
Abstract
1. Budesonide is a glucocorticoid used in the treatment of several respiratory and gastrointestinal inflammatory diseases. Glucocorticoids have been demonstrated to induce cytochrome P450 (CYP) 3A and the efflux transporter P-glycoprotein (P-gp). This study aimed to evaluate the potential of budesonide to act as a perpetrator or a victim of transporter- or CYP-mediated drug–drug interactions (DDIs).
2. In vitro studies were conducted for P-gp, breast cancer resistance protein and organic anion and cation transporters (OATP1B1, OATP1B3, OAT1, OAT3, OCT2) in transporter-transfected cells. Changes in mRNA expression in human hepatocytes and enzyme activity in human liver microsomes by budesonide were determined for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A.
3. The data indicated that budesonide is a substrate of P-gp but is not a substrate or an inhibitor of the other transporters investigated. Budesonide is neither an inducer nor an inhibitor of major CYP enzymes. The effect of P-gp on budesonide disposition is anticipated to be low owing to CYP3A-mediated clearance.
4. Collectively, our data indicate there is a low risk of budesonide perpetrating clinical DDIs mediated by the transporters or CYPs studied.
Introduction
Budesonide is a glucocorticoid used for the treatment of several inflammatory diseases in the respiratory and gastrointestinal tracts (Ceylan, Citation2006; Silverman & Otley, Citation2011). It is available in different formulations, including an inhalation powder (Pulmicort Flexhaler®, AstraZeneca, Södertälje, Sweden), a nebulised suspension (Pulmicort Respules™, AstraZeneca UK Ltd, Luton, UK), an aerosol nasal spray (Rhinocort Aqua®, AstraZeneca UK Ltd, Luton, UK), oral tablets (Entocort® EC, AstraZeneca, Södertälje, Sweden; Uceris®, Salix Pharmaceuticals, Raleigh, NC) and a rectal foam (Uceris®, Salix Pharmaceuticals, Raleigh, NC).
Budesonide is available as a 1:1 mixture of two epimers with 22R and 22S configurations, and is rapidly absorbed and extensively metabolised from the different formulations. The 22R epimer has a binding affinity for the glucocorticoid receptor that is two to three times higher than that of the 22S epimer; stereoselective metabolism of the 22R epimer has also been reported (Dahlberg et al., Citation1984; Ryerfeldt et al., Citation1984). Plasma concentrations of budesonide increase in a linear fashion; following a single oral dose of 9 mg, a maximum plasma concentration (Cmax) of ∼5 nmol/L is attained after 0.5–10 hours (US Food and Drug Administration, Citation2009). On average, 60–80% of an oral dose (capsules) is absorbed in the ileum and colon (Edsbäcker et al., Citation2003; Lundin et al., Citation2001). Once absorbed, budesonide is cleared through extensive first-pass metabolism by cytochrome P450 (CYP) 3A4 and, to a lesser extent, CYP3A5 in the liver and intestinal wall, resulting in low systemic bioavailability (Silverman & Otley, Citation2011). This has been shown to vary depending on the specific formulation with 10–15% systemic bioavailability reported for oral and rectal formulations compared with 13–29% bioavailability for nasally administered formulations (Danielsson et al., Citation1993; Silverman & Otley, Citation2011; Thorsson et al., Citation1999). Budesonide is primarily metabolised to 16α-hydroxyprednisolone and 6β-hydroxybudesonide () (Jonsson et al., Citation1995). The activity of these metabolites is less than 1% of that of the parent compound, and they do not contribute further to the therapeutic effect of budesonide (Silverman & Otley, Citation2011). In vitro studies of the phase II metabolism of budesonide have demonstrated that it undergoes sulfation by cytosolic sulfotransferase enzyme 1A1 (Meloche et al., Citation2002); studies in lung tissue slices have also demonstrated the formation of fatty acid esters from budesonide (Nave et al., Citation2007). Budesonide is eliminated in the urine (60%) and faeces (40%) as metabolites (US Food and Drug Administration, Citation2009).
Figure 1. Main metabolic pathway of budesonide. Figure reproduced from Jonsson et al. (Citation1995) with permission from the American Society for Pharmacology and Experimental Therapeutics (ASPET).
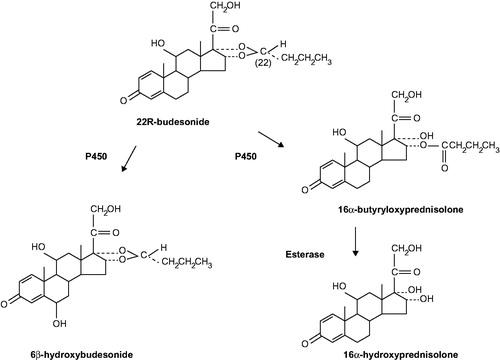
Co-administration of oral budesonide (10 mg, single dose) with oral ketoconazole (a known inhibitor of CYP3A; 100 mg, twice daily) has been shown to lead to an eightfold increase in systemic budesonide exposure (US Food and Drug Administration, Citation2009). Furthermore, co-administration of oral budesonide (4 mg, single dose) with cimetidine (a potent inhibitor of CYP3A4; 1 g daily) resulted in a 52% and a 31% increase in budesonide peak plasma concentration and area under the curve, respectively (US Food and Drug Administration, Citation2009). In vitro co-incubation of human liver microsomes (HLM) with budesonide and ketoconazole resulted in the in vitro inhibition of budesonide metabolism (concentration of inhibitor at 50% inhibition [IC50] ∼ 0.1 μM) (Jonsson et al., Citation1995). This effect was also observed with other known inhibitors of CYP3A family enzymes, including troleandomycin, erythromycin and cyclosporine with respective IC50 values of ∼1 μM, ∼100 μM and ∼100 μM (Jonsson et al., Citation1995). With this knowledge, a reduction of budesonide dose may need to be considered if concomitant treatment with known inhibitors of CYP3A is indicated.
It has been established that glucocorticoids are inducers of CYP3A4 in human hepatocytes (Pascussi et al., Citation2001) and in vivo (McCune et al., Citation2000), which may have an effect on systemic exposure to co-administered drugs. The increased expression of CYP3A4 mRNA in LS180 colon carcinoma cells after budesonide treatment (25 μM) has been reported in vitro (Zimmermann et al., Citation2009). A threefold induction of the murine homologue Cyp3a11 was also observed in mice administered oral budesonide (25 mg/kg) for four days (Zimmermann et al., Citation2009). It is therefore possible that budesonide could induce its own metabolism, and that of concomitant medications via the induction of CYP3A4 expression, provided both are absorbed in the same region of the gastrointestinal tract. Furthermore, budesonide (10 μM and 25 μM) has been demonstrated to induce the expression of the efflux transporter P-glycoprotein (P-gp) in LS180 intestinal cells in a dose-dependent manner (Maier et al., Citation2007). This increased expression could potentially have an effect on the bioavailability of co-administered therapeutic drugs.
A budesonide oral suspension for the treatment of eosinophilic esophagitis is currently being developed (Dellon et al., Citation2016,Citation2017). The objectives of this in vitro drug–drug interaction (DDI) study were (1) to evaluate the substrate and/or inhibitor potential of budesonide for the following transporters: P-gp, breast cancer resistance protein (BCRP), hepatic organic anion-transporting polypeptides 1B1 and 1B3 (OATP1B1 and OATP1B3), renal organic anion transporters 1 and 3 (OAT1 and OAT3) and organic cation transporter 2 (OCT2); and (2) to evaluate the potential of budesonide either to induce CYP1A2, CYP2B6 and CYP3A4 in human hepatocytes, or to inhibit seven major CYPs (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A) in HLM.
Materials and methods
Suppliers of materials used in transporter and CYP studies are detailed in Supplemental Material S1. Budesonide concentrations were selected for each study type based on the European Medicines Agency (EMA) and US Food and Drug Administration (FDA) guidelines for DDI studies (European Medicines Agency, Citation2012; US Food and Drug Administration, Citation2012). Assays were not performed with the individual epimers of budesonide because they both undergo the same metabolic pathway (metabolism by CYP3A4); stereochemical effects on potential DDIs were therefore not expected.
Uptake transporter studies
Uptake transporter-related assays were conducted with vector control- and transporter-transfected human embryonic kidney cells. The cell culture conditions are detailed in Supplemental Material S2. Budesonide, probe substrates and known inhibitors were prepared in HBSSg_7.4 buffer. The uptake of a probe substrate of each transporter was conducted in parallel with separate cells of the same batch of vector control- and transporter-transfected cells.
For inhibition assessment, the tested concentrations of budesonide were 0.03 μM (for OAT1, OAT3 and OCT2; approximately 50 times the unbound Cmax) and 1.1 μM (for OATP1B1 and OATP1B3; approximately 25 times the maximum unbound inhibitor concentration in portal venous blood, [I]u, inlet, max), respectively. The incubation period was either 2 min (OCT2) or 5 min (all other transporters) with 500 μL of probe substrate, with or without budesonide or a known inhibitor.
For substrate assessment, the tested concentrations of budesonide were 0.002, 0.2 or 2 μM, with incubation periods of 2, 5 and 10 min, respectively. The lowest concentration tested (0.002 μM) was eightfold higher than the lower limit of quantification of budesonide (0.00025 μM) for this assay, and was therefore considered high enough to accumulate sufficiently in the cells. After incubation, the dosing solutions were aspirated, and the cells were rinsed twice with ice-cold HBSSg_7.4 buffer. The cells were then lysed in 400 μL of acetonitrile (ACN):water (3:1, v/v) containing an internal standard (IS) and lysates were collected for determination of the probe substrate concentration (inhibitor assessment) or probe substrate and budesonide concentrations (substrate assessment) using LC–MS/MS. The analytical conditions are described in Supplemental Material S3.
Probe substrates for OATP1B1- and OATP1B3-transfected cells, OAT1-transfected cells, OAT3-transfected cells and OCT2-transfected cells were atorvastatin (0.15 μM), para-aminohippurate (PAH, 10 μM), furosemide (5 μM) and 1-methyl-4-phenylpyridinium iodide (MPP+, 5 μM), respectively. Known inhibitors were rifamycin SV (10 μM), probenecid (100 μM), probenecid (100 μM) and imipramine (300 μM), respectively.
Efflux transporter studies
Efflux transporter-related assays were conducted with cell monolayers via bidirectional permeability: apical-to-basolateral (AP-to-BL) and basolateral-to-apical (BL-to-AP). P-gp inhibition and substrate assessment were conducted with Caco-2 (C2BBe1) cells and multidrug resistance protein (MDR1)-transfected Madin–Darby canine kidney (MDCK) epithelial cells, and BCRP inhibition and substrate assessment with C2BBe1 and BCRP-transfected MDCK cells. Cell culture conditions are detailed in Supplemental Material S2. All incubations were performed in a humidified incubator at 37 °C with 5% CO2. Probe substrates, known inhibitors and budesonide were freshly prepared in Hanks’s balanced salt solution supplemented with 10 nM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) and 15 mM d-glucose (HBSSg_7.4 buffer).
For the inhibition assays, the tested concentration of budesonide was 2 μM (the estimated Cmax reflecting the concentrations at the luminal side of the gastrointestinal wall; obtained by dividing the highest dose given in a phase II paediatric trial (Gupta et al., Citation2015) by a volume of 250 mL). Digoxin and cladribine were used as probe substrates for P-gp and BCRP, respectively; valspodar and Ko143 were used as inhibitors of P-gp and BCRP, respectively. The transport of digoxin and cladribine was measured in the absence and presence of budesonide (with both probe substrates), valspodar (1 μM, with digoxin only) and Ko143 (0.5 μM, with cladribine only). A 30-min pre-incubation with budesonide or known inhibitor was performed on both the AP and BL sides to pre-load the cells, the pre-incubation solution was aspirated, and the following procedures were performed: for AP-to-BL transport, 0.5 mL of fresh dosing solution of a probe substrate with or without budesonide or known inhibitor was added to the AP side (donor), and 1.5 mL of buffer with or without budesonide or known inhibitor was added to the BL side (receiver). For BL-to-AP transport, 1.5 mL of dosing solution of a probe substrate with or without budesonide or known inhibitor was added to the BL side (donor), and 0.5 mL of buffer with or without budesonide or known inhibitor was added to the AP side (receiver). At pre-selected time points (donor at 0 and 120 min and receiver at 120 min), 300 μL aliquots were taken from the receiver and replaced with the same volume of fresh buffer with or without the appropriate inhibitor. No replacement/withdrawal occurred on the donor side.
For the substrate assays, the concentrations of budesonide were 0.2, 2 and 20 μM. The lowest concentration of budesonide tested was increased from 0.002 μM (used in the uptake transporter assays) to 0.2 μM following insufficient accumulation of budesonide in the cells during the uptake transporter assays. This increased concentration allowed sufficient accumulation of budesonide in the receiver compartment for determination of the efflux rate ratio. For AP-to-BL transport, 0.5 mL of budesonide dosing solution with or without known inhibitor was added to the AP side (donor), and 1.5 mL of buffer with or without known inhibitor was added to the BL side (receiver). For BL-to-AP transport, 1.5 mL of dosing solution of budesonide with or without known inhibitor was added to the BL side (donor), and 0.5 mL of buffer with or without known inhibitor was added to the AP side (receiver). The sampling profiles were as described above.
The concentrations of probe substrate in the collected dosing solution, donor and receiver samples, as well as budesonide in the dosing solution, were analysed by liquid chromatography with tandem mass spectrometry (LC–MS/MS). The analytical conditions are described in Supplemental Material S3 and Supplemental Table S1.
CYP studies
CYP induction
Before induction experiments, fresh human hepatocytes (Supplemental Table S2) were recovered by incubation with hepatocyte culture medium in a 95% air/5% CO2 incubator at 37 °C for 24 h. Hepatocytes were then treated with hepatocyte culture medium fortified with budesonide at three concentrations (0.00345, 0.010 and 0.0345 μM for CYP1A2 and CYP2B6, equal to 1 × total Cmax, 3 × total Cmax and 10 × total Cmax, respectively; 0.0186, 0.186 and 1.86 μM for CYP3A, equal to 0.001 × dose/250 mL, 0.01 × dose/250 mL and 0.1 × dose/250 mL, respectively). Positive controls were treated in parallel with hepatocyte culture medium fortified with a known inducer of each CYP: omeprazole (50 μM) for CYP1A2, phenobarbital (1000 μM) for CYP2B6 and rifampicin (50 μM) for CYP3A. Vehicle controls were treated with hepatocyte culture medium containing 0.1% dimethyl sulfoxide (DMSO). The hepatocyte incubation was conducted in a 95% air/5% CO2 incubator at 37 °C for three days (72 h) with daily replacement of the hepatocyte culture medium containing budesonide, positive inducer or vehicle.
CYP enzyme activity was determined by measuring the formation of CYP probe substrate metabolite. Briefly, the wells were washed with Dulbecco’s phosphate-buffered saline and incubated with 200 μL of Williams’s Medium E containing an individual CYP probe substrate at 37 °C for 1 h in a 95% air/5% CO2 incubator. After the incubation, 150 μL of the CYP incubation mixture from each well was transferred into a 96-well plate containing 150 μL of ice-cold ACN per well. The solutions were mixed and centrifuged at 1640×g for 10 min. The supernatants were transferred to a high-performance liquid chromatography (HPLC) autosampler plate, and the concentrations of CYP probe metabolite were analysed by LC–MS/MS. The analytical conditions are described in Supplemental Material S3 and Supplemental Table S3.
The viability of cells, expressed as the percentage of [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium, inner salt] absorbance relative to vehicle control, was measured spectrophotometrically using the CellTiter 96® AQueous One Solution Cell Proliferation Assay following the manufacturer’s protocols.
Total RNA was isolated from the treated cells using an RNeasy® mini kit, and treated with RNase-free DNase following the manufacturer’s protocols. The concentration of RNA was determined using a Qubit™ Fluorometer with a Qubit™ RNA HS assay kit. cDNA was synthesised from up to 1 μg of the total RNA harvested from the cells using a QuantiTect® Reverse Transcription kit. Analysis of CYP gene expression by quantitative polymerase chain reaction (Supplemental Table S4) was performed using a LightCycler® 480 II System.
The concentration of budesonide in the hepatocyte culture medium, at 0, 4, 8 and 24 h post-dose on day 3 of the incubation period, was measured by LC–MS/MS (Supplemental Material S3).
CYP reversible inhibition
Budesonide, at eight concentrations (0–10 μM; the maximal concentration was based on 0.2×Igut ≈ 7.3 μM, Igut = molar dose/250 mL), was incubated with HLM (0.25 mg protein/mL) in phosphate buffer (100 mM, pH 7.4) containing MgCl2 (5 mM), reduced nicotinamide adenine dinucleotide phosphate (NADPH) (1 mM) and an individual CYP probe substrate (at close to or below the Michaelis–Menten constant [Km], ). The total organic solvent content in the final incubation was less than 1% (DMSO ≤ 0.1%, other organic solvent ≤ 1%). The reaction mixture without NADPH was equilibrated in a shaking water bath at 37 °C for 5 min. The reaction was initiated by the addition of NADPH, followed by incubation at 37 °C for 10–30 min depending on the individual CYP isoform. The reaction was terminated by the addition of two volumes of ACN containing an IS (stable isotope-labelled CYP probe metabolite). Negative (vehicle) controls were conducted without budesonide. Positive controls were performed in parallel using known CYP inhibitors (). After the removal of protein by centrifugation at 1640×g for 10 min at 4 °C, the supernatants were transferred to an HPLC autosampler plate. The formation of individual CYP probe metabolite (expressed as the peak area ratio of CYP probe metabolite to IS) was determined by LC–MS/MS (Supplemental Material S3).
Table 1. Probe substrates, metabolites, positive inhibitors and known inhibitors for CYP inhibition in human liver microsomes.
CYP time-dependent inhibition (TDI)
CYP TDI was evaluated by a 30-min pre-incubation of budesonide with HLM in the presence and absence of NADPH, followed by the CYP enzyme activity assay. This approach allowed the calculation of an IC50 shift, which is used to determine whether or not a compound is a time-dependent inhibitor (Berry & Zhao, Citation2008; Fowler & Zhang Citation2008; Obach et al., Citation2007; Perloff et al., Citation2009).
The CYP reaction was performed using an incubation volume of 200 μL. Briefly, budesonide, at eight concentrations (0–10 μM), was pre-incubated at 37 °C for 30 min with HLM (0.25 mg protein/mL) in phosphate buffer (100 mM, pH 7.4) containing MgCl2 (5 mM) in the presence and absence of NADPH (1 mM). The total organic solvent content in the final incubation was less than 1% (DMSO ≤ 0.1%, other organic solvent ≤ 1%). The CYP reaction was initiated by adding an individual CYP probe substrate (at close to or below Km, ) with (when NADPH was not added in the pre-incubation step) or without (when NADPH was added in the pre-incubation step) the addition of NADPH. The reaction mixture was incubated at 37 °C for 10–30 min depending on the individual CYP isoform. The reaction was terminated with ice-cold ACN containing an IS. Negative (vehicle) controls were conducted using the incubation mixture without budesonide. Positive controls were performed in parallel using known CYP time-dependent inhibitors (). After the removal of protein by centrifugation at 1640×g for 10 min at 4 °C, the supernatants were transferred to an HPLC autosampler plate. The formation of individual CYP probe metabolite (expressed as the peak area ratio of CYP probe metabolite to IS) was determined by LC–MS/MS (Supplemental Material S3).
Data analysis
Data from transporter and CYP assays were used to calculate the following parameters.
Efflux ratio and percentage inhibition for efflux transporter studies
where dCR/dt = the slope of the cumulative concentration in the receiver compartment versus time (μM/s); VR = volume of the receiver compartment (mL); A = cell membrane surface area (1.13 cm2); CDin = initial donor concentration (μM).
where TA = test article (Giacomini et al., Citation2010).
Influx rate ratio for uptake transporter studies
where CS = substrate concentration in the cell lysate (μM); VS = substrate assessment cell lysate volume (mL); CP = protein concentration in the cell lysate (mg/mL); VP = protein content determination cell lysate volume (mL).
where TF = transporter-transfected cells; VC = vector control-transfected cells.
CYP induction
CYP induction was calculated as described previously (Hewitt et al., Citation2007).
CYP inhibition IC50 shift
The IC50 shift for CYP TDI was calculated using the following equation:
where IC50 (−NADPH) and IC50 (+NADPH) are the IC50 values with 30-min pre-incubation in the absence and presence of NADPH, respectively.
Results
P-gp and BCRP inhibition assessments
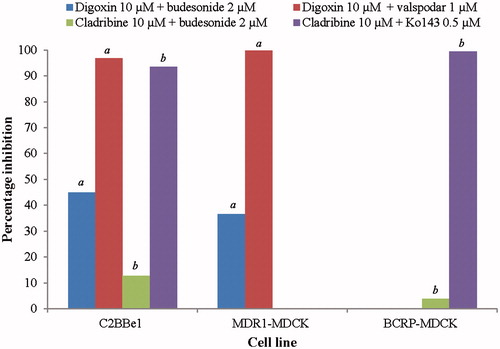
Less than 50% inhibition of P-gp or BCRP was observed in the presence of budesonide (2 μM) in both C2BBe1 and MDCK cells (). The extrapolated IC50 for budesonide was therefore greater than 2 μM. In the presence of known efflux inhibitors valspodar and Ko143 (controls), the percentage inhibition values were greater than 95% for P-gp and greater than 90% for BCRP ().
Figure 2. P-gp and BCRP inhibition assessments. aP-gp inhibitor assessment. bBCRP inhibitor assessment. BCRP-MDCK: breast cancer resistance protein-transfected Madin–Darby canine kidney epithelial cells; C2BBe1: Caco-2 cells; MDR1-MDCK: multidrug resistance protein-transfected Madin–Darby canine kidney epithelial cells; NA: not applicable; P-gp: P-glycoprotein.
P-gp and BCRP substrate assessments
The corrected efflux ratios of budesonide (2 μM) for P-gp and BCRP were 26.9 and 1.61, respectively (). The P-gp inhibitor valspodar caused a decrease in the efflux ratio from 26.9 to 0.0874 (>50%) (). The BCRP inhibitor Ko143 caused a decrease in the efflux ratio from 1.61 to 1.21 (<50%) ().
Table 2. P-gp and BCRP substrate assessments.
Uptake transporter inhibition assessment
No inhibition of any of the uptake transporters evaluated was observed in the presence of budesonide (1.1 μM for OATP1B1 and OATP1B3; 0.03 μM for OAT1, OAT3 and OCT2) (). The extrapolated IC50 values for budesonide were therefore greater than 1.1 μM for OATP1B1 and OATP1B3, and greater than 0.03 μM for OAT1, OAT3 and OCT2. In the presence of known inhibitors as controls, the percentage inhibition values were at least 89.2% ().
Table 3. Uptake of known substrates in the presence/absence of budesonide or a known inhibitor.
Uptake transporter substrate assessment
At a concentration of 0.002 μM, the respective permeability values for budesonide were below the limit of quantification (0.00025 μM), and influx rate ratios could not be determined (). At budesonide concentrations of 0.2 and 2 μM, the influx rate ratios were less than 2.00 for all uptake transporters at 2-, 5- and 10-min time points (). The influx rate ratios for known substrates used as controls were at least 6.39 ().
Table 4. Uptake of budesonide and known substrates at 2, 5 and 10 min.
CYP enzyme activity and mRNA induction assays
The effect of budesonide on cell viability was determined following CYP induction and enzyme activity assays. Hepatocytes remained viable following both assays (data not shown). The budesonide concentration in the hepatocyte cell culture medium decreased after hepatocyte exposure in all tested donors according to concentration measurements made on day 3 of hepatocyte treatment (data not shown).
Budesonide did not meaningfully increase CYP1A2 mRNA expression or enzyme activity in any of the three evaluated donors at any concentration tested (). In donor 1, budesonide caused a 2.8-, 3.2- and 3.5-fold induction of CYP2B6 mRNA at 0.00345, 0.010 and 0.0345 μM, respectively. Such increases were not observed in donors 2 and 3 (). A 3.7-fold and 6.3-fold induction of CYP3A mRNA was observed in donor 1 and donor 4, respectively, following exposure to 1.86 μM budesonide, but not at lower concentrations (0.0186 μM and 0.186 μM) (). No meaningful increases in mRNA induction of CYP3A were observed in donor 2 ().
Table 5. Fold induction of CYP mRNA and enzyme activity in human hepatocytes by budesonide and known inducers.
CYP inhibition assays
The IC50 values of budesonide for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 and CYP2D6 in HLM were all greater than 10 μM (). The IC50 of budesonide for CYP3A was 4.7 μM when midazolam was the probe substrate, and greater than 10 μM when testosterone was the probe substrate (). An IC50 shift for budesonide could not be determined for any of the seven CYPs because the IC50 values were greater than 10 μM under both reversible (30 min pre-incubation without NADPH) and irreversible (30 min pre-incubation with NADPH) conditions (). The differences in CYP inhibition between reversible and irreversible conditions were less than 10% at 10 μM budesonide for all seven CYPs. The IC50 and IC50 shift values for known CYP inhibitors are shown in Supplemental Table S6. Enzyme activity versus budesonide concentration curves for CYP reversible inhibition and CYP TDI are shown in Supplemental Figure 1 and Supplemental Figure 2, respectively.
Table 6. Inhibition of CYP enzyme activity in HLM by budesonide.
Discussion
The objectives of this in vitro DDI study were (1) to evaluate the substrate/inhibitor potential of budesonide for the transporters P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3 and OCT2; and (2) to evaluate the induction/inhibition potential of budesonide for seven major CYP enzymes.
Budesonide is not an inhibitor of P-gp or BCRP
Less than 50% inhibition of P-gp and BCRP was observed in the presence of budesonide (2 μM) in both C2BBe1 and MDCK cell models. The extrapolated IC50 for budesonide was therefore greater than 2 μM. The FDA drug interaction study guidelines propose two [I]/IC50 ratios: [I]1/IC50 reflects systemic exposure, where [I]1 is the estimated total plasma Cmax; and [I]2/IC50, where [I]2 reflects the concentration at the luminal side of the gastrointestinal wall (oral administration) and is obtained by dividing the highest dose by 250 mL. It has previously been shown that [I]1 for budesonide is 0.0035 μM and [I]2 is 18.6 μM following a 2 mg oral dose (Shire data; Shire, Citation2008). The results of the current study showed that the [I]/IC50 ratios are 0.0035/(>2) (i.e. <0.1) and 18.6/(>2) (i.e.<10). Budesonide is therefore not considered a likely perpetrator of clinical P-gp- or BCRP-mediated DDIs.
Budesonide is a substrate of P-gp but not a substrate of BCRP
The decrease in efflux ratio from 26.9 to 0.0874 (>50%) in the presence of a P-gp inhibitor observed in the present study indicated that budesonide is a substrate of P-gp (European Medicines Agency, Citation2012; US Food and Drug Administration, Citation2012). Budesonide, however, is not a substrate of BCRP, as shown by the efflux ratio of 1.61 and a decrease to 1.21 (<50%) in the presence of Ko143. Our findings are in agreement with those of Dilger et al. (Citation2004) who investigated basal to apical transport of budesonide in monolayers of MDR1-transfected LLC-PK1 kidney cells, in the presence and absence of the P-gp inhibitor valspodar; budesonide transport was inhibited in presence of valspodar, identifying it as a substrate of P-gp. Budesonide is cleared primarily by CYP3A4 metabolism (US Food and Drug Administration, Citation2009) and is highly permeable; therefore, co-administration with P-gp inhibitors is anticipated to have a minimal effect on budesonide clearance. Although the role of P-gp in budesonide absorption is not entirely clear, given the low overall systemic exposure and relatively high safety margin identified for budesonide (US Food and Drug Administration, Citation2009), the safety risk with respect to potentially increased systemic exposure as a result of P-gp inhibition at the gastrointestinal level is considered to be low.
Budesonide is neither a substrate nor an inhibitor of OAT1, OAT3, OCT2, OATP1B1 or OATP1B3
In uptake transporter inhibition assessments, the extrapolated IC50 for budesonide was greater than 0.03 μM for OAT1, OAT3 and OCT2; and greater than 1.1 μM for OATP1B1 and OATP1B3. The EMA and FDA drug interaction study guidelines differ in their assessment of OAT1, OAT3 and OCT2 inhibition potential: if unbound [I]1/IC50 is less than 0.02 (European Medicines Agency, Citation2012) or less than 0.1 (US Food and Drug Administration, Citation2012), a clinical DDI study is not required. It has previously been shown that the unbound [I]1 of budesonide is 0.0005 μM (Shire data; Shire, Citation2008). Data from the present study showed that the [I]1/IC50 ratios for OAT1, OAT3 and OCT2 were 0.0005/(>0.03) (i.e. <0.02). For OATP1B1 and OATP1B3, the [I]/IC50 ratios were 0.0035/(>1.1) (i.e. <0.1). In contrast, Konig et al. (Citation2011) reported the inhibition of OATP1B1- and 1B3-mediated mesalazine uptake by budesonide in transporter-transfected human embryonic kidney cells; however, the budesonide concentrations tested in that study (10 μM and 100 μM) were much higher than those used here (0.002, 0.2 and 2 μM). Clinical DDI studies with budesonide are therefore considered not to be required based on clinically relevant concentrations.
In uptake transporter substrate assessments at budesonide concentrations of 0.2 μM and 2 μM, the influx rate ratios were less than 2.00 for all uptake transporters. This result demonstrated that budesonide was not a substrate of any of the transporters at the tested concentrations.
There is a low risk of induction or inhibition of seven major CYPs by budesonide
A greater than twofold increase in mRNA expression or enzyme activity compared with the vehicle control is considered clinically significant (European Medicines Agency, Citation2012). An in vitro study is considered positive for enzyme induction if a greater than 100% concentration-dependent increase in mRNA expression is observed. An mRNA increase of less than 100% is a negative finding only when the increase in mRNA is less than 20% of the response of the positive control (European Medicines Agency, Citation2012).
In this study, budesonide (0.00345, 0.010 and 0.0345 μM) was not found to be an inducer of CYP1A2 mRNA expression or enzyme activity in any of the three donors tested. In the case of CYP2B6, budesonide caused 2.8-, 3.2- and 3.5-fold inductions in mRNA expression at 0.00345, 0.010 and 0.0345 μM, respectively, in donor 1; however, inductions of less than twofold and less than 20% of the positive control value were observed in donors 2 and 3. In addition, budesonide did not increase CYP2B6 enzyme activity at any of the three tested concentrations in any of the three donors; the risk of CYP2B6 induction by budesonide may therefore be considered low.
In donor 1 and donor 4, 3.7-fold and 6.7-fold inductions of CYP3A4 mRNA were observed, respectively, at 1.86 μM budesonide, but not at lower concentrations (0.0186 μM and 0.186 μM). Both fold inductions were less than 20% of the positive control value (76 for donor 1 and 734 for donor 2), indicating that budesonide was not an inducer of CYP3A. These findings contrast with those of Zimmermann et al. (Citation2009) who reported a significant, dose-dependent induction of CYP3A4 mRNA expression in intestinal cells following budesonide exposure. However, the concentrations at which significant increases in mRNA expression were observed (10 μM and 25 μM) were considerably higher than those tested in the studies presented here (0.0186, 0.186 and 1.86 μM), and may not be clinically relevant. Further comparative studies in hepatocytes and intestinal cells using clinically relevant budesonide concentrations will provide additional data on the effect of budesonide exposure on CYP3A4 mRNA expression. Despite some observed effects of budesonide on mRNA induction in this study, the data indicated that budesonide is not an inducer of CYP1A2, CYP2B6 or CYP3A4. Budesonide is therefore considered to be an unlikely mediator of DDIs via the induction of increased CYP expression.
The IC50 of budesonide was greater than 10 μM for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 and CYP2D6 in HLM. For CYP3A, the IC50 values were 4.7 μM with midazolam as the probe substrate and greater than 10 μM with testosterone as the probe substrate. An IC50 of 4.7 μM is more than 1000-fold higher than the total Cmax of budesonide (0.0035 μM) and is therefore unlikely to be clinically relevant. TDI is the result of irreversible covalent binding or quasi-irreversible, noncovalent tight binding of a reactive metabolite to the enzyme that metabolises the test article (US Food and Drug Administration, Citation2012). An IC50 shift of greater than or equal to 1.5 is suggestive of TDI (Berry & Zhao, Citation2008; Grimm et al., Citation2009). Despite the fact that an IC50 shift for budesonide could not be determined, the differences in CYP inhibition between reversible and irreversible conditions were less than 10% at 10 μM for all seven tested CYPs. This result indicated that budesonide is not a time-dependent inhibitor of any CYP tested. Based on the CYP data generated, and low clinical exposure with 85–90% protein binding (US Food and Drug Administration, Citation2000), it is unlikely that budesonide is a mediator of DDIs via either reversible or irreversible inhibition of CYP activity.
Summary
This study has generated non-clinical DDI data for budesonide using a wide range of in vitro assays, performed using different cell types. The interaction of budesonide with seven major CYPs involved in therapeutic drug metabolism has been investigated, as well as key efflux and uptake transporters. Based on the in vitro data from this study, there is a low potential risk of budesonide acting as a perpetrator of metabolic DDIs through the induction/inhibition of efflux transporters, uptake transporters or CYP enzymes. These data add to our knowledge of budesonide pharmacokinetics and suggest that in vivo DDI studies may not be needed for the continued clinical development of budesonide. Further studies in intestinal cell lines may provide relevant additional data, given the high exposure of oral budesonide formulations to the intestinal system. This study also furthers our knowledge of the lack of potential corticosteroid-induced DDIs; such information may prove useful to clinicians when prescribing corticosteroids as concomitant medications.
Declaration of interest
Funding for this study was provided by Shire Human Genetic Therapies, Inc. Nancy Chen, Richard D. Finkelman and Devin Welty are full-time employees of Shire. Donghui Cui was an employee of Shire at the time of the study. Qing Wang and Zhiming Wen are full-time employees of Absorption Systems, which received funding from Shire Human Genetic Therapies Inc. to perform this study.
Supplementary material available online
IXEN_1349868_Supplementary_Material.pdf
Download PDF (425.8 KB)Acknowledgements
The authors would like to thank Chris Bode, Elizabeth Henry, Zabrina Gallagher and Joseph Rager (Absorption Systems, Exton, PA) for providing technical support. Medical writing support was provided by Dr Laura Knapp of PharmaGenesis London, London, UK with funding provided by Shire International GmbH.
Related Research Data
References
- Berry LM, Zhao Z. (2008). An examination of IC50 and IC50-shift experiments in assessing time-dependent inhibition of CYP3A4, CYP2D6 and CYP2C9 in human liver microsomes. Drug Metab Lett 2:51–9
- Ceylan E. (2006). Budesonide-formoterol (inhalation powder) in the treatment of COPD. Int J Chron Obstruct Pulmon Dis 1:115–22
- Dahlberg E, Thalen A, Brattsand R, et al. (1984). Correlation between chemical structure, receptor binding, and biological activity of some novel, highly active, 16 alpha, 17 alpha-acetal-substituted glucocorticoids. Mol Pharmacol 25:70–8
- Danielsson Å, Edsbäcker S, Löfberg R, et al. (1993). Pharmacokinetics of budesonide enema in patients with distal ulcerative colitis or proctitis. Aliment Pharmacol Ther 7:401–7
- Dellon ES, Katzka DA, Collins MH, et al. (2016). Safety and efficacy of oral budesonide suspension for maintenance therapy in eosinophilic esophagitis. Gastroenterology 150:S188
- Dellon ES, Katzka DA, Collins MH, et al. (2017). Budesonide oral suspension improves symptomatic, endoscopic, and histologic parameters compared with placebo in patients with eosinophilic esophagitis. Gastroenterology 152:776–86.e5
- Dilger K, Schwab M, Fromm MF. (2004). Identification of budesonide and prednisone as substrates of the intestinal drug efflux pump P-glycoprotein. Inflamm Bowel Dis 10:578–83
- Edsbäcker S, Bengtsson B, Larsson P, et al. (2003). A pharmacoscintigraphic evolution of oral budesonide given as controlled-release (Entocort) capsules. Aliment Pharmacol Ther 17:525–36
- European Medicines Agency. 2012. Guideline on the investigation of drug interactions [Online]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf [last accessed 23 Mar 2017]
- Fowler S, Zhang H. (2008). In vitro evaluation of reversible and irreversible cytochrome P450 inhibition: current status on methodologies and their utility for predicting drug–drug interactions. AAPS J 10:410–24
- Giacomini KM, Huang SM, Tweedie DJ, et al. (2010). Membrane transporters in drug development. Nat Rev Drug Discov 9:215–36
- Grimm SW, Einolf HJ, Hall SD, et al. (2009). The conduct of in vitro studies to address time-dependent inhibition of drug-metabolizing enzymes: a perspective of the pharmaceutical research and manufacturers of America. Drug Metab Dispos 37:1355–70
- Gupta SK, Vitanza JM, Collins MH. (2015). Efficacy and safety of oral budesonide suspension in pediatric patients with eosinophilic esophagitis. Clin Gastroenterol Hepatol 13:66–76e3
- Hewitt NJ, de Kanter R, LeCluyse E. (2007). Induction of drug metabolizing enzymes: a survey of in vitro methodologies and interpretations used in the pharmaceutical industry—do they comply with FDA recommendations? Chem Biol Interact 168:51–65
- Jonsson G, Astrom A, Andersson P. (1995). Budesonide is metabolized by cytochrome P450 3A (CYP3A) enzymes in human liver. Drug Metab Dispos 23:137–42
- Konig J, Glaeser H, Keiser M, et al. (2011). Role of organic anion-transporting polypeptides for cellular mesalazine (5-aminosalicylic acid) uptake. Drug Metab Dispos 39:1097–102
- Lundin P, Edsbäcker S, Larsson P, et al. (2001). Targeted delivery of oral budesonide (Entocort® capsules) in healthy volunteers and Crohn’s disease patients. Gastroenterology 120:A749
- Maier A, Zimmermann C, Beglinger C, et al. (2007). Effects of budesonide on P-glycoprotein expression in intestinal cell lines. Br J Pharmacol 150:361–8
- McCune JS, Hawke RL, LeCluyse EL, et al. (2000). In vivo and in vitro induction of human cytochrome P4503A4 by dexamethasone. Clin Pharmacol Ther 68:356–66
- Meloche CA, Sharma V, Swedmark S, et al. (2002). Sulfation of budesonide by human cytosolic sulfotransferase, dehydroepiandrosterone-sulfotransferase (DHEA-ST). Drug Metab Dispos 30:582–5
- Nave R, Fisher R, McCracken N. (2007). In vitro metabolism of beclomethasone dipropionate, budesonide, ciclesonide, and fluticasone propionate in human lung precision-cut tissue slices. Respir Res 8:65–74
- Obach RS, Walsky RL, Venkatakrishnan K. (2007). Mechanism-based inactivation of human cytochrome P450 enzymes and the prediction of drug–drug interactions. Drug Metab Dispos 35:246–55
- Pascussi JM, Drocourt L, Gerbal-Chaloin S, et al. (2001). Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur J Biochem 268:6346–58
- Perloff ES, Mason AK, Dehal SS, et al. (2009). Validation of cytochrome P450 time-dependent inhibition assays: a two-time point IC50 shift approach facilitates kinact assay design. Xenobiotica 39:99–112
- Ryerfeldt A, Edsbäcker S, Pauwels R. (1984). Kinetics of the epimeric glucocorticoid budesonide. Clin Pharmacol Ther 35:525–30
- Shire. (2008). Dose-ranging study of oral viscous budesonide in pediatrics with eosinophilic esophagitis [Online]. Available from: https://clinicaltrials.gov/ct2/show/NCT00762073?term=NCT00762073&rank=1 [last accessed 13 Dec 2016]
- Silverman J, Otley A. (2011). Budesonide in the treatment of inflammatory bowel disease. Expert Rev Clin Immunol 7:419–28
- Thorsson L, Borgá O, Edsbäcker S. (1999). Systemic availability of budesonide after nasal administration of three different formulations: pressurized aersosol, aqueous pump spray, and powder. J Clin Pharmacol 47:619–24
- US Food and Drug Administration. (2000). Pulmicort Respules™ (budesonide inhalation suspension) 0.25 mg and 0.5 mg [Online]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2000/20929lbl.pdf [last accessed 7 Mar 2017]
- US Food and Drug Administration. (2009). Entocort® EC (budesonide) capsules product label. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/021324s008lbl.pdf [last accessed 7 Mar 2017]
- US Food and Drug Administration. (2012). Guidance for industry drug interaction studies – study design, data analysis, implications for dosing, and labeling recommendations. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf [last accessed 7 Mar 2017]
- Zimmermann C, van Waterschoot RA, Harmsen S, et al. (2009). PXR-mediated induction of human CYP3A4 and mouse Cyp3a11 by the glucocorticoid budesonide. Eur J Pharm Sci 36:565–71