
Abstract
Bexagliflozin is a C-aryl glucoside inhibitor of human sodium-glucose linked transporter 2 (SGLT2) that undergoes oxidation and glucuronidation to form six principal metabolites in humans.
In vitro metabolism by human liver microsomes and recombinant enzymes is primarily mediated by CYP3A4 and UGT1A9. Three major oxidation products and three major glucuronides have been identified in vivo. Metabolism by rats is mostly by oxidation whereas metabolism by monkeys and humans is mostly by glucuronidation. Metabolism by monkeys closely resembles metabolism by humans and all metabolites found in humans are also found in monkeys. A greater diversity of metabolites has been identified among human in vivo specimens than among in vitro reaction products.
Following oral dosing of humans with 14C-bexagliflozin, the 3′-O-glucuronide contributed 32% of the parent AUC and all other metabolites contributed <10%. Of the 91.6% of input radioactivity recovered, 51.1% was in faeces, predominantly as bexagliflozin, and 40.5% was in urine, largely as the 3′-O-glucuronide. Unidentified metabolites contributed 0.27% of the input radiolabel.
A quantitative accounting for the metabolism and disposition of bexagliflozin in vivo has been developed.
Introduction
Type 2 diabetes mellitus (T2DM) is a disease of increasing incidence and prevalence worldwide (CDC, Citation2017; WHO, Citation2018). It is the leading cause of blindness, renal failure and non-traumatic amputation in American adults, and is associated with an increased risk for cardiovascular diseases and death.
Pharmacotherapy for T2DM typically consists of an initial course of metformin, followed by additional oral hypoglycaemic agents or parenteral medications as the disease worsens. Antidiabetic agents that promote insulin release (secretagogues), that intensify insulin action (sensitisers) or insulin itself, largely act by increasing the rate of diversion of plasma glucose to adipocytes. However, the aetiology of the disease is widely considered to be secondary to a loss of weight control leading to a hypertrophic adipose reservoir that is increasingly incapable of responding to insulin effectively. In this setting, the action of agents that promote glucose assimilation by adipocytes exacerbates the causative factors for disease emergence and makes restoration of euglycemic homeostasis more difficult. SGLT2 inhibitors represent attractive alternatives in this respect because their pharmacodynamic effect is exerted in the kidney.
The kidney functions by a default elimination process in which all of the contents of the plasma below ∼50 kDa in mass are excreted, followed by the selective reabsorption of the constituents that are beneficial to retain. The latter consist of nearly all of the water and electrolytes, as well as metabolically important small molecules, such as sugars, vitamins and amino acids. Glucose is a sufficiently important resource that the kidneys devote a tandem two-stage recovery process to its retention. In the first stage, in the pars convoluta of the proximal tubule (Vallon et al., Citation2011; Vrhovac et al., Citation2015), SGLT2 retrieves glucose by concentrative co-transport of one glucose molecule and one Na+ ion from the filtrate into the tubular epithelium (Ghezzi et al., Citation2018). Because there is a substantial transmembrane electrochemical potential for Na+ ions, glucose can be concentrated in the tubular epithelial cells even when found in a lower concentration in the extracellular fluid than in the epithelial cytoplasm (Ghezzi et al., Citation2018). In the second stage of reabsorption, in the pars recta of the proximal tubule (Vrhovac et al., Citation2015), the remaining glucose is taken up by the related transporter SGLT1, which co-transports one glucose molecule for every two Na+ ions (Ghezzi et al., Citation2018).
Humans lacking SGLT2 by genetic mutation of SLC5A2, the gene encoding SGLT2, are generally healthy but exhibit varying degrees of glucosuria (van den Heuvel et al., Citation2002; Santer et al., Citation2003). Humans lacking sodium-glucose linked transporter 1 (SGLT1, by mutation of SLC5A1) present as infants with a profound diarrhoea that is fatal unless strict dietary limitation of glucose and galactose is enforced (Wright, Citation1998). Affected individuals do not show substantial glucosuria (Rieg & Vallon, Citation2018).
SGLT2 inhibitors have become an important new treatment option for adults with T2DM (ADA, Citation2019; Scheen, Citation2015). Unlike insulins, their secretagogues and sensitisers, SGLT2 inhibitors divert glucose to urine, causing a daily metabolic wasting of up to 500 kcal and offering an insulin-independent mechanism for restoration of euglycaemia. SGLT2 pharmacotherapy has been increasingly documented to have ancillary benefits, such as reduction in cardiovascular risks (Radholm et al., Citation2018; Wiviott et al., Citation2019; Zinman et al., Citation2015) and preservation of renal function (Perkovic et al., Citation2019), beyond the salutary effects of improved glycaemic control.
Bexagliflozin (EGT0001442) is a C-aryl glucoside SGLT2 inhibitor with a favourable therapeutic profile. It has been found to produce clinically meaningful decreases in glycated haemoglobin (HbA1c) in diabetic adults with stage 3B chronic kidney disease (Allegretti et al., Citation2019), and consistently induces weight loss as well as substantial reductions of blood pressure in clinical trial populations (Allegretti et al., Citation2019; Halvorsen et al., Citation2019). This article presents the principle conclusions regarding the metabolism of bexagliflozin in rats, monkeys and humans.
Materials and methods
Reagents and standards
Reagents were of analytical grade or higher unless otherwise noted. Bexagliflozin was provided by Theracos Inc. for clinical experimentation or by Egret Pharma (Shanghai) Ltd. for non-clinical studies. [14-C]-bexagliflozin with a specific activity of 39 μCi mg−1 (20.18 mCi [0.75 GBq]/mmol, 99.8% radiochemical purity) was prepared from uniformly 14C-labeled glucose by PerkinElmer Life and Analytical Sciences (Boston, MA). Internal standard EGT0001479 (bexagliflozin in which each of the hexose carbons were replaced by 13C) and all metabolites used as chromatographic standards (EGT0001301, EGT0001494, EGT0001663, EGT0001684, EGT0001685, EGT0002147, EGT0002148, and EGT0002149) were synthesised and characterised by Egret Pharma (Shanghai) Ltd. Methyl-α-D-[U-14C]-glucopyranoside (AMG) was purchased from PerkinElmer. The individual compounds listed in were provided by Egret Pharma (Shanghai). EGT0001663 is a racemic mixture of compounds resulting from hydroxylation of the methylene bridge of the substituted diphenylmethane moiety, individually identified as EGT0001684 [(S)-EGT0001663] and EGT0001685 [(R)-EGT0001663]. If the chromatographic methods could not resolve these compounds, they are referred to as EGT0001663. The nomenclature for glucuronides is based on the glucose numbering system, not the IUPAC (pyran-based) system. Thus EGT0002147, 2148 and 2149 () are referred to here as the 2′-, 6′- and 3′-O-glucuronides, respectively.
Table 1. In vitro activity of bexagliflozin and its principal human metabolites.
In vitro assays
SGLT2 uptake assays were performed as described (Zhang et al., Citation2019), by measuring the sodium-dependent uptake of methyl-α-d-[U-14C]-glucopyranoside in cells expressing recombinant human SGLT2.
Following pilot studies to optimise conditions, human liver microsome reactions were conducted at a microsomal protein concentration of 2 mg mL−1 and at a single concentration of bexagliflozin (1 mM) for 2 h in a reaction mixture containing reduced nicotinamide adenine dinucleotide phosphate (NADPH; 2.0 mM when present), uridine diphosphoglucuronic acid (UDPGA; 2.0 mM when present), MgCl2 (10 mM) and EDTA (2 mM) in a total volume of 0.5 mL phosphate buffered saline (100 mM, pH 7.4). Control experiments were performed by excluding either NADPH or UDPGA or both from the incubation mixtures. Reactions were terminated by adding ice-cold acetonitrile to the reaction mixtures. Samples were stored frozen at −70 °C for analysis.
Recombinant human UGT reaction mixtures contained 50 mM Tris (pH 7.5), 10 mM MgCl2, 5 mM UDPGA, 0.025 mg mL−1 alamethicin, 0.35 mg mL−1 UGT membrane protein and 100 μM bexagliflozin. Samples were incubated at 37 °C for 2 h in a 200 μL volume. Reactions were terminated by the addition of 100 μL of acetonitrile, centrifuged at 10,000g for 3 min and the supernatants transferred to fresh tubes. Prior to analysis samples were centrifuged at 10,000g for 5 min. The supernatants were collected and an aliquot (10 μL) was injected directly into an HPLC-PDA/MS system for analysis.
Bexagliflozin (10 and 100 μM) was incubated in duplicate with a panel of recombinant human CYP enzymes (Bactosomes, Cypex Ltd., Dundee, UK) rCYP1A2, rCYP2B6, rCYP2C8, rCYP2C9, rCYP2C19, rCYP2D6, rCYP3A4, at 10 pmol CYP per incubation. Incubations were conducted at 37 ± 1 °C for 0.5 h in 0.2 mL incubation mixtures with an NADPH-generating system. Reaction samples were analysed by LC/MS/MS to quantify the formation of EGT0001301, EGT0001494 and EGT0001663.
Animals
Male Sprague–Dawley rats, 240–270 g, were purchased from Shanghai Laboratory Animal Co., Ltd. (Shanghai, China) or Charles River UK, Ltd. Cynomolgus monkeys were provided by Aptuit Ltd., Edinburgh UK. All animal studies were performed in accordance with applicable laws and regulations and following protocols approved by the respective animal care and use committees of the participating institutions. Bexagliflozin was delivered by oral gavage as a solution in 10% polyethylene glycol of mean molecular weight ∼400 Da (PEG400) for radioisotope studies and in 30% PEG400 for non-isotopic studies. The form of bexagliflozin used in non-isotopic studies was a 2:1 l-proline: bexagliflozin co-crystal, identified as EGT0001474.
Rats were delivered [14C]-bexagliflozin at a target dose level of 3 mg kg−1 (135 μCi [5 MBq] kg−1). For analysis of excreta the urine and faeces from three animals individually housed in glass metabolic cages were collected up to 120 h post-dose. At the time of faecal collection, the cages were washed with water to collect any residual radioactivity. After aqueous washing, each cage was rinsed with methanol, and the methanol wash separately subjected to analysis. Faeces were homogenised in water. Any cage debris was collected on a daily basis to be pooled by animal over the entire period of collection. Because all the input radioactivity was recovered from the excreta, the cage debris samples were not analysed. Expired CO2 was collected into 2 serial solvent traps containing a CO2 absorbing solution of 2-ethoxyethanol: ethanolamine, 7:3 (v/v). Collections of expired air were discontinued at 48 h post-dose, after it was established that <0.5% of the dose was recovered in the previous 24 h collection period. For pharmacokinetic sampling, serial blood specimens (∼200 µL) were drawn from the tail vein and transferred into K3EDTA tubes at pre-dose, 0.5, 1, 2, 4, 8, 12 and 24 h post-dose. Samples were thoroughly mixed and placed on wet ice prior to centrifugation at ∼4 °C within 1 h of collection, and the plasma was removed for analysis. For analysis of metabolites, nine animals were dosed and cohorts of three each underwent euthanasia and exsanguination at 0.5, 2 and 8 h post-dose.
Four male non-naïve cynomolgus monkeys each underwent a single oral delivery of [14C]-bexagliflozin at a target dose level of 3 mg kg−1 and 131 μCi (4.83 MBq) kg−1. Following administration of the test article, urine and faeces were collected up to 168 h post-dose. Whole blood specimens were collected pre-dose and at 0.5, 1, 2, 4, 8, 12 and 24 h following administration. For focussed analyte profiling, male and female cynomolgus monkeys were dosed at 60 mg kg−1 and plasma samples collected in the context of a toxicokinetic sampling programme.
Animal sample preparation
Plasma
The pooled plasma samples were deproteinised by the addition of acetonitrile (1.5 mL/mL plasma) and vortex mixed. The precipitated protein was removed by centrifugation (1800g for 15 min). The supernatant was collected and evaporated to dryness under a stream of nitrogen at 35 °C and the residue dissolved in methanol (500 µL). The methanol was centrifuged at ∼4300g for 10 min. Duplicate aliquots (20 µL) were taken from the centrifuged samples to determine the recovery efficiency.
Urine
Urine samples, pooled by species and time point were prepared by removing ∼10% of the original weights to generate pools over the time period of 0–6 h. These samples were centrifuged at 1800g for 10 min to remove particulate matter. Duplicate aliquots (20 µL) were taken from the centrifuged samples to determine the recovery efficiency.
Faeces
Faeces samples, pooled by species and time point, were prepared by collecting ∼10% of the original homogenate weight to generate pools over the time periods of 0–24 h and 24–48 h (monkey only). Approximately five volumes of methanol were added to the pooled faeces. The samples were vortex mixed to suspend the sample and mechanically disrupted with a rotary homogeniser. The suspension was vortex mixed, sonicated for ∼30 min and further vortex mixed, after which it was centrifuged at 1800g for 15 min. The supernatant was removed and evaporated to dryness under nitrogen. Additional methanol was added and used to re-suspend the residual pellet by vortex mixing, sonication and further vortex mixing prior to centrifugation. The supernatant was removed and evaporated to dryness under nitrogen. The extraction procedure of the faecal pellet was repeated two additional times with the solvents from each extraction combined into a single pooled sample. The percent recovery in the solvent extract was calculated by taking an aliquot from the pooled extracts for liquid scintillation counting (LSC) (20 µL) and comparing the values to the calculated disintegrations per min in the total weight of faeces taken for extraction. The solvent was then removed by evaporation under nitrogen at 35 °C and the residue dissolved in methanol. Duplicate aliquots (20 µL) were taken from the samples to estimate recovery efficiency.
Human subjects
Male volunteers between the ages of 18 and 55 years, inclusive, in good health based on their medical histories, physical examinations, ECGs, and routine laboratory tests (blood chemistry, haematology, urinalysis and drug screen), and having a body mass index between 18 and 30 kg m−2, inclusive, who were willing and able to be confined to the test facility premises, to comply with the requirements of the study, able to communicate well with investigators and to comprehend and provide written informed consent in accordance with institutional and regulatory guidelines were eligible. Participants were required not to have smoked within 6 months of admission, to have passed an alcohol abuse screen, and to either be sterile or agree to a stringent form of contraception for at least 45 days following incarceration.
Prospective subjects were ineligible if they had a medical history incompatible with participation, a heart rate >100 bpm, QRS >120 ms, QTcB >450 ms, PR >220 ms or any rhythm disturbance considered clinically significant, a history of alcohol consumption exceeding 14 drinks/week, a blood pressure above 140/90, a blood donation in excess of 500 mL within 60 days, a positive result for HBV, HCV or HIV, a history of drug, vitamin, herbal or dietary supplement use within 14 days, exempting acetaminophen at <2 g/day; if they had been exposed to an investigational drug within 30 days, had previously been exposed to bexagliflozin, had a history of drug abuse, were not willing to abstain from consuming products containing caffeine or alcohol from 48 h prior to and for the duration of the study, or grapefruit, grapefruit juice or related products for 14 days prior to and for the duration of the study, had a febrile illness at the time of dosing, had inadequate venous access, had been vaccinated within 30 days, had a history of incontinence or bladder dysfunction, had a history of recurrent urinary tract infections, had participated in more than one other radiolabelled investigational study drug trial within 12 months or any radiolabelled study within 6 months or any study such that the total exposure would have exceeded 5 rem in a period of one year, or did not have regular bowel movements.
The study was carried out according to the International Conference on Harmonisation tripartite guideline for Good Clinical Practice and in compliance with local laws and regulations and the principles of the Declaration of Helsinki (1996). The protocol and informed consent were reviewed and approved by the sponsor and by an Institutional Review Board prior to initiation of study conduct. Each subject provided written informed consent prior to participation in any study procedures.
Human study design
Absorption, distribution, metabolism and excretion were measured in a phase 1, single dose, open-labelled, non-randomised single-centre study. Following an overnight fast of 10 or more hours, six healthy adult male participants ingested a single oral dose of 50 mg of [14C]-bexagliflozin containing 100 μCi of total radioactivity in 120 mL of 5% ethanol solution, followed by two 50 mL ingestions of water. A standardised breakfast was provided 30 min later. Plasma and whole blood samples were collected within 30 min prior to dosing, and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 36 and 48 h after dosing. Samples were collected every 24 h thereafter to 192 h post-dose, but contained negligible radioactivity from hour 48 on. The total collection time varied with radioactivity recovery. Urine samples were collected at pre-dose (−12 to 0 h) and in 12 h batches for 48 h followed by 24 h collections on subsequent days for up to 9 days post-dosing. Faecal samples were collected every 24 h. For both urine and stool specimens, collection was to continue until radioactivity levels in specimens from 2 consecutive collection intervals were ≤1% of the administered dose and the total excreted radioactivity levels had exceeded 90% of the administered radioactivity or until 9 days post-dose, whichever occurred first. The final collection was at 192 h post-dose.
Human sample preparation
Plasma
Approximately 2–5 g of each pooled plasma sample was combined with acetonitrile (sample: solvent, 1:3, w:v), vortex mixed, sonicated, centrifuged and the supernatants were removed. The extraction was repeated twice, and the supernatants were combined. Duplicate aliquots were analysed by liquid scintillation counting (LSC) to determine extraction recoveries, which ranged from 86.4 to 100%. The combined supernatants were evaporated to dryness and reconstituted in 350 µL of acetonitrile: water: methanol (3:3:1, v:v:v). Samples were vortex mixed, sonicated, centrifuged, and duplicate aliquots were analysed by LSC to determine reconstitution recoveries, which ranged from 91.4 to 100%. The reconstituted samples were analysed by HPLC to determine the metabolite profiles, with fractions collected at 10 s intervals and analysed by solid scintillation counting.
Urine
Approximately 1 mL of each pooled urine sample was centrifuged in a microcentrifuge, and duplicate aliquots were analysed by LSC to determine recoveries of radioactivity, which were 100%. Pooled urine samples were analysed by HPLC to determine the metabolite profiles, with fractions collected at 10 s intervals and analysed by solid scintillation counting.
Faeces
Approximately 1–2 g of each pooled faeces sample was combined with acetonitrile (sample: solvent, 1:5, w:v), vortex mixed, sonicated, centrifuged and the supernatants were removed. The extraction was repeated twice, and the supernatants were combined. Duplicate aliquots were analysed by LSC to determine extraction recoveries, which ranged from 92.8% to 100%. The combined supernatants were evaporated to dryness and reconstituted in 1 mL of acetonitrile: water (50:50, v:v). Samples were vortex mixed, sonicated, centrifuged and duplicate aliquots were analysed by LSC to determine reconstitution recoveries, which ranged from 94.6% to 100%. The reconstituted samples were analysed by HPLC to determine the metabolite profiles, with fractions collected at 10 s intervals and analysed by solid scintillation counting.
Bioanalytical methods
Radioisotopic analysis, rats and monkeys
Samples were fractionated by a Jasco HPLC system (Jasco (UK) Ltd, Great Dunmow, UK) and a LabLogic ß-RAM model 3 radioactivity flow monitor (LabLogic Systems Ltd, Sheffield, UK). Chromatographic data were captured and evaluated using a validated software package, Chromeleon (v4.30; Dionex (UK) Ltd, Macclesfield). Regions of radioactivity and UV peaks were assigned manually on each chromatogram and quantified by peak area integration. Radioactivity in bulk samples was determined in a Tri-Carb Series (PerkinElmer LAS UK Ltd) liquid scintillation analyser. Quench correction was checked using quenched radioactive reference standards (PerkinElmer Life Sciences UK Ltd). Samples were counted to a Poisson error of 0.5% or for 5 min (whichever was attained first). Appropriate liquid scintillant blanks were also counted to establish background levels of radioactivity. Blank counts were subtracted from quench-corrected sample counts. The limit of quantification was taken as twice the background count level.
Analysis of unlabelled rat plasma samples
An Agilent 1100 liquid chromatographic system equipped with a G1379A vacuum degasser, a G1311A quaternary pump, a G1316A column oven, a G1313A autosampler (Agilent, Waldbronn, Germany) was used. Detection was performed on an API 4000 triple quadrupole mass spectrometer equipped with atmospheric pressure chemical ionisation interface (Applied Biosystems, Concord, Ontario, Canada) and was operated in multiple reaction monitoring (MRM) mode. The samples were chromatographed by using a Capcell Pak C18 MG column (100 × 4.6 mm, 5 μm, Shiseido, Tokyo, Japan).
The mobile phase consisted of acetonitrile: methanol: water: 10 mM ammonium acetate (450:275:275:5.5, v/v/v/v). The flow rate was 0.7 mL/min. The precursor → product transition pair was (m/z) 482.3 → 167.1 for bexagliflozin and m/z 488.2 → 169.0 for internal standard EGT0001479 ([13C]-bexagliflozin). An optimised collision energy of 37 eV was used. For analysis of EGT0001494, the mobile phase consisted of acetonitrile:methanol:water:10 mM ammonium acetate (164:100:588:2, v/v/v/v). The flow rate was maintained at 0.7 mL/min. The precursor → product transition pair was (m/z) 456.2 → 343.0 for EGT0001494. An optimised collision energy of 22 eV was used. For analysis of EGT0001301 plasma samples the mobile phase consisted of acetonitrile:methanol:water:10 mM ammonium acetate (450:275:677:5.5, v/v/v/v). The flow rate was 0.7 mL/min. The precursor → product transition pair was (m/z) 442.2 → 329.1 for EGT0001301. An optimised collision energy of 19 eV was used.
Analysis of unlabelled monkey samples
A Shimadzu LC-20A liquid chromatographic system equipped with a DGL-20A vacuum degasser, a dual pump and an SIL-20A autosampler (Shimadzu) was used. Detection was performed on an API 4000 LC-MS/MS mass spectrometer equipped with TurboIonSpray (ESI) Interface (Applied Biosystems, Concord, Ontario, Canada). Analyst 1.5 software packages (Applied Biosystems) were used to control the LC-MS/MS system, as well as the data acquisition and processing. Chromatographic separation was achieved on a Waters Nova-Pak® C18 (150 × 3.9 mm, 4 μm) column. The column temperature was maintained at ambient temperature (25 °C). A post-column diverter valve was used to direct HPLC column elute to a waste container for the first 4.0 min of the chromatographic run and then to the ionisation source. The flow rate was maintained at 0.8 mL/min and the following mobile phases were used: A: 0.1% formic acid in 0.5 mM NH4Ac under positive mode and 5 mM NH4Ac under negative mode; B: 0.1% formic acid in mixture of acetonitrile/methanol (50/50). The fraction of A was 85% to 2 min, 50% to 4.5 min and 15% to 5.6 min, thereafter 85% to 8 min (end). The ESI (+) conditions were: 5500 V spray voltage, source temperature 550 °C, sheath gas nitrogen gas flow 60 psi. The ESI (−) conditions were the same as the ESI (+) conditions except that −4500 V spray voltage was used. Quantification was performed using multiple reaction monitoring (MRM). The positive ion mode precursor → product transition pairs with collision energies (eV) were m/z 482.3 → 167.1 (37 eV) for EGT0001442, 488.2 → 169.0 (37 eV) for internal standard EGT0001479, 442.4 → 167.1 (39 eV) for EGT0001301, 456 → 191.3 (38 eV) for EGT0001494, 498.4 → 379.4 (38 eV) for EGT0001663. The negative ion mode transition pairs were 639.3 → 385 (−45 eV) and 639.3 → 373.3 (−47 eV) for EGT0002147, EGT0002148 and EGT0002149; and 469.3 → 376 (−24 eV) for the internal standard EGT0001479.
Additional metabolites in unlabelled human samples
Plasma samples collected at 2, 4, 8, 12, 24 and 48 h post dose were extracted with methanol and evaporated to dryness under nitrogen. Urine samples were collected at 0–12, 12–24, 24–36 and 36–48 h post dose. To 10 mL of urine was added 4 mL of methanol and the mixture vortex mixed and centrifuged at 10,000 rpm for 10 min. The supernatant was removed and evaporated to dryness under nitrogen. Faecal samples collected at 0–24 and 24–48 h post dose from 4 human volunteers were individually homogenised (∼3 times the faecal weight with a 50:50 ethanol:water solution) before preparation. Faecal homogenate samples were then extracted with methanol (1:3, sample:solvent) and the resulting supernatants were evaporated to dryness as determined by the following method: A weighed (∼10 g, or appropriate amount) aliquot of faecal homogenate was extracted by the addition of 30 mL (or appropriate volume) of methanol, vortex-mixed, sonicated for ∼30 min, and vortex-mixed prior to centrifugation at ∼4000 rpm for 10 min. The supernatant from each sample was decanted into a Corning centrifuge tube (50 mL). Each sample was extracted twice more with 30 mL (or appropriate volume) of methanol and the supernatant fractions were pooled and evaporated to dryness under a gentle stream of nitrogen. A total of 48 dried plasma samples, 32 dried urine samples and 8 dried faecal homogenate samples were shipped on dry ice. At reception, the samples were stored at −70 ± 5 °C and protected from light until analysis. To each dried urine sample, 400 μL of methanol was added and vortex-mixed, sonicated for 15 min, and vortex-mixed prior to centrifugation at 3000 rpm for 10 min. To investigate metabolites of bexagliflozin, plasma and urine samples from volunteers were pooled. The pooling strategy was summarised as follows: samples from subjects who received placebo were pooled into one sample at each time points post dose; samples from subjects who received bexagliflozin were pooled into one sample at each time points post dose. According to the pooling strategy, a 50 μL-aliquot of the supernatant from each urine sample was pooled, then evaporated to dryness at 40 °C under a stream of nitrogen and reconstituted in 500 μL of the acetonitrile/water/formic acid (50/50/0.05). A 10 μL aliquot of the resulting solution was injected onto the UPLC-Q/TOF MS system for analysis. To each dried plasma samples, 200 μL of the acetonitrile/water/formic acid (50/50/0.05, v/v/v) were added, and sonicated for 5 min. The sample was vortex-mixed prior to centrifugation at 3000 rpm for 10 min. According to the pooling strategy, a 50 μL aliquot of the supernatant from each sample was pooled and vortex-mixed. A 10 μL aliquot of the resulting solution was injected onto the UPLC-Q/TOF MS system for analysis. To the dried faecal homogenate samples, 1000 μL of the acetonitrile/water/formic acid (50/50/0.05) were added and the mixture sonicated for 10 min. The sample was vortex-mixed prior to centrifugation at 3000 rpm for 10 min and a 50 μL aliquot of the supernatant was diluted 10-fold with the acetonitrile/water/formic acid (50/50/0.05). A 10-μL aliquot of the resulting solution was injected onto the UPLC-Q/TOF MS system for analysis on a Waters ACQUITY UPLC System (Waters Corporation, Milford, MA). The column was an ACQUITY HSS T3 C18 Column with 1.7 μm particle size (100 × 2.1 mm). The column temperature was 40 °C and the flow rate was 0.5 mL/min. The mobile phase was 10 mM ammonium acetate (A) and acetonitrile with 0.05% formic acid (B). The gradient used was 5–70% B linear in 15 min, then to 100% B linear from 15 min to 17 min and finally 5% B linear. The system equilibration time was 2.5 min and total run time was 20 min. The injection volume was 10 μL. Analytes were detected by a Synapt Q-TOF MS (Waters Corporation, Milford, MA) by positive or negative electrospray. The data acquisition range was 80–1000 Da. Leucine enkephalin was used as the lock mass (m/z 556.2771). The source temperature was 120 °C, the desolvation gas temperature was 400 °C, the capillary voltage was 3.0 kV and the cone voltage was 20 V. The desolvation gas was nitrogen, and the collision gas was argon. Data acquisition was performed by Masslynx V4.1 (Waters Corporation, Milford, MA). Data processing was carried out with Metabolynx using an enhanced set of biotransformations after application of a customised mass defect filter (MDF). The structural elucidation was performed using MassFragment™.
Results
Metabolism in vitro
Metabolism by human liver microsomes and recombinant enzymes
Following incubation with human liver microsomes, HPLC-PDA/MS analysis identified the presence of the 3′-O-glucuronide, EGT0002149. Three phase 1 metabolites, M1 (EGT0001301), M2 (EGT0001494), and M4 (EGT0001663), were found and characterised by HPLC-PDA/MS (for structures, please refer to ). Other glucuronides and glucuronide conjugates of phase 1 metabolites were not observed in any samples. Following incubation of bexagliflozin with human liver microsomes in the presence of UDPGA, the glucuronide EGT0002149 was identified. Following incubation of bexagliflozin with human liver microsomes in the absence of UDPGA, no glucuronide conjugates were observed. No other potential metabolites were found in the sample. Formation of the three oxidative metabolites was NADPH-dependent.
Following incubation of bexagliflozin with individual UGTs (UGT1A4, UGT1A6, UGT1A9, UGT1A10 and UGT2B7), glucuronide conjugation of bexagliflozin was only observed following incubation with recombinant UGT1A9. No glucuronide formation was detected following incubation in the absence of enzyme. By comparison of the retention time and mass spectrum, the glucuronide conjugate of bexagliflozin following incubation with recombinant UGT1A9 and UDPGA was identified as the 3′-O-glucuronide EGT0002149. In a subsequent testing, small amounts of EGT0002149 were also found to be formed following incubation with recombinant human UGT1A1.
The formation of EGT0001301 () by recombinant CYP3A4, CYP2C8 and CYP2D6 was approximately 40-fold, 3-fold and 3-fold of the negative control values, respectively. The rate of EGT0001301 formation by all other recombinant human CYP enzymes evaluated was similar to that of negative controls. Bexagliflozin was converted to EGT0001494 only by recombinant human CYP3A4 and only at a bexagliflozin concentration of 100 μM. Bexagliflozin at 10 and 100 μM was converted to EGT0001663 by CYP3A4. Formation of EGT001663 was also observed following incubation of bexagliflozin at 10 μM with CYP2D6. The rate of EGT0001663 formation was ∼10-fold higher by CYP3A4 than by CYP2D6. EGT0001663 was not formed by the remaining human CYP enzymes evaluated.
Pharmacological activity of metabolites
The pharmacological activity of the metabolites was assessed by an assay measuring the sodium-dependent uptake of the non-metabolizable substrate, methyl-α-d-[U-14C]-glucopyranoside, by cells expressing recombinant human SGLT2 (Zhang et al., Citation2019). presents the results of the evaluations. A substantial stereospecific effect was seen for isomers resulting from bridging methylene oxidation. With the exception of EGT0001684, all of the metabolites had <10% of the activity of the parent compound ().
Metabolism in vivo
Metabolism by rats
presents the distribution of radioactivity as a percentage of input dose (for urine and faeces) or of the percentage of plasma radioactivity (for plasma abundance).
Table 2. Distribution of radiolabel among parent compound and metabolites in rats, monkeys and humans.
Plasma radioactivity
For all samples, the majority of radioactivity (ranging from 73% to 86%) was detected at a retention time similar to that of bexagliflozin. Material coeluting with EGT0001494 ranged from 4% to 8% of sample radioactivity. Material eluting at a retention time similar to that of EGT0001301 was detected in the 2 h plasma sample only and accounted for 4% of sample radioactivity, and label eluting at a retention time similar to that of EGT0001663 was detected in the 0.5 h plasma sample only and accounted for 2% of sample radioactivity. An unidentified substance (VM11) eluting at 27.3 min accounted for 2–11% of sample radioactivity.
Urine radioactivity
The largest percentage of radioactivity in urine (∼58%) was detected at the retention time of EGT0001494. Label eluting with the retention time of bexagliflozin, EGT0001301 and EGT0001663 accounted for 24%, 2% and 6% of urine radioactivity, respectively. Unidentified metabolite VM2 accounted for ∼5% of urine radioactivity.
Faeces radioactivity
The largest fraction of faecal radioactivity (∼71%) eluted with the retention time of bexagliflozin. Substances having retention times similar to those of EGT0001301, EGT0001494 and EGT0001663 accounted for ∼12%, 7% and 4% of faecal radioactivity, respectively. Unidentified metabolite VM7 accounted for ∼5% of faecal radioactivity.
Focussed analyte detection
To improve the precision of metabolite quantitation, the amount of circulating EGT0001301 and EGT0001494 was determined by an HPLC/MS/MS method in male rats administered a single oral dose of 6.7 mg kg−1 of bexagliflozin. The AUC0–∞ values for EGT0001301 and EGT0001494 were 517 ± 24 and 516 ± 60 ng h mL−1, respectively, under conditions in which the corresponding value for the parent compound was 21885 ± 3623 ng h mL−1. The ratios of exposure to bexagliflozin by AUC were each 0.0236.
Metabolism by monkeys
presents the distribution of radioactivity as a percentage of input dose (for urine and faeces) or of the percentage of plasma radioactivity (for plasma abundance).
Plasma radioactivity
The majority of radioactivity in plasma (ranging from 63% to 76%) was detected at a retention time similar to that of bexagliflozin. Material coeluting with EGT0001494 ranged from 7% to 9% of sample radioactivity.
Urine radioactivity
From urine, 29.2% of the delivered dose was recovered, although the proportion rose to 41.4% with the inclusion of label recovered from cage wash fluids. The largest proportions of urine radioactivity, 38% and 42%, eluted with the retention times of EGT0001494 and bexagliflozin, respectively. Isotope co-eluting with EGT0001663 accounted for ∼11% of urine radioactivity. Unidentified compound VM7 accounted for ∼6% of urine radioactivity. Substances coeluting with EGT0001301 were not detected.
Faeces radioactivity
The majority of radioactivity in faeces (∼61%) had the elution behaviour of bexagliflozin. Substances eluting with the retention times of EGT0001301, EGT0001494 and EGT0001663 comprised 16–18%, 11–12% and approximately 1% of the faecal radioactivity, respectively. Unidentified substance VM1 contributed 6–8% of faecal radioactivity.
Focussed analyte detection
The amount of circulating metabolites was determined by an HPLC/MS/MS method for samples from male and female cynomolgus monkeys administered 60 mg kg−1 of bexagliflozin. presents the ratios of the calculated AUC0–∞ values for individual metabolites to those of the parent compound. Metabolism by male and female monkeys did not show significant sex-dependent differences.
Table 3. Relative proportions of metabolites in male and female cynomolgus monkeys.
Clinical experimentation
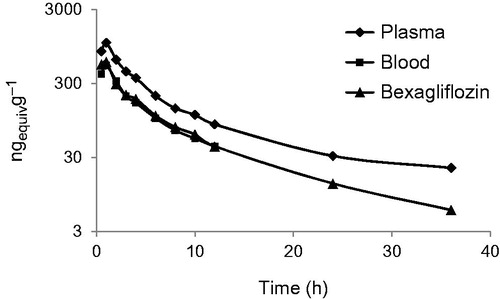
Following administration of a dose of 50 mg of bexagliflozin containing 100 μCi of 14 C-labelled compound to healthy adult male subjects, median Tmax occurred at 1 h post-dose and the Cmax (±SD) for total radioactivity in plasma, 1070 ± 156 ng mL−1, was approximately twice the Cmax for total radioactivity in blood, 541 ± 128 ng mL−1, indicating that little of the label partitioned into erythrocytes. The majority of excretion of total radioactivity occurred principally in the first 36 h after administration and was essentially complete by 48 h post-dose. By the time sample collection ceased, 91.6% of the input radioactivity had been recovered. Faecal excretion accounted for 51.1% and urinary excretion for 40.5% of the input label. depicts the total radioactivity in blood and plasma, and as bexagliflozin in plasma, as a function of time from dose administration. Supplementary Figure 1 presents radiochromatograms for plasma metabolites and displays the representative radiochromatograms spanning all of the individually enumerated metabolites in plasma and excreta.
Figure 1. Plasma concentration of total radioactivity in plasma, blood and as bexagliflozin in plasma as a function of time from dose administration in humans.
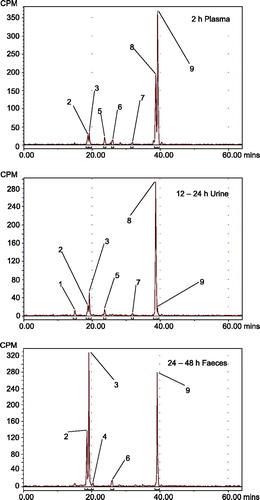
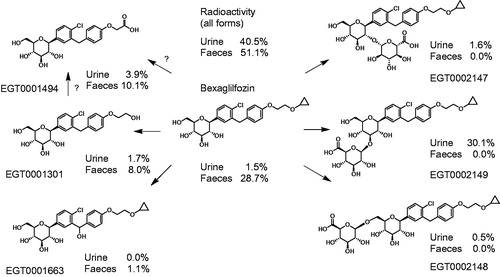
In urine, bexagliflozin accounted for 1.5% of the input dose. Most of the radioactivity was in the form of the 3′-O-glucuronide EGT0002149, which accounted for 30.1% of the input label. The remaining radioactivity was distributed among EGT0001301 (1.69%), EGT0001494 (3.88%), EGT0002147 (1.63%), EGT0002148 (0.45%) and an unidentified metabolite (0.15% of input). and Supplementary Figure 2 present radiochromatograms for urine metabolites. Nearly complete coverage was achieved with only six synthetic standards.
The majority of the radioactivity in faeces was attributable to bexagliflozin, accounting for 28.7% of the administered dose. EGT0001301 and EGT0001494 were the principal metabolites in faeces, accounting for 8.0% and 10.1% of the dose, respectively, by the time sample collection had ended. EGT0001663 was also identified in faeces, accounting for 1.06% of the dose through 144 h post-dose. An unidentified metabolite was detected in the 24- to 48-h sample only and accounted for 0.12% of the input label through 144 h. No glucuronides were detected in faeces, but the possible action of microbial glucuronidases could not be excluded. and Supplementary Figure 3 present radiochromatograms for the faecal metabolites. Nearly complete coverage by the synthetic standards was realised. Supplementary Figure 4 presents the postulated structures of additional, less abundant metabolites that were identified by UPLC/MS/MS of unlabelled human specimens.
The inferred pharmacokinetic parameters resulting from the administration of radiolabelled bexagliflozin in an oral solution are presented in . The largest contribution to exposure by metabolites was conferred by the 3′-O-glucuronide EGT0002149, at 32.2%. The next largest contribution was from the oxidative metabolite EGT0001494, at 6.8%. Collectively, oxidative metabolites contributed 14.1% and glucuronides contributed 35.8% of the parent exposure. Taken in conjunction with the activity data of , the predicted pharmacodynamic effect attributable to the action of metabolites was <1%.
Table 4. Pharmacokinetic parameters for bexagliflozin and its principal metabolites in humans, as inferred by distribution of 14C-labelled material.
summarises the metabolite prevalence estimated from the studies above for rats, monkeys and humans. Human and monkey metabolite patterns are closely similar, with an excess of oxidative metabolites seen in the monkey. With the exception of the principal human metabolite EGT0002149, all metabolites had greater abundance by AUC in monkey than in human plasma. Cynomolgus monkeys therefore represent an appropriate species for preclinical toxicology.
Figure 2. Representative radiochromatograms of human samples. Radiochromatograms depicting the nine discrete human specimen peaks are shown. Peak identifications: 1, metabolite HM1; 2, EGT0001301; 3, EGT0001494; 4, metabolite HM2; 5, EGT0002147; 6, EGT0001663; 7, EGT0002148; 8, EGT0002149; 9, bexagliflozin.
summarises the disposition of dose into human urine and faeces according to metabolite. All of the metabolites were present in urine, but only the oxidative metabolites were in faeces.
Figure 3. Metabolic fate of bexagliflozin in rats, monkeys and humans. The principal metabolites of the parent compound bexagliflozin in SD rats, cynomolgus monkeys and healthy human subjects are displayed above. The percentages represent the plasma AUC proportionality relative to bexagliflozin.
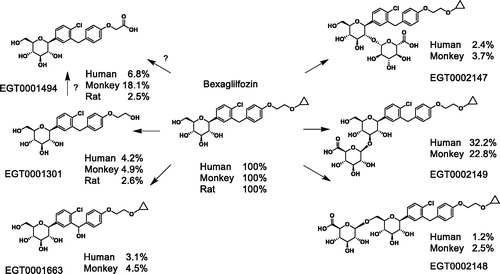
Figure 4. Relative disposition of bexagliflozin in human excreta. The percentages shown represent the proportion of input radioactivity recovered in the specified form.
Discussion
In this article, the principal findings of several studies investigating the metabolism of bexagliflozin have been collected. In vitro studies have identified CYP3A4 and UGT1A9 as the major isoenzymes acting on bexagliflozin. Additional contributions by CYP2C8, CYP2D6 and UGT1A1 have been detected.
The pattern of metabolite formation in vivo indicates that glucuronidation is the predominant pathway for biotransformation of bexagliflozin by humans. The predominant elimination product found in urine is the major circulating metabolite, the 3′-O-glucuronide. UGT1A9 is more highly expressed in kidneys than in liver, whereas UGT1A1 is found in liver but not kidneys (Knights et al., Citation2013). Hence renal glucuronidation may represent a significant source of clearance.
Approximately half of the dose is eliminated in urine and half in faeces. The material in faeces does not contain glucuronides, and is mostly parent compound, accompanied by the two most prevalent oxidative metabolites, EGT0001301 and EGT0001494. Although some of the bexagliflozin in faeces may represent unabsorbed compound, it would be expected, given the high concentration of circulating glucuronides, that at least some glucuronide would have been eliminated in bile, as this is a well-established pathway for xenobiotic disposition. That glucuronides were not found suggests the possibility that enteric microbial glucuronidases may have caused their degradation. The relatively high concentration of bexagliflozin in faeces, coupled with the relatively short terminal half-life in plasma, indicates that the contribution of colonic absorption/re-absorption to plasma persistence can be neglected.
The glucuronides have negligible activity in vitro, and of the oxidative metabolites only EGT0001684 has an activity within a factor of 10 of the activity of bexagliflozin. The fractional pharmacodynamic activity contributed by metabolites can be estimated by multiplying the relative abundance of each metabolite by its relative in vitro potency and summing over all metabolites. By this method of estimation, EGT0001494 contributes 0.17%, EGT0001301, 0.02%, and EGT0001663, 0.025–0.40% of the activity of bexagliflozin, the latter depending on the degree of stereospecificity of metabolism.
Metabolism by rats is dominated by oxidation whereas metabolism by monkeys is dominated by glucuronidation. The pattern of metabolism by monkeys aligns well with the pattern in humans, making monkeys an appropriate species for investigating the preclinical toxicology of bexagliflozin. The relative proportions of dose recovered in faeces and urine (with the inclusion of the cage wash contribution) are essentially the same for monkeys and humans. There are no metabolites found in substantial amounts in humans that are not also found in monkeys, and in all but one case, the abundance in monkeys exceeds that in humans.
In general, the coverage of human metabolites by the six characterised reference standards was excellent, and only 0.27% of input radioactivity was recorded in the form of unidentified compounds, 0.15% in urine and 0.12% in faeces. Although this is probably an underestimate given detection limitations, it nonetheless highlights the relative simplicity of the metabolism and adequacy of the metabolite coverage by synthetic standards. More metabolites were found by analysis of in vivo specimens than by analysis of in vitro reaction products.
Although compound EGT0001494 is a major in vivo metabolite in all three species, it is produced only sporadically and in small amounts in vitro. As a result, it is not clear whether it is principally produced directly from bexagliflozin or as a secondary oxidation product of EGT0001301, and the metabolic charts ( and ) reflect this uncertainty. It may also be that the EGT0001494 results from extrahepatic metabolic contributions not captured by the in vitro methodology. Two frequently cited extrahepatic sites for xenobiotic oxidation are the small bowel and kidneys.
Conclusion
Bexagliflozin undergoes oxidation and glucuronidation in humans to six major metabolites. The most prominent circulating metabolite is the 3′-O-glucuronide, which in vitro studies suggest is largely formed by the action of UGT1A9. The most prominent oxidative metabolites arise by cleavage of the cyclopropyl ether and by ether cleavage followed by oxidation of the resulting primary alcohol to a carboxylic acid, or by direct formation of the carboxylic acid. In vitro studies suggest that CYP3A4 is predominantly responsible for the formation of the oxidative metabolites. Metabolism by humans is closely similar to metabolism by monkeys, and all metabolites found in humans are also found in monkeys, and in all but one case, in greater relative abundance in monkeys than in humans. The contribution by metabolites to the pharmacodynamic action of bexagliflozin in humans can be estimated to be <1%.
Supplemental Material
Download PDF (130 KB)Supplemental Material
Download PDF (299.2 KB)Supplemental Material
Download PDF (117.1 KB)Supplemental Material
Download PDF (77.7 KB)Disclosure statement
A.W. has an equity interest in Theracos Inc. The remaining authors declare no financial interests.
Additional information
Funding
References
- ADA. (2019). Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes-2019. Diabetes Care 42:S90–S102.
- Allegretti AS, Zhang W, Zhou W, et al. (2019). Safety and effectiveness of bexagliflozin in patients with type 2 diabetes mellitus and stage 3a/3b CKD. Am J Kidney Dis. doi: 10.1053/j.ajkd.2019.03.417
- CDC. (2017). National Diabetes Statistics Report, 2017. https://www.cdc.gov/diabetes/data/statistics/statistics-report.html.
- Ghezzi C, Loo DDF, Wright EM. (2018). Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 61:2087–97.
- Halvorsen YC, Walford GA, Massaro J, et al. (2019). A 24-week, randomized, double-blind, active-controlled clinical trial comparing bexagliflozin to sitagliptin as an adjunct to metformin for the treatment of type 2 diabetes in adults. Diabetes Obes Metab. doi:10.1111/dom.13833
- Knights KM, Rowland A, Miners JO. (2013). Renal drug metabolism in humans: the potential for drug-endobiotic interactions involving cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT). Br J Clin Pharmacol 76:587–602.
- Perkovic V, Jardine MJ, Neal B, et al. (2019). Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 380:2295–306.
- Radholm K, Figtree G, Perkovic V, et al. (2018). Canagliflozin and heart failure in type 2 diabetes mellitus. Circulation 138:458–68.
- Rieg T, Vallon V. (2018). Development of SGLT1 and SGLT2 inhibitors. Diabetologia 61:2079–86.
- Santer R, Kinner M, Lassen CL, et al. (2003). Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol 14:2873–82.
- Scheen AJ. (2015). Pharmacodynamics, efficacy and safety of sodium-glucose co-transporter type 2 (SGLT2) inhibitors for the treatment of type 2 diabetes mellitus. Drugs 75:33–59.
- Vallon V, Platt KA, Cunard R, et al. (2011). SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22:104–12.
- Van Den Heuvel LP, Assink K, Willemsen M, Monnens L. (2002). Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet 111:544–7.
- Vrhovac I, Balen Eror D, Klessen D, et al. (2015). Localizations of Na(+)-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch 467:1881–98.
- WHO, 2018. Diabetes. https://www.who.int/news-room/fact-sheets/detail/diabetes.
- Wiviott SD, Raz I, Bonaca MP, et al. (2019). Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 380:347–57.
- Wright EM. (1998). I. Glucose galactose malabsorption. Am J Physiol 275:G879–82.
- Zhang W, Welihinda A, Mechanic J, et al. (2019). EGT1442, a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. Pharmacol Res 63:284–93.
- Zinman B, Wanner C, Lachin JM, et al. (2015). Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373:2117–28.