control-SilensomesTM in the presence and absence of ketoconazole (0.3 μM). The incubation conditions were similar to those described above for SilensomesTM except for the CYP3A substrates and microsomal protein concentrations (10 μM midazolam with 0.3 mg protein/mL and 20 μM nifedipine with 0.3 mg protein/mL). Metabolites are measured after 5–60 min. For these assays, terminated incubation mixtures were treated and analyzed as described above for SilensomesTM incubations.
Intrinsic clearance measurement in CYP3A4-SilensomesTM and control-SilensomesTM
Bortezomib, loperamide, midazolam, mirtazapine, nifedipine, buspirone, domperidone, indinavir, ranolazine, simvastatine, tamsulosin, and omeprazole at 0.1 μM were incubated in triplicates with CYP3A4-SilensomesTM and control-SilensomesTM at protein concentrations between 0.02 and 2 mg/mL, allowing optimal measurement of their intrinsic clearance (linearity with protein concentration). The disappearance of drugs was determined after 0, 7, 17, 30 and 60 min. Incubation conditions, treatment of samples and analysis were similar to those described above for SilensomesTM incubations.
Intrinsic clearance measurement in rhCYP450 and determination of relative activity factor
In order to normalize formation rates obtained with rhCYP450, the relative activity factor (RAF) was used (Crespi & Penman, Citation1997; Venkatakrishnan et al., Citation1998) and was calculated with the following equation:
(1)
RhCYP450s were screened for their metabolic activities on phenacetin (CYP1A2) at 4.5 μM, paclitaxel (CYP2C8) at 4 μM, diclofenac (CYP2C9) at 4 μM, omeprazole (CYP2C19), at 5 μM, dextromethorphan at 5 μM, testosterone at 30 μM and nifedipine at 5 μM for CYP3A4. The following RAF values were established for CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 (determined respectively with testosterone and nifedipine): 90, 69, 3.1, 75, 26, 9.8 and 49.
Drugs described above (0.1 μM) were incubated with rhCYP450. The concentration of bactosomal protein incubated with drugs was corrected by RAF. Intrinsic drug clearances (substrate disappearance) were determined after 0, 7, 17, 30 and 60 min. Treatment of samples and analysis were similar to those described above for SilensomesTM incubations.
Data analysis for estimation of KI and kinact
The natural logarithm of the percentage of remaining activity was plotted against pre-incubation time for each concentration of inhibitor tested. The slopes of the linear portion of each plot were determined (kobs) and the slope vs. inhibitor concentration data set was fitted to determinate KI and kinact as previously described with the following equation using XLfit (Perloff et al., Citation2009):
(2)
where [I] is the inhibitor concentration, kobs is the inactivation rate for the corresponding [I], kinact is the maximal inactivation rate constant, and KI is the inhibitor concentration that produces half the maximal rate of inactivation.
Data analysis for estimation of Clint
The in vitro intrinsic clearance (Clint, mL/min mg protein) was determined by substrate depletion (where substrate concentration gave the maximal clearance), using the following equation:
(3)
where “Slope” is the elimination rate constant (min−1) for exponential substrate loss, “Vol” is the incubation volume (mL), and “prot” is the microsomal protein (mg) in incubation.
Estimation of CYP3A4 contribution to drug metabolism
For the SilensomesTM approach, the CYP3A4 contribution to drug metabolism was estimated by the ratio of intrinsic clearance values in CYP3A4-SilensomesTM (Clint 3A4SiL, mL/min g HLM protein) and in homologous control-SilensomesTM (Clint cSiL, mL/min g HLM). The relative contribution of CYP3A4 was estimated with the following equation:
(4)
The “limit of detection” for CYP3A4 contribution depends both on the high selectivity of CYP3A4-silensomes and the experimental uncertainty. In the present work, the experimental variability (both from the in vitro incubation and analytical method) for the measurement of the intrinsic clearances was set at 20%. Therefore, one can consider that at the lowest fm, CYP3A4 silensomes can be reliably determined to be 20% (“limit of detection” for CYP3A4 contribution). Any CYP3A4 inhibition lower than 20% was not deemed as significant to consider a reasonable net CYP3A4 contribution.
For the rhP450 approach, the contribution of CYP3A4 to drug metabolism was calculated by the ratio of intrinsic clearance values in rhCYP3A4 (Clint 3A4, mL/min mg protein) and the sum of all rhCYP450 tested (Clint all CYP’s mL/min mg protein) CYP1A2 + CYP2C8 + CYP2C9 + CYP2C19 + CYP2D6 + CYP3A4.
(5)
Estimation of fm in vivo
The in vivo CYP3A4 contributions (fm, CYP3A4) to the metabolism of the drugs selected for the validation of the CYP3A4-SilensomesTM were calculated using the Rowland and Matin equation (Rowland & Matin, Citation1973):
(6)
where [I] is the inhibitor concentration available to the enzyme, KI the constant of inhibition for the inhibitor, and subscript I indicates the presence of the inhibitor. It was assumed that the extrahepatic clearance of the drugs selected was negligible.
Assuming that the design of the clinical studies allowed complete inhibition of CYP3A4, the above equation could be simplified as:
(7)
Therefore
(8)
The AUC ratio (R) of each drug in the presence vs. absence of a strong CYP3A4 inhibitor were obtained from the literature (). For nifedipine, as no drug–drug interaction study with a strong CYP3A4 inhibitor has been reported, the moderate CYP3A4 inhibitor, diltiazem, was used.
Statistical analysis
Quantitative data were expressed as means ± standard error to the mean (SEM). They were statistically analyzed using Student’s t-test. Criteria of significance were: * for p < 0.05; ** for p < 0.01; *** for p < 0.001. All statistical analyses were performed using GraphPad Prism software 5.0 (GraphPad Software, Inc., San Diego, CA).
The accuracy of the prediction was assessed and compared from root mean-squared error (RMSE) and average fold error, calculated with the following equations (Obach et al., Citation1997; Sheiner & Beal, Citation1981):
(9)
(10)
Results
Preparation of the CYP3A4-SilensomesTM and control-SilensomesTM
The in vitro experimental conditions were set up to prepare batches of CYP3A4-SilensomesTM corresponding to HLM devoid of CYP3A4 activity. The first step consisted in choosing a specific MBI able to fully and rapidly inactivate the CYP3A4 in a batch of HLM. A comparison of the different MBIs reported in the literature according to their KI, kinact and specificity regarding inhibition of other CYP450 led to the selection of azamulin (). Among the different types of CYP3A4 MBI, azamulin and CYP3cide had higher kinact parameters, allowing quick inactivation. However, a larger range of enzyme specificity data is available for azamulin, with 18 CYP450s tested in a competitive inhibition model and 7 CYP450s tested in a pre-incubation model.
Table 3. Comparison of KI, kinact and reported specificity of the more potent CYP3A4 MBIs.
Table 4. Individual contribution of CYP3A4 pathways to oxidative metabolism of 12 drugs using CYP3A4-Silensomes and CYP3A4 recombinant models.
First, the azamulin concentration was optimized to allow maximal CYP3A inhibition without inhibition of the other major CYP450s (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 2E1). Second, azamulin pre-incubation time was extended to ensure maximal CYP3A inhibition, but was kept short enough to maintain all CYP450 activities at their initial levels. With this aim, the inhibition parameters, KI and kinact of azamulin were measured using midazolam as a CYP3A substrate. Experimental conditions were chosen to ascertain proper characterization of MBI (limiting residual direct reversible inhibition of azamulin) and following the general rules described by Zimmerlin and colleagues (Zimmerlin et al., Citation2011), in particular, a sufficient dilution of the inactivator used in the pre-incubation, a short incubation time of midazolam, and a high-substrate concentration (here respectively at least 1/20 dilution, 7 min and 50 μM of midazolam, i.e. >10-fold the Km for CYP3A-dependent midazolam-1′-hydroxylase activity). The microsomal protein concentration during the pre-incubation period was fixed at 2 mg/mL to mimic SilensomesTM future production.
From the time- and concentration-dependent loss of midazolam-1′-hydroxylase activity curves, initial inactivation rates (kobs) were calculated and plotted against azamulin concentrations (). The non-linear regression analysis of EquationEquation (2)(2) (see “Material and methods” section) allowed calculation of a KI and kinact for azamulin for CYP3A-dependent midazolam-1′-hydroxylase activity of 0.920 ± 0.378 μM and 0.449 ± 0.048 min−1, respectively. Perloff et al. (Citation2009) measured a similar kinact value for CYP3A4, confirming our choice of azamulin for the generation CYP3A4-SilensomesTM, which display potent and sustainable CYP3A inhibition.
Figure 1. Mechanism based inactivation of CYP3A4-1′-hydroxymidazolam activity by azamulin in HLM. CYP3A4 midazolam-1′-hydroxylase remaining activity was measured following various pre-incubation times with azamulin at different concentrations and incubation with midazolam (50 μM). KI and kinact were determined as described in materiel and methods. Points are means ± SEM of three independent experiments by fitting these data with the equation kobs= (kinact*I)/(I+KI).
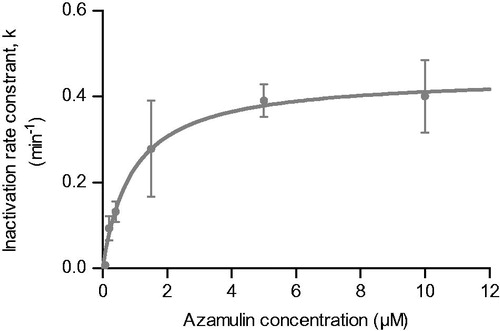
From these inhibition parameters and using EquationEquation (2)(2) , the percentages of remaining CYP3A activity were estimated for various scenarios of azamulin concentrations and pre-incubation times in order to select the best conditions, i.e. a 15 min pre-incubation of 5 μM azamulin. In an on-line, sequential process (MBI pre-incubation followed by substrate incubation), this led to an effective inhibition of CYP3A midazolam-1′-hydroxylase activity compared to control microsomes, i.e. 77% (data not shown).
These conditions appear appropriate since Stresser et al. (Citation2004) showed that a pre-incubation of azamulin at 5 μM for 10 min in HLM did not impact the activities of CYP1A2, 2C8, 2C9, 2C19, 2D6 and 2E1. Therefore, these experimental conditions were retained for the preparation of the CYP3A4-SilensomesTM.
The concept behind SilensomesTM is to obtain a batch of HLM that is chemically silenced for one CYP450, here CYP3A4, and ready-to-use for the turnover measurement of a NCE in comparison with the control batch. The final product should therefore be at a standard microsomal concentration close to 20 mg/mL. In order to minimize nonspecific azamulin binding, the inactivation step was performed at a lower concentration, i.e. 2 mg/mL. Subsequently, the product of HLM inactivation was concentrated to reach the usual HLM protein concentration of 20 mg/mL by successive filtration/centrifugations and ultracentrifugation, which also allowed washing of the SilensomesTM. Optimization of this process led to good reproducibility (CV = 8%, n = 5).
After protein precipitation, the total azamulin concentration remaining at the end of the preparation was quantified by LC-MS/MS and represented approximately 10% of the initial azamulin concentration (i.e. 0.467 ± 0.064 μM, n = 3), thus minimizing potential reversible inhibition. In parallel, control-SilensomesTM were also prepared using the same process, except for the addition of azamulin during the inactivation step.
SilensomesTM preparation maintained non-targeted CYP450 activity
The maintenance of CYP450 activity during the process was studied by comparing the activity of native HLM and control-SilensomesTM for CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4 by incubating their substrates at a concentration close to their respective Km. shows that the whole process of SilensomesTM production did not significantly modify CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2E1 and CYP3A4. For CYP2C19 and CYP2D6, a weak (less than 25%) yet statistically significant change in activity was observed. Therefore, it was concluded that the process described here allows good preservation of CYP450 activity.
Figure 2. Impact of the SilensomesTM preparation process on native CYP450 activities. The different CYP450 activities of control-SilensomesTM were compared to native HLM by incubating CYP450-specific substrates. The substrates were incubated at concentrations close to their respective Km: phenacetin at 4.5 μM (CYP1A2), bupropion at 50 μM (CYP2B6), paclitaxel at 4 μM (CYP2C8), diclofenac at 4 μM (CYP2C9), omeprazole at 5 μM (CYP2C19), dextromethorphan at 5 μM (CYP2D6), chlorzoxazone at 40 μM (CYP2E1) and for nifedipine (CYP3A4 [a]) at 10 μM, testosterone (CYP3A4 [b]) at 30 μM, and midazolam (CYP3A4 [c]) at 0.5 μM. (1) One-way Anova test comparing CYP3A4 inhibition rate to the other CYP450 inhibition rates. (2) t-test comparing the non-CYP3A4 inhibition rate to zero.
![Figure 2. Impact of the SilensomesTM preparation process on native CYP450 activities. The different CYP450 activities of control-SilensomesTM were compared to native HLM by incubating CYP450-specific substrates. The substrates were incubated at concentrations close to their respective Km: phenacetin at 4.5 μM (CYP1A2), bupropion at 50 μM (CYP2B6), paclitaxel at 4 μM (CYP2C8), diclofenac at 4 μM (CYP2C9), omeprazole at 5 μM (CYP2C19), dextromethorphan at 5 μM (CYP2D6), chlorzoxazone at 40 μM (CYP2E1) and for nifedipine (CYP3A4 [a]) at 10 μM, testosterone (CYP3A4 [b]) at 30 μM, and midazolam (CYP3A4 [c]) at 0.5 μM. (1) One-way Anova test comparing CYP3A4 inhibition rate to the other CYP450 inhibition rates. (2) t-test comparing the non-CYP3A4 inhibition rate to zero.](/cms/asset/30c5af1c-7b78-4734-9cfe-2c51dd4001d0/ixen_a_1208854_f0002_b.jpg)
CYP3A inhibition potency and specificity
CYP3A4-SilensomesTM were compared with control-SilensomesTM for CYP3A4 activity, using several CYP3A4 substrates (testosterone, midazolam and nifedipine), and for the major non-CYP3A4 CYP450 activity to check both CYP3A inhibition potency and specificity, two critical parameters for this type of CYP450-phenotyping model. shows that testosterone-6β-hydroxylase, midazolam-1′-hydroxylase and nifedipine-oxydase activities were inhibited by 96%, 83%, and 81%, respectively, in the CYP3A4-SilensomesTM compared to control-SilensomesTM, when the CYP3A-specific substrates were incubated at concentrations close to their respective Km. Similarly, testosterone-6β-hydroxylase, midazolam-1′-hydroxylase and nifedipine-oxydase activities were inhibited by 100%, 81% and 86%, respectively, when the substrates were incubated at a saturating concentration ().
Figure 3. Impact of CYP3A4 quenching by azamulin in CYP3A4-SilensomesTM towards CYP450 activities measured at initial rate (A) and saturating conditions (B). The different CYP450 activities of CYP3A4-SilensomesTM and its counterpart control were compared using the following CYP450 probe substrates: phenacetin for CYP1A2, bupropion for CYP2B6, paclitaxel for CYP2C8, diclofenac for CYP2C9, omeprazole (A) or S-mephenytoin (B) for CYP2C19, dextromethorphan for CYP2D6, chlorzoxazone for CYP2E1, and nifedipine (a), testosterone (b) and midazolam (c) for CYP3A4 used at either concentrations close to Km (A) (see details in ) or Vmax (B) (see details in ). Inhibition percentages are means ± SEM of experiments performed on two (B) or five (A) independent SilensomesTM batches in duplicate (A) or triplicate (B). (1) is the results of Anova’s tests comparing non-CYP3A4 inhibitions to be different from CYP3A4 inhibitions. (2) corresponds to statistically testing inhibition rates to be different from 0.
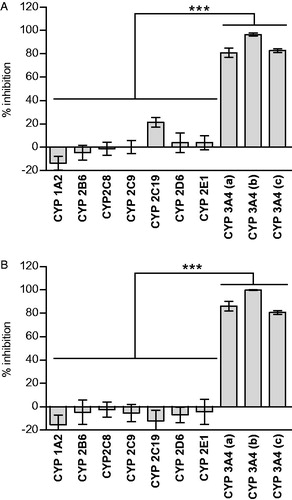
Then, to check the selectivity/specificity of the CYP3A4 inhibition, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP2E1 activities were measured in CYP3A4-SilensomesTM and their control counterparts using specific substrates at initial rate and saturating conditions. For each CYP450, inhibitions in CYP3A4-SilensomesTM vs. control-SilensomesTM were calculated. At concentrations close to their Km, except for omeprazole, all inhibitions were not significantly different from zero (). Omeprazole-5-hydroxylase activity displayed significant inhibition, which was explained by a CYP3A4 contribution to its metabolism ()(McGinnity et al., Citation2000). In contrast, CYP2C19-dependent S-mephenytoin-6-hydroxylase activity at saturating conditions () was not significantly impacted in CYP3A4-SilensomesTM, confirming the specificity of the model towards CYP2C19. In the same way, at saturating concentrations, no inhibitions were significantly different from zero (). Thus, we concluded that CYP3A4 activity was significantly inhibited (more than 80%) and non-CYP3A4 activity was preserved in CYP3A4-SilensomesTM.
CYP3A4 vs. CYP3A5 specificity
As no specific CYP3A5 substrate was available, the inhibition selectivity of azamulin for CYP3A4 vs. CYP3A5 was assessed using recombinant human CYP3A4 and CYP3A5 bactosomes (rhCYP3A4 and rhCYP3A5) that were pre-incubated with azamulin and next diluted by 1/10 to mimic preparation conditions of the CYP3A4-SilensomesTM. These pre-incubates were then incubated with midazolam (50 μM) and nifedipine (50 μM) (no significant CYP3A5 testosterone 6β-hydroxylase activity, as already reported in the literature (Patki et al., Citation2003)) at different time points in order to measure the intrinsic clearance of the CYP3A-specific reactions (midazolam-1′-hydroxylase and nifedipine-oxydase). As expected, both the midazolam-1′-hydroxylase and nifedipine-oxydase CYP3A-specific activities were fully inhibited when azamulin was pre-incubated with CYP3A4 bactosomes, showing that the preparation conditions of the SilensomesTM ensured maximal inhibition of CYP3A4. Conversely, midazolam-1′-hydroxylase activity was not significantly inactivated by azamulin in CYP3A5 bactosomes. CYP3A5-dependent activity of nifedipine was only inhibited by 25% on average ().
Figure 4. Specificity of azamulin for CYP3A4 vs. CYP3A5. (A) Azamulin was pre-incubated (5 μM, 15 min) with recombinant CYP3A4 and CYP3A5, after a 1/10 dilution, inhibition of CYP3A4 and CYP3A5-mediated midazolam-1′-hydroxylase and nifedipine-oxydase activities were measured using midazolam (50 μM) and nifedipine (50 μM). Midazolam (10 μM) (B) and nifedipine (20 μM) (C) disappearance kinetics at concentration close to their respective Km were followed in CYP3A4-SilensomesTM and control-SilensomesTM in the presence or absence of ketoconazole (0.3 μM).
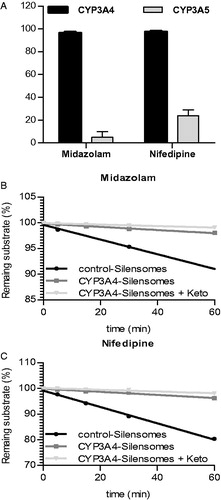
Inhibition of CYP3A4 in CYP3A4-SilensomesTM could be considered as complete on the basis of CYP3A4-dependent testosterone metabolism (). Consequently remaining midazolam-1′-hydroxylase and nifedipine-oxydase activities in CYP3A4-SilensomesTM (from 15% to 20%; ) were related to non-CYP3A4 activity and more likely CYP3A5 activity. In this context, as already described by Tseng et al. (Citation2014), the overall CYP3A contribution to substrate metabolism could be determined by the impact of ketoconazole on these latter substrates. The intrinsic clearances of midazolam-1′-hydroxylase and nifedipine-oxydase activities were measured in CYP3A4-SilensomesTM in the presence or absence of ketoconazole at concentrations known to only inhibit CYP3A4 and CYP3A5 (Gibbs et al., Citation1999; Stresser et al., Citation2004). show an additional 15% and 22% inhibition in the activities of midazolam-1′-hydroxylase and nifedipine-oxydase in the presence of ketoconazole. Thus, the majority of the remaining non-CYP3A4 activity in CYP3A4-SilensomesTM is most likely to be related to the contribution of the CYP3A5 that was not mechanistically inhibited by azamulin.
Altogether these results demonstrate the specificity of azamulin for CYP3A4 vs. CYP3A5 in the CYP3A4-SilensomesTM manufacturing process, and this can be used to differentiate CYP3A4 and CYP3A5.
CYP3A4 irreversible inhibition is maintained in SilensomesTM in storage conditions at −80 °C
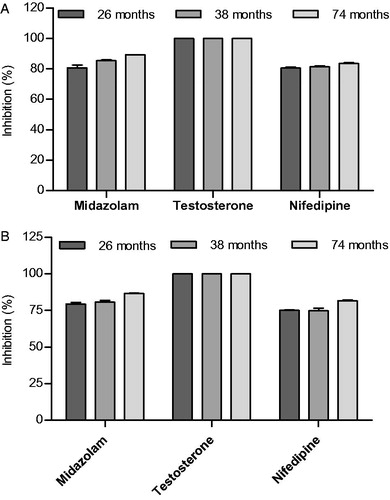
For the confident use of SilensomesTM in phenotyping assays, maintenance of both the enzyme activity of non-targeted CYP450 and inactivation of CYP3A4 is expected to last for several years storage at −80 °C. It is well known that activities of CYP450s are well conserved over many years with cryopreservation (Yamazaki et al., Citation1997). It was critical to prove that CYP3A4 inactivation was also maintained under such conditions. Testosterone-6β-hydroxylase, midazolam-1′-hydroxylase and nifedipine-oxydase activities were measured at initial rate and at saturating conditions after 26, 38 and 74 months of storage at −80 °C. shows that CYP3A4 inhibition did not change up to six years after SilensomesTM cryopreservation for all three enzyme activities, and these were equivalent to the values measured on day one after SilensomesTM preparation ().
Figure 5. Preservation of CYP3A4 inactivation in CYP3A4-SilensomesTM measured at initial rate (A) and saturating conditions (B). Batches of CYP3A4-SilensomesTM and homologous control-SilensomesTM were prepared and cryopreserved at −80 °C for 26, 38 and 74 months. Nifedipine-oxydase, testosterone-6ß-hydroxylase, midazolam-1′-hydroxylase CYP3A4 activities were measured on each of these batches by incubating nifedipine (10 μM and 200 μM), testosterone (30 μM and 100 μM) and midazolam (0.5 μM and 50 μM) at concentrations close to their respective Km (A) and at Vmax (B). For every batch, the percentage of CYP3A4 activity inhibition was determined by comparison of the CYP3A4-SilensomesTM and its homologous control-SilensomesTM.
Comparison of relative contribution of CYP3A4 for 12 reference drugs using CYP3A4-SilensomesTM and rhCYP450 models
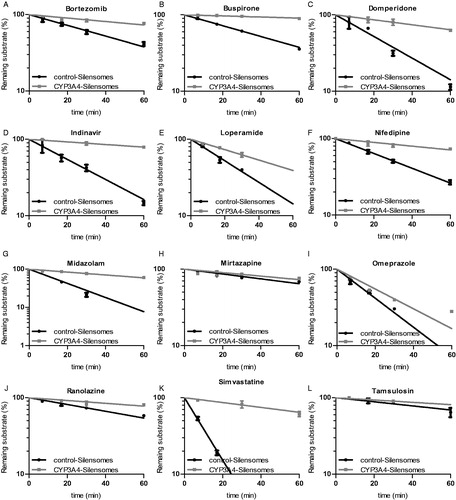
Based on in vitro and/or in vivo literature data, bortezomib, loperamide midazolam, mirtazapine, nifedipine, buspirone, domperidone, indinavir, ranolazine, simvastatine, tamsulosin, and omeprazole were selected as test substrates to validate the CYP3A4-SilensomesTM model under phenotyping assays conditions. These substrates were thoroughly characterized in previous in vitro and/or clinical studies and known to have low (omeprazole, mirtazapine), intermediate (loperamide, ranolazine, domperidone, tamsulosin) and high (nifedipine, midazolam, bortezomib, buspirone, indinavir, simvastatine) CYP3A4 contributions to their hepatic metabolism ().
The intrinsic clearance of the twelve drugs was measured in CYP3A4-SilensomesTM and control-SilensomesTM based on a substrate-disappearance kinetics. The contribution of CYP3A4 for each drug was deduced from the ratio of both clearances ( and ). As expected, the lowest CYP3A4 contribution was found for mirtazapine (24%) and omeprazole (37%). Loperamide, ranolazine, domperidone, tamsulosin displayed intermediate CYP3A4 contributions, with respectively, 53%, 57%, 65%, 45% inhibition of the intrinsic clearance in CYP3A4-SilensomesTM compared to its control counterpart. Bortezomib, nifedipine, midazolam, buspirone, indinavir and simvastatine were the most sensitive CYP3A4 substrates with 73%, 77%, 88%, 90%, 86% and 91% CYP3A4 contributions to their oxidative metabolism, respectively.
Figure 6. Drug-disappearance kinetics in CYP3A4-SilensomesTM and homologous control SilensomesTM. The indicated drugs were incubated at 0.1 μM and their disappearance followed for 60 min in CYP3A4-SilensomesTM and control-SilensomesTM. Percentages of remaining substrates are mean ± SEM of three independent experiments and were linearly fitted to determine clearance.
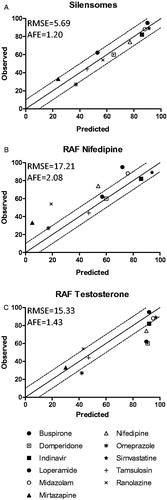
The same 12 drugs were also analyzed by the conventional rhCYP450 model normalized with the RAF approach (Crespi & Penman, Citation1997; Venkatakrishnan et al., Citation1998). We established that RAF values are compound specific, with an RAF of 9.8 for testosterone-6β-hydroxylase and 49 for nifedipine-oxydase activity. Both values were used for contribution calculations (). Overall, the nifedipine RAF led to a lower CYP3A4 contribution than the testosterone RAF.
Finally, the predictions from the three strategies were compared to values obtained in vivo (for 11 drugs) in order to identify the most accurate method (). As illustrated in , SilensomesTM enable a contribution prediction with less than 10% error and a correlation slope not significantly different from 1. In contrast, for rhCYP3A4, whatever the RAF used (i.e. nifedipine or testosterone), the prediction was far less precise, with more than 10% error for 4 of the 11 compounds using the testosterone RAF and for 5 of the 11 compounds using the nifedipine RAF. RMSE values also confirm that SilensomesTM lead to more accurate predictions than rhCYP3A4 (). In conclusion, CYP3A4-SilensomesTM were revealed to be, under test conditions, the most accurate models for easily determining in vitro the in vivo CYP3A4 contribution to drug clearance.
Figure 7. Correlation between observed in vivo and predicted CYP3A4 contribution to drug metabolism using the SilensomesTM direct quantitative model (A) or indirect rhCYP3A4 approach with the relative activity factors (RAFs) of nifedipine (B) and testosterone (C). Observed in vivo CYP3A4 contributions were plotted according to the contributions predicted in vitro with SilensomesTM (A) and rhCYP3A4 normalized with nifedipine RAF (B) or with testosterone RAF (C). All contributions values are from . The solid line indicates the line of perfect correlation and the dotted line the ±10% error interval.
Discussion
For drug approval, regulatory agencies − such as the Food and Drug Administration (FDA), European Medicines Agency (EMA) and Pharmaceuticals and Medical Devices Agency (PMDA) − require that major transporters and metabolizing enzymes, especially CYP450s, be identified and their respective contribution known in order to predict potential drug–drug interactions with co-medications.
Several models are available to phenotype CYP450s, each presenting advantages and drawbacks which have been largely described in the literature (Parmentier et al. Citation2007; Ogilvie et al., Citation2008; Wienkers & Stevens, Citation2003). Among them, HLM and rhCYP450 are the most used, but none is recognized as the gold standard and usually agencies, such as the EMA and PMDA, recommend the use of both models.
In addition, the specific contribution of the different CYP450s necessitates the use of chemical or biological inhibitors. For competitive inhibitors, relative KI and Km can impact the result (i.e. inhibition decreasing with increasing substrate concentration), and inhibitor metabolism and microsomal binding also have to be taken into account. Therefore, for constant and complete inhibition, the incubation conditions must be adapted to each NCE/inhibitor couple (Wang et al., Citation2000). For example, the inhibition of CYP3A4 by ketoconazole, a recommended specific inhibitor, is highly dependent on microsomal concentration due to its very low fu, mic (Raungrut et al., Citation2010). Alpha-naphthoflavone, a recommended CYP1A2 inhibitor, is highly metabolized and its use is not appropriate in co-incubation with a low turnover substrate (Lee et al., Citation1994). Moreover, these chemical inhibitors are usually not completely specific, nor satisfactory. For example, quercetine, a CYP2C8 inhibitor recommended by the FDA, also inhibits CYP3A4. The use of antibodies as biological inhibitors often results in incomplete inactivation; antibodies are not available for all CYP450s, and cross-reactions can impact several CYP450s. For example, there is no antibody able to distinguish CYP3A4 and CYP3A5. Finally, due to all their respective drawbacks, the co-incubation of chemical or biological inhibitors with a NCE is time-consuming and costly requiring parallel assays under identical conditions with reference CYP450 substrates.
A correlation between the turnover of NCE and reference CYP450 activities across a bank of HLM prepared from several individual donor livers can also be used instead of inhibitors in a CYP450 phenotyping assay. However, this method is only qualitative (Parmentier et al., Citation2007).
Direct scaling of the amount of CYP450 present in the rhCYP450 preparation compared to its abundance in liver fails to predict CYP450-mediated clearance. RAF determined from intrinsic clearance is considered to be a better normalization procedure (Emoto & Iwasaki, Citation2007). The lack of correlation between enzyme amount and activity in rhCYP450 compared to liver and HLM can be explained by several factors, such as non-human hepatocyte membrane environment, expression ratios with co-enzyme and co-factor, absence of cooperativity between several CYP450 within hetero-oligomers and the absence of post-translational modifications. All these points lead to substrate-dependent Vmax, but also variations in Km values between rhCYP450 and HLM. This is exemplified in with several CYP3A4 substrates characterized for the Km of specific reactions in HLM and rhCYP3A4. Consequently, for each rhCYP450 preparation, RAF between HLM and rhCYP450 has to be determined on a characterized, specific substrate and then used to extrapolate the NCE’s clearance from rhCYP450 to HLM. However, several articles relate that for the same CYP450, the activity of rhCYP450 and HLM is not always impacted in the same manner for all substrates. For example, for CYP1A2, Venkatakrishnan and colleagues reported a 4-fold difference in the RAF when comparing methoxyresorufin or phenacetin (Lipscomb & Poet, Citation2008; Venkatakrishnan et al., Citation2000). The same phenomenon was identified for CYP2C9 using diclofenac, tolbutamine and S-warfarin (Crewe et al., Citation2011; Kumar et al., Citation2006; Locuson et al., Citation2007) and with CYP3A4, a 2-fold difference between nifedipine and testosterone (Emoto & Iwasaki, Citation2007; Patki et al., Citation2003) is evident. In our study, a 5-fold difference was observed (RAF of 9.8 for testosterone and 49 for nifedipine). Such a difference might be related to the substrate-dependent inhibition observed with CYP3A4 (Kenworthy et al., Citation1999) and to the fact that CYP3A4 is an enzyme that acts at multiple sites.
Table 5. Difference CYP3A4 substrates’ Km on rhCYP3A4 vs. HLM.
The use of “testosterone RAF” allowed was in agreement with in vivo literature data for 7 over the 11 drugs tested, but sometimes “nifedipine RAF” was more accurate, as in the case of loperamide. SilensomesTM results are easier to interpret. Furthermore, it was demonstrated that the sum of normalized clearances for loperamide, bortezomib and nifedipine in each individual CYP450 tested was unexpectedly higher than the drug clearance measured in native HLM (no issue of linearity with time and proteins, ). All these examples tend to prove that, although useful, rhCYP450 model is not as reliable in predicting the in vivo situation as HLM models and can lead to inconclusive results.
Finally, it is important to remember that RAF measurements have to be repeated for new batches of rhCYP450 or HLM and can only be measured if a CYP450-specific substrate is available, which is not the case for some CYP450s such as CYP3A5 or CYP1A1.
In this context, the main aim of this article is to propose an original, convenient and ready-to-use tool: SilensomesTM. SilensomesTM consist of HLM chemically silenced for individual CYP450s, thanks to a specific MBI that allows the maintenance of CYP450 activities. SilensomesTM display both the specificity and selectivity of rhCYP450s together with the physiologically representative nature of HLM. The proof of concept was demonstrated with CYP3A4, the predominant CYP450 involved in the metabolism of around more than 50% of marketed drugs.
A full characterization of the CYP3A4-SilensomesTM was performed using conventional probe substrates for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. Three substrates were used to characterize CYP3A4 activity because of the dependent inhibition observed with CYP3A4 (Kenworthy et al., Citation1999; Stresser et al., Citation2004).
In summary, the selected conditions of inactivation (15 min pre-incubation with 5 μM azamulin) and the different steps of the washing/concentration process allow numerous criteria essential for a proper in vitro model used in CYP450 phenotyping study to be met:
CYP3A4-SilensomesTM ensured complete inactivation of CYP3A4 enzyme as shown in , where the testosterone, midazolam and nifedipine CYP3A4-dependent metabolism was fully abolished in accordance with Stresser et al.’s results, but in a more conventional on-line sequential process. CYP3A4 inactivation was maintained in CYP3A4-SilensomesTM even at saturating substrate concentrations as shown in and, as expected with MBI properties, where there is no protection by the substrate contrary to competitive inhibitors. Using this model, CYP3A4 contribution can therefore be measured at all substrate concentrations.
Full CYP3A4 inactivation was maintained for up to at least 6 years when CYP3A4-SilensomesTM were stored at −80 °C () showing that the covalent binding of the azamulin intermediate metabolite to CYP3A4 is fully stable for a long time and even after freezing/thawing cycles (Yamazaki et al., Citation1997). This property is particularly important for SilensomesTM to be stored and ready-to-use as a standard batch of HLM.
CYP3A4-SilensomesTM were selective towards at least seven other major CYP450 (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 2E1) as shown in . Azamulin selectivity has already been studied for 18 CYP450s in a competitive inhibition model using rhCYP450 (Stresser et al., Citation2004). The lowest IC50 (other than for CYP3A4) was for CYP3A5 with 0.38 μM. The author pointed out that the IC50 ratio between CYP3A4 and CYP3A5 would probably not be sufficient to differentiate these two enzymes in a phenotyping assay. The preparation of the SilensomesTM includes a filtration/ultracentrifugation step that allows washing the inactivated HLM to get rid of the majority of the pre-incubate constituents (solvent, co-factors, etc.), especially free azamulin that could act as a reversible inhibitor towards other CYP450s during the incubation of the test items. Consequently, only 10% of the initial azamulin concentration (5 μM) remains at the end of the process. The expected azamulin concentration in an incubation at 2 mg/mL of HLM is therefore 0.05 μM (even less as free azamulin), which is far lower than the IC50 for CYP3A5. This explains the specificity of SilensomesTM for CYP3A4 shown in and and highlights the value of this model when compared to conventional on-line sequential co-incubations with CYP450 inhibitors.
As shown in and , SilensomesTM allow the determination of CYP3A4 clearance and contribution for substrates with high (midazolam or simvastatin) to low (mirtazapine or tamsulosin) metabolic clearance and with high (nifedipine, midazolam, bortezomib, buspirone, indinavir, simvastatine), moderate (loperamide, ranolazine, domperidone, tamsulosin) and low (mirtazapine and omeprazole) CYP3A4 contribution. In addition, contributions determined with SilensomesTM correlated well with either in vitro in HLM or in vivo data. For the 11 substrates studied, the contribution displayed a ±10% accuracy, which was not the case for contributions obtained from rhCYP3A4 with either nifedipine or testosterone RAF ().
Conclusions
In this article, we showed that CYP3A4-SilensomesTM displayed many advantages compared to current phenotyping assays (microsomes ±CYP inhibitors, recombinant human CYP450, etc.). First, due to the properties of suicide inhibitors, the high potency of CYP3A4 inactivation, and the high selectivity azamulin, CYP3A4-SilensomesTM allowed a direct quantification of CYP3A4 contribution to the oxidative metabolism of a NCE as it was shown for 12 drugs with various CYP3A4 fm (ranging from 24% to 91%). As human liver microsomes, they are more representative of the in vivo situation than recombinant human CYP450. Finally, as they are priorly prepared and characterized for their CYP3A4 inactivation and CYP450 selectivity, they are ready- and easy-to-use and reduce time and cost of experiments (no positive control nor CYP3A4 actvity measurement required). Thus, in this proof of concept we have made an original, and reliable, tool to ensure an accurate prediction of the pharmacokinetic drug–drug interaction risk. This model is fully validated for CYP3A4 and the same strategy can be applied to other CYP450 with adapted specific MBI.
Declaration of interest
The authors report no declarations of interest.
References
- Böttiger Y, Tybring G, Götharson E, Bertilsson L. (1997). Inhibition of the sulfoxidation of omeprazole by ketoconazole in poor and extensive metabolizers of S-mephenytoin. Clin Pharmaco. Ther 62:384–91
- Boyce MJ, Baisley KJ, Warrington SJ. (2012). Pharmacokinetic interaction between domperidone and ketoconazole leads to QT prolongation in healthy volunteers: a randomized, placebo-controlled, double-blind, crossover study. Br J Clin Pharmacol 73:411–21
- Burt HJ, Pertinez H, Säll C, et al. (2012). Progress curve mechanistic modeling approach for assessing time-dependent inhibition of CYP3A4. Drug Metab Dispos Biol Fate Chem 40:1658–67
- Crespi CL, Penman BW. (1997). Use of cDNA-expressed human cytochrome P450 enzymes to study potential drug–drug interactions. Adv Pharmacol San Diego CA 43:171–88
- Crewe HK, Barter ZE, Yeo KR, Rostami-Hodjegan A. (2011). Are there differences in the catalytic activity per unit enzyme of recombinantly expressed and human liver microsomal cytochrome P450 2C9? A systematic investigation into inter-system extrapolation factors. Biopharm Drug Dispos 32:303–18
- Deb S, Chin MY, Adomat H, Guns EST. (2014). Ginsenoside-mediated blockade of 1α,25-dihydroxyvitamin D3 inactivation in human liver and intestine in vitro. J Steroid Biochem Mol Biol 141:94–103
- Emoto C, Iwasaki K. (2007). Approach to predict the contribution of cytochrome P450 enzymes to drug metabolism in the early drug-discovery stage: the effect of the expression of cytochrome b(5) with recombinant P450 enzymes. Xenobiotica Fate Foreign Compd Biol Syst 37:986–99
- Gibbs MA, Thummel KE, Shen DD, Kunze KL. (1999). Inhibition of cytochrome P-450 3A (CYP3A) in human intestinal and liver microsomes: comparison of Ki values and impact of CYP3A5 expression. Drug Metab Dispos Biol Fate Chem 27:180–7
- Hashizume T, Mise M, Terauchi Y, et al. (1998). N-Dealkylation and hydroxylation of ebastine by human liver cytochrome P450. Drug Metab Dispos Biol Fate Chem 26:566–71
- Hsu A, Granneman GR, Cao G, et al. (1998). Pharmacokinetic interaction between ritonavir and indinavir in healthy volunteers. Antimicrob. Agents Chemother 42:2784–91
- Jerling M, Huan BL, Leung K, et al. (2005). Studies to investigate the pharmacokinetic interactions between ranolazine and ketoconazole, diltiazem, or simvastatin during combined administration in healthy subjects. J Clin Pharmacol 45:422–33
- Kenworthy KE, Bloomer JC, Clarke SE, Houston JB. (1999). CYP3A4 drug interactions: correlation of 10 in vitro probe substrates. Br J Clin Pharmacol 48:716–27
- Kim KA, Chung J, Jung DH, Park JY. (2004). Identification of cytochrome P450 isoforms involved in the metabolism of loperamide in human liver microsomes. Eur J Clin. Pharmacol 60:575–81
- Kivistö KT, Lamberg TS, Kantola T, Neuvonen PJ. (1997). Plasma buspirone concentrations are greatly increased by erythromycin and itraconazole. Clin Pharmacol Ther 62:348–54
- Krishna G, Moton A, Ma L, et al. (2009). Effects of oral posaconazole on the pharmacokinetic properties of oral and intravenous midazolam: a phase I, randomized, open-label, crossover study in healthy volunteers. Clin Ther 31:286–98
- Kumar V, Rock DA, Warren CJ, et al. (2006). Enzyme source effects on CYP2C9 kinetics and inhibition. Drug Metab Dispos Biol Fate Chem 34:1903–8
- Lee HS, Jin CB, Chong HS, et al. (1994). Involvement of P4503A in the metabolism of 7,8-benzoflavone by human liver microsomes. Xenobiotica Fate Foreign Compd Biol Syst 24:1053–62
- Lipscomb JC, Poet TS. (2008). In vitro measurements of metabolism for application in pharmacokinetic modeling. Pharmacol Ther 118:82–103
- Locuson CW, Wienkers LC, Jones JP, Tracy TS. (2007). CYP2C9 protein interactions with cytochrome b(5): effects on the coupling of catalysis. Drug Metab Dispos Biol Fate Chem 35:1174–81
- McGinnity DF, Parker AJ, Soars M, Riley RJ. (2000). Automated definition of the enzymology of drug oxidation by the major human drug metabolizing cytochrome P450s. Drug Metab Dispos 28:1327–34
- Neuvonen PJ, Kantola T, Kivistö KT. (1998). Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol. Ther 63:332–41
- Obach RS, Baxter JG, Liston TE, et al. (1997). The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther 283:46–58
- Ogilvie BW, Usuki E, Yerino P, Parkinson A. (2008). In Vitro approaches for studying the inhibition of drug-metabolizing enzymes and identifying the drug-metabolizing enzymes responsible for the tabolism of drugs (reaction phenotyping) with emphasis on cytochrome P450. In: Rodrigues AD, ed. Drug–drug interactions. New York: Informa Healthcare, 231–358
- Parmentier Y, Bossant MJ, Bertrand M, Walther B. (2007). In vitro studies of drug metabolism. Chem Mol Sci Chemical Eng 5:231–57
- Patki KC, Von Moltke LL, Greenblatt DJ. (2003). In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: role of cyp3a4 and cyp3a5. Drug Metab Dispos Biol. Fate Chem 31:938–44
- Perloff ES, Mason AK, Dehal SS, et al. (2009). Validation of cytochrome P450 time-dependent inhibition assays: a two-time point IC50 shift approach facilitates kinact assay design. Xenobiotica 39:99–112
- Proctor NJ, Tucker GT, Rostami-Hodjegan A. (2004). Predicting drug clearance from recombinantly expressed CYPs: intersystem extrapolation factors. Xenobiotica 34:151–78
- Raungrut P, Uchaipichat V, Elliot DJ, et al. (2010). In vitro-in vivo extrapolation predicts drug–drug interactions arising from inhibition of codeine glucuronidation by dextropropoxyphene, fluconazole, ketoconazole, and methadone in humans. J. Pharmacol Exp Ther 334:609–18
- Rowland M, Matin SB. (1973). Kinetics of drug–drug interactions. J Pharmacokinet Biopharm 1:553–67
- Sheiner LB, Beal SL. (1981). Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm 9:503–12
- Stresser DM, Broudy MI, Ho T, et al. (2004). Highly selective inhibition of human CYP3Aa in vitro by azamulin and evidence that inhibition is irreversible. Drug Metab Dispos Biol Fate Chem 32:105–12
- Suzuki A, Iida I, Hirota M, et al. (2003). CYP isoforms involved in the metabolism of clarithromycin in vitro: comparison between the identification from disappearance rate and that from formation rate of metabolites. Drug Metab Pharmacokinet 18:104–13
- Tateishi T, Ohashi K, Sudo T, et al. (1989). Dose dependent effect of diltiazem on the pharmacokinetics of nifedipine. J Clin Pharmacol 29:994–7
- Tayrouz Y. (2001). Ritonavir increases loperamide plasma concentrations without evidence for P-glycoprotein involvement. Clin Pharmacol Ther 70:405–14
- Troost J, Tatami S, Tsuda Y, et al. (2011). Effects of strong CYP2D6 and 3A4 inhibitors, paroxetine and ketoconazole, on the pharmacokinetics and cardiovascular safety of tamsulosin. Br J Clin Pharmacol 72:247–56
- Tseng E, Walsky RL, Luzietti RA, et al. (2014). Relative contributions of cytochrome CYP3A4 versus CYP3A5 for CYP3A-cleared drugs assessed in vitro using a CYP3A4-selective inactivator (CYP3cide). Drug Metab Dispos 42:1163–73
- Venkatakrishnan K, Moltke LLV, Greenblatt DJ. (1998). Human cytochromes P450 mediating phenacetin O-deethylation in vitro: validation of the high affinity component as an index of CYP1A2 activity. J Pharm Sci 87:1502–7
- Venkatakrishnan K, von Moltke LL, Harmatz JS, et al. (2000). Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: ratios of accessory proteins as sources of discrepancies between the approaches. Drug Metab Dispos 28:1493–504
- Walsky RL, Obach RS. (2004). Validated assays for human cytochrome P450 activities. Drug Metab. Dispos. Biol. Fate Chem 32:647–60
- Wang RW, Newton DJ, Liu N, et al. (2000). Human cytochrome P-450 3A4: in vitro drug–drug interaction patterns are substrate-dependent. Drug Metab. Dispos. Biol. Fate Chem 28:360–6
- Wienkers LC, Stevens J. (2003). Cytochrome P450 reaction phenotyping. In: Lee J, Scott Obach R, Fisher MB, eds. Drug metabolizing enzymes: cytochrome P450 and other enzymes in drug discovery and development. London: CRC Press – Taylor and Francis group,, 255–310
- Wienkers LC, Heath TG. (2005). Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov 4:825–33
- Yamazaki H, Urano T, Hiroki S, Shimada T. (1996). Effects of erythromycin and roxithromycin on oxidation of testosterone and nifedipine catalyzed by CYP3A4 in human liver microsomes. J Toxicol Sci 21:215–26
- Yamazaki H, Inoue K, Turvy CG, et al. (1997). Effects of freezing, thawing, and storage of human liver samples on the microsomal contents and activities of cytochrome P450 enzymes. Drug Metab Dispos Biol Fate Chem 25:168–74
- Zimmerlin A, Trunzer M, Faller B. (2011). CYP3A time-dependent inhibition risk assessment validated with 400 reference drugs. Drug Metab Dispos Biol Fate Chem 39:1039–46