
Abstract
Viloxazine is currently being developed as a treatment for attention deficit/hyperactivity disorder (ADHD). The aim of these studies is to update the understanding of the rat and human metabolism and the in vitro drug–drug interaction profile of viloxazine to a degree where it meets current regulatory standards for such investigations.
In vivo absorption-distribution-metabolism-excretion (ADME) studies demonstrated that in humans 5-hydroxylation followed by glucuronidation is the major metabolic route. This route was also seen as a minor route in rats where the major route is O-deethylation with subsequent sulfation.
In humans, the 5-hydoxylation pathway is mediated by CYP2D6. An estimate for the fraction of the metabolism via this pathway suggests a PM/EM difference of <2-fold, making it highly unlikely that this will be an issue of clinical significance.
Viloxazine forms a unique N-carbamoyl glucuronide in humans. The chemical reactivity characteristics of this metabolite are similar to stable glucuronide conjugates and dissimilar from acyl glucuronides; therefore, it is regarded as a stable Phase II conjugate.
In vitro drug–drug interaction (DDI) testing indicates that viloxazine is not a significant inhibitor or inducer of CYPs and transporters with the exception of CYP1A2.
Introduction
During the 1970s, viloxazine (Emovit®, Vivalan®, Vivarin®, Vicilan®; ) was approved in Italy, Belgium, the United Kingdom, Ireland, Germany, Portugal, Spain, the former Yugoslavia, France, and Slovakia for the treatment of major depressive disorder (MDD). At that time, its mechanism of action (MoA) was described as norepinephrine reuptake inhibition. Later studies, however, found a more complex pharmacologic profile (Pinder et al., Citation1977).
Figure 1. Structure of viloxazine.
Supernus Pharmaceuticals, Inc. (Supernus) is currently developing SPN-812 (viloxazine extended released), as a treatment for attention-deficit/hyperactivity disorder (ADHD). SPN-812 is a multimodal serotonergic and noradrenergic modulating agent (SNMA) with demonstrated activity at serotonin receptors and the norepinephrine transporter. In vivo, viloxazine has been shown to increase serotonin (5-HT), norepinephrine (NE), and dopamine (DA) levels in the prefrontal cortex (Garcia-Olivares et al., Citation2020), a region strongly implicated in ADHD pathophysiology. In vitro, viloxazine exhibits antagonistic activity at 5-HT2B receptors and agonistic activity at 5-HT2C receptors (data on file, Supernus Pharmaceuticals, Inc.), although the downstream effects of this activity remains to be fully elucidated. Viloxazine also increases NE and DA levels through norepinephrine transporter (NET) inhibition.
Current FDA-approved medications for ADHD include CNS stimulants and nonstimulant medications such as norepinephrine reuptake inhibitors and alpha2-adrenergic receptor agonists (INTUNIV®, Citation2019; KAPVAY®, Citation2017; STRATTERA®, Citation2017).
When viloxazine was originally developed by Imperial Chemical Industries in the 1970s, its metabolism in rat, dog, and human was determined as part of the development package. These studies used the most up-to-date techniques and technology at the time, including paper chromatography and thin layer chromatography, and produced a comprehensive understanding of viloxazine metabolism in animal species (mouse, rat, rabbit, dog, and monkey) and humans (Case et al., Citation1975; Case & Reeves, Citation1975). In the animal species investigated, renal elimination was the major route of excretion of drug and metabolites. Viloxazine was shown to be extensively metabolized in rats with the major pathways being O-deethylation and sulfation (Case et al., Citation1975). In dogs, the major metabolic pathways were found to be hydroxylation of the phenyl ring, followed by subsequent conjugation with either glucuronic acid or sulfate, N-methylation, formation of the N-methyl-N-oxide, and oxidation of the morpholine ring (Case et al., Citation1975).
In humans, renal elimination was also found to be the major route of excretion of viloxazine and its metabolites, with more than 90% of the administered radioactivity recovered in the urine over 96 h (Case & Reeves, Citation1975). As such, the human metabolite profile resembled what was seen in dogs as opposed to rats. Consequently, the major rat O-deethylation pathway was nearly absent in humans and the major metabolic routes appeared to be hydroxylation and glucuronidation of the phenyl ring, N-methylation, and oxidation of the morpholine ring. Metabolism on the morpholine ring was minor compared to the metabolism on the phenyl ring. This study identified the glucuronide of the 5-hydroxy metabolite as the major radioactive component excreted in human urine with minor amounts of 5-hydroxy metabolite, along with the 4-hydroxy metabolite and its glucuronide (Case & Reeves, Citation1975).
The studies reported here aim to update our understanding of the rat and human metabolism of viloxazine using modern techniques and technologies. Moreover, characterization of the in vitro drug-drug interaction profile of viloxazine will help facilitate its continued safe administration in humans receiving concomitant medications, as well as meeting current regulatory standards for such investigations.
Materials and methods
Radiolabeled drug substance
[14C]-viloxazine was synthesized by Quotient Chemistry, Rushden, UK. The chemical and radiochemical purity was >99% (with no unspecified impurity >1.0%) as verified by high-pressure liquid chromatography (HPLC).
Nonradiolabeled materials
3-Hydroxyviloxazine, 4-hydroxyviloxazine, 5-hydroxyviloxazine, and 6-hydroxyviloxazine were synthesized by Quality Chemical Laboratories, Wilmington, NC.
In vitro metabolism
Primary fresh hepatocytes from Sprague Dawley rats, dogs, and humans (n = 2 male donors) and cryopreserved (n = 1 male donor) were obtained from CellzDirect, Pittsboro, NC. [14C]-viloxazine was incubated with the hepatocytes (0.75 × 106 cells/mL) at 37 °C in Williams’s E medium (final volume 1 mL). Incubations were initiated by the addition of 10 µL of [14C]-viloxazine aqueous solutions (0.3 and 3 µg/mL) and terminated at 0, 30, 60, and 120 min by the addition of 2 mL acetonitrile. The samples were vortex mixed, and then stored on ice before centrifugation (1400×g, 10 min, 4 °C) to precipitate the protein. The supernatants were removed and evaporated to dryness and then reconstituted in 460 µL of mobile phase A (0.2% formic acid in water) and 40 µL of mobile phase B (0.2% formic acid in acetonitrile (ACN):isopropanol (95:5)) before being analyzed by HPLC with radiometric detection to determine the metabolite profiles. Incubations with [14C]-7-ethoxycoumarin showed that CYP enzyme, glucuronidation, and sulfation activities were present in the hepatocyte incubations.
In vitro CYP phenotyping
In vitro incubation conditions
Microsomal incubations were performed in a total volume of 0.25 mL. Pooled human hepatic microsomes (mixed gender pool CellzDirect, Austin, TX) were diluted with 0.1 M potassium phosphate buffer, pH 7.4, containing 1 mM EDTA and 12.5 µL [14C]-viloxazine stock solution to give final protein concentration of 0.5 mg microsomal protein/mL. Samples were preincubated at 37 °C for 3 min, followed by the addition of NADPH (1 mM final concentration). Incubations were terminated at the designated time by the addition of 0.5 mL of ice-cold ACN and being vortex mixed. Incubation mixtures were centrifuged at 1400×g for 10 min at 4 °C. Supernatants were transferred to separate vials and stored at −20 °C until analysis. All incubations were performed in triplicate.
Optimization of in vitro incubation conditions
[14C]-viloxazine (final concentration of 10 µM) was incubated with 0.1, 0.25, or 0.5 mg protein/mL of human hepatic microsomes for 0, 5, 10, 20, and 30 min. Following analysis, the experiment was repeated under the following conditions: 0.5 mg/mL of human hepatic microsomes for 0, 15, 30, 45, 60, and 90 min. Control incubations were performed at 0 and 90 min. Incubation conditions that produced a linear rate of metabolite formation were used as assay conditions for subsequent experiments.
Kinetic analysis using pooled human hepatic microsomes
The in vitro incubation conditions detailed above were followed for the kinetic analysis experiment and used to determine the kinetic values (Km and Vmax) with the following exception: pooled human hepatic microsomes were diluted with assay buffer and [14C]-viloxazine (final incubation concentrations of 10, 25, 50, 75, 100, 250, 500, 750, and 1000 µM).
Chemical inhibition analysis using pooled human hepatic microsomes
The in vitro incubation conditions detailed above were used for the chemical inhibition analysis experiment with the following exceptions: (1) Sulfaphenazole (3 µM) for CYP2C9, (2) MAB-1A2 Monoclonal Antibody Inhibitory to CYP1A2, (3) MAB-2B6 Monoclonal Antibody Inhibitory to CYP2B6, (4) IH-MAB-2C19 Monoclonal Antibody Inhibitory to CYP2C19, (5) IH-MAB-2D6 Monoclonal Antibody Inhibitory to CYP2D6, (6) MAB-2E1 Monoclonal Antibody Inhibitory to CYP2E1, and (7) Anti-Rat CYP3A2 (used for human CYP3A4) were selected as enzyme-specific inhibitors to assess the involvement of CYP1A2, 2B6, 2C9, 2C19, 2D6, 2E1, and 3A4 in the metabolism of [14C]-viloxazine. Pooled human hepatic microsomes were diluted with buffer, inhibitor, and [14C]-viloxazine (40 µM). Control incubations were performed by adding the equivalent amount of solvent with antibodies (Tris buffer) and with sulfaphenazole (in ACN), as applicable.
Incubations with cDNA-expressed human cytochromes P450 (SupersomesTM)
The in vitro incubation conditions detailed above were used with the following exceptions. Incubation mixtures contained SupersomesTM expressing recombinant human CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, or vector-treated controls (Gentest, Woburn, MA). These were diluted with buffer, [14C]-viloxazine (40 µM), and NADPH (1 mM) to give a final cDNA-expressed CYP concentration of 50 pmol CYP/mL. The exception was CYP2E1, which was at 100 pmol CYP/mL. For CYP2C9 (and the appropriate vector control), the assays were run in 0.1 M Tris buffer (pH 7.4). Incubations were terminated at 30 min as described above.
HPLC analysis of viloxazine from in vitro studies
The metabolic profiles following incubation of [14C]-viloxazine were generated using HPLC with radio-detection (Waters Acquity HPLC system, Waters Corporation, Milford MA). Separation was achieved using Phenomenex Polar-RP 80 A, 4 µm, 4.6 × 250 mm column (Phenomenex, Torrance, CA) maintained at 30 °C by using two mobile phases: A (0.2% formic acid in water) and B (0.2% formic acid in ACN:isopropanol (95:5)). A linear gradient at a flow rate of 1 mL/min was set as follows: 8% B (0 min) → 8% B (10 min) → 60% B (18 min) → 60% B (30 min) → 100% B (32 min) → 8% B (33 min) → 8% (38 min). The column elute was introduced into a Shimadzu SPD-10A UV detector set at 254 nm and a Packard Radiomatic 500 Series radio-detector. Incubations with [14C]-7-ethoxycoumarin showed that CYP enzyme, glucuronidation, and sulfation activities were present in the hepatocyte incubations.
In vitro CYP inhibition
Viloxazine was incubated with a pool of human liver microsomes (20 mg/ml, mixed gender Corning®, Woburn, USA) in the presence of CYP-selective substrates at final concentrations of 0, 3.37, 10.1, 33.7, 101, 303, and 1010 µM for CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5, and at concentrations of 0, 0.001, 0.003, 0.1, 0.3, 1, and 10 µM for CYP1A2 only. The following probe CYP substrates were used: (1) phenacetin for CYP1A2, (2) bupropion for CYP2B6, (3) amodiaquine for CYP2C8, (4) tolbutamide for CYP2C9, (5) (S)-mephenytoin for CYP2C19, (6) dextromethorphan for CYP2D6, (7) midazolam, and (8) testosterone for CYP3A4/5. Incubations were performed in triplicate using 96-well plates at 37 °C. Incubation mixtures contained viloxazine, human liver microsomes (0.3 mg/ml for CYP2C9 and 2C19, 0.2 mg/ml for CYP2D6, 0.1 mg/ml for CYP1A2, 2B6, and 3A4 with testosterone and 0.025 mg/ml for CYP2C8 and 3A4/5 with midazolam), and 100 mM phosphate buffer, pH 7.4. Samples were preincubated for 0 or 30 min with and without cofactor (1 mM NADPH) prior to the addition of CYP-selective substrate and cofactor (if not already present in the preincubation). The final incubation volume was 200 µL with a solvent concentration of 0.25%. Positive control incubations contained a chemical inhibitor selective for each CYP enzyme in place of the viloxazine. Positive controls for reversible inhibition were as follows: (1) α-naphthoflavone for CYP1A2, (2) phencyclidine for CYP2B6, (3) quercetin for CYP2C8, (4) sulfaphenazole for CYP2C9, (5) modafinil for CYP2C19, (6) quinidine for CYP2D6, and (7) ketoconazole for CYP3A4/5. For the time-dependent inhibition, the positive controls were as follows: (1) furafylline for CYP1A2, (2) phencyclidine for CYP2B6, (3) phenelzine for CYP2C8, (4) tienilic acid for CYP2C9, (5) ticlopidine for CYP2C19, (6) paroxetine for CYP2D6, and (7) azamulin for CYP3A4. The positive control inhibitors where incubated over concentration ranges that produced acceptable IC50 values. Microsomal reactions were terminated after a 10 min incubation period by the removal of an aliquot (50 µL) into methanol (100 µL) containing the appropriate internal standard. The terminated samples were analyzed LC-MS/MS. Where >50% inhibition of metabolite formation was observed, an IC50 value was calculated using GraFit v7.0.2 (Erithacus Software Ltd, Surrey, UK).
In vitro CYP induction
Methods for mRNA study
The potential of viloxazine to induce CYP1A2, 2B6, and 3A4 was studied in cryopreserved human hepatocytes from three individual donors (BioreclamationIVT, Baltimore, MD). The hepatocytes from each donor were poured directly into a tube containing 5 mL of pre-warmed (37 °C) InVitroGRO CP medium (BioreclamationIVT, Baltimore, MD) supplemented with Torpedo antibiotic mix for each vial thawed. The cell suspension was then diluted to 0.7 × 106 cells/mL with InVitroGRO CP medium supplemented with Torpedo antibiotic mix. The 24-well collagen coated plates were seeded at 500 µL (∼0.35 × 106 cells) per well and incubated at 37 °C in a humidified atmosphere containing 5% carbon dioxide for approximately 4 h. The plates were assessed for attachment and a Matrigel overlay was applied. The InVitroGRO CP medium was replaced with ice cold supplemented Williams’ Medium E containing Matrigel at a final concentration of 0.25 mg/mL. Plates were returned to the incubator overnight. Upon study initiation, the supplemented Williams’ Medium E was removed and replaced with fresh pre-warmed supplemented Williams’ Medium E containing either viloxazine at final concentrations of 1.4, 3.5, 14, 35, 70, 140, and 350 µM in water, 50 µM omeprazole (positive control for CYP1A2 induction), 1 mM phenobarbital (positive control for CYP2B6 induction), 25 µM rifampicin (positive control for CYP3A4 induction), or 25 µM flumazenil (negative control for CYP1A2, 2B6, and 3A4 induction). Appropriate solvent controls – water for viloxazine, methanol for omeprazole, phenobarbital, rifampicin and flumazenil at 0.5% of the final incubation volume were also included. Following exposure, RNA was extracted from the cells and analyzed for levels of mRNA of the target genes using quantitative RT-PCR methods. When concentration-dependent increases in mRNA levels were greater than 2-fold, the data were analyzed by non-linear regression using Grafit 7.0.2 to determine Emax and EC50 values.
Methods for probe substrate study
Freshly plated human hepatocytes (CellzDirect, Austin, TX) were purchased and, upon receipt, the transport medium was removed and replaced with room temperature supplemented hepatocyte maintenance medium (HMM from Lonza, Walkersville, MD). These were then maintained in an incubator set at 37 °C, 5% CO2, and 95% relative humidity for at least 2 days. The medium was changed at least every 48 h prior to study initiation. Upon study initiation, HMM was removed from hepatocytes and replaced with supplemented HMM containing either viloxazine at final concentrations of 0.73, 7.3, and 73 µM in HMM, 25 µM omeprazole (positive control for CYP1A2 induction), 1 mM phenobarbital (positive control for CYP2B6 induction), or 50 µM rifampicin (positive control for CYP2C9, CYP2C19, and CYP3A4 induction). Appropriate solvent controls – HMM for viloxazine, DMSO for omeprazole, phenobarbital and rifampicin 0.5% of the final incubation volume were also included. Media containing viloxazine and the positive control inducers were replaced with fresh solutions after 24 and 48 h, for a total of 72 h of treatment. Approximately 24 h after the last dose, media containing viloxazine and the positive control inducers were removed and replaced with warmed (37 °C) HMM containing either 5 μM 7-ethoxyresorufin, 500 μM bupropion, 100 μM diclofenac, 150 μM S-mephenytoin, or 250 μM testosterone. The final concentration of organic solvent in each cytochrome P450 incubation was ≤1% (v/v). Hepatocytes were returned to the incubator and maintained at 37 °C for one hour. Incubations were terminated by the addition of 0.25 mL acetonitrile containing an appropriate internal standard, and changes in enzyme activity were assessed by fluorescence for CYP1A2 and LC-MS/MS for CYP2B6, CYP2C9, CYP2C19, and CYP3A4.
In vitro P-gp, BCRP, OATP1B1, OATP1B3, MATE1, and MATE2-K transporter inhibition assessment
Kidney (MDCKII) cells were supplied as PreadyPort™ Ready-to-use Transfected Monolayer Kits by ReadyCell S.L., Barcelona, Spain. Viloxazine was incubated with cells at concentrations of 0, 0.3, 1, 3, and 10 mM (in triplicate) in the presence of the probe substrates of P-gp and BCRP, [3H]-digoxin and [3H]-prazosin, respectively, over a period of 120 min. At the end of the incubation, probe substrate concentrations in the samples (donor compartment sampled at 120 min; receiver compartment sampled at 60, 90, and 120 min) were quantified by radioactivity measurement using liquid scintillation counting (LSC). Viloxazine (at concentrations of 0, 0.462, 1.386, 4.62, and 13.86 mM) was assessed as a potential time-dependent inhibitor of OATP1B1 and OATP1B3 using the TransportoCells™ (Corning®, Bedford, MA) system. A 30 min pre-incubation of viloxazine or the reference inhibitor, cyclosporine A, with cells was conducted prior to incubations of viloxazine or cyclosporine A in the presence of the probe substrate, [3H]-estradiol 17β-glucuronide. Viloxazine (at concentrations of 0, 3.3, 6.6, 33, 66, 330, 1100, and 3300 µM) was assessed as a potential inhibitor of MATE1 and MATE2-K using the TransportoCells™ (Corning®, Bedford, MA) system. Incubations of viloxazine were conducted in the presence of the probe substrate, [14C]-metformin. [14C]-metformin was also incubated in the absence and presence of the reference inhibitor, pyrimethamine, in parallel as a positive control. At the end of the appropriate incubation period, the incubations were stopped by placing the culture plates on ice, aspirating the compound solutions, and rinsing the cells twice with assay buffer before cell lysis. Probe substrate concentrations were quantified by radioactivity measurement using LSC.
In vitro transporter OATP1B1 and OATP1B3 substrate assessment
Viloxazine (at concentrations of 0.659, 2.20, 6.59, and 19.8 µM) was assessed as a potential substrate of OATP1B1 and OATP1B3 by incubating with HEK293 cells transiently transfected with the transporters (TransportoCells™, Corning®, Bedford, MA). Incubations contained either viloxazine or the OATP1B probe substrate [3H]-estradiol 17β-glucuronide ([3H]-E17βGluc; 2 μM) with and without the reference inhibitor cyclosporine A (10 μM). Viloxazine was assessed with cyclosporine A at the lowest and highest test compound concentration only. [3H]-E17βGluc was assessed with and without cyclosporine A at a single concentration to determine the extent of inhibition and to demonstrate a fully functioning assay system. After 5 min, the incubation was terminated by immediately placing the culture plates on ice (4 °C), aspirating all incubation buffer from the cells, and rinsing twice with TransportoCells™ buffer (Corning®, Bedford, MA) prior to cell lysis. Probe substrate concentrations were quantified by radioactivity measurement using LSC and viloxazine concentrations were measured by LC-MS/MS.
Rat ADME
Mass balance
[14C]-viloxazine was administered by oral gavage (10 mL/kg) to groups (n = 3/group) of male and female Sprague Dawley rats at a target dose of 100 mg/kg and a radioactive dose of approximately 160 µCi/kg. Individual animals were housed in metabolic cages to allow for the separation and collection of urine and feces. Excreta were collected for a period of up to 168 h post-dose. Total radioactivity was measured in urine by direct analysis using LSC, whereas analysis of feces required homogenization with water:ethanol (80:20 v/v) prior to combustion and measurement by LSC. At the end of the collection period, the carcasses were retained and analyzed for any residual radioactivity. Selected urine and feces samples were retained for metabolite profiling.
In vivo metabolism
Male and female Sprague Dawley rats received [14C]-viloxazine at a target dose of 100 mg/kg and a radioactive dose of approximately 160 µCi/kg. Plasma samples collected at 1, 4, and 8 h post-dose were pooled by gender and time point. Each pooled plasma sample was extracted with acetonitrile (1:5 v/v) twice and then finally with methanol (1:5 v/v). All the supernatants were combined and analyzed by LSC to determine an extraction recovery, which ranged from 74.8 to 99.9%. The supernatants were evaporated to dryness and reconstituted in 150 µL of 0.2% formic acid. The recovery from reconstitution ranged from 94 to 110%. Urine samples (0–8 and 8–24 h post-dose) were centrifuged for approximately 20 min and then the supernatant taken for metabolite profiling. Fecal homogenates (8–24 h post-dose) were extracted twice with acetonitrile (1:5 v/v) and the supernatants combined. LSC analysis of the acetonitrile supernatants determined the extraction recovery of radioactivity to be 41.4 and 54.9% for male and female feces, respectively. The residual pellet was further extracted twice with methanol (1:5 v/v) and the supernatants combined. LSC analysis of the methanol supernatants indicated that an additional 7.26% and 4.82% of the radioactivity could be recovered for male and female feces, respectively. All the supernatants (ACN and methanol) were combined, evaporated to dryness, and then reconstituted in 150 µL of 0.2% formic acid in water and 150 µL of formic acid in ACN:isopropanol (95:5). There was no loss of radioactivity upon reconstitution. Each reconstituted sample was analyzed using the same HPLC conditions as detailed in the in vitro metabolism section with the exception that the analysis was performed by collecting 10-second fractions using a Foxy fraction collector (Teledyne ISCO, Lincoln, NE) and measurement by TopCount (PE Life Sciences, UK) to determine the radioactive profile.
Radiolabeled human metabolism study
Clinical study design and study medication
The study was approved by the local ethics and radiation safety committees and conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All subjects gave written informed consent prior to participation. The study population comprised seven healthy male subjects aged 30–55 years, body weight of at least 60 kg, body mass index (BMI) of 18.5–32 kg/m2, and in good health as determined by past medical history, physical examination, vital signs, electrocardiogram (ECG), and laboratory tests at screening. Subjects were excluded from the study if they had been exposed to radiation (excluding background radiation) exceeding 5 mSv in the last 12 months, or 10 mSv in the last 5 years, including that from the present study, diagnostic X-rays, and other medical exposures. Occupationally exposed workers, as defined in the Ionising Radiation Regulations 1999, were not allowed to participate in the study. Subjects were required to fast overnight prior to the dose and until two hour after dose administration. Each subject received a single oral dose of aqueous solution (20 mL) containing 100 mg viloxazine hydrochloride (as free base) and no more than 8.7 MBq (236 μCi) of radioactive [14C]-viloxazine.
Each subject received the single 100 mg oral dose of [14C]-viloxazine on the morning of Day 1. Subjects were confined to the study center from the evening of Day 1 before administration of the study drug until up to 11 days post-dose. Subjects were scheduled for release from the study as a group on the morning of Day 11, or earlier if either of the following was achieved prior to Day 11: (1) the daily total excreted radioactivity was no more than 1% of the administered dose on 2 consecutive days, or (2) 90% of the administered radioactivity was recovered in the urine and feces.
Sample collection
Blood was collected at pre-dose, and at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 18, 24, 30, 36, and 48 h post-dose, as well as at 24 h intervals until the end of the study by either direct venipuncture or an indwelling cannula inserted in a forearm vein. Venous blood was collected at each time point in heparinized tubes. Separate aliquots were used for the various study assessments; e.g., total radioactivity in blood and plasma, chiral and achiral PK, as well as metabolite profiling and identification. The remaining whole blood was transferred into polypropylene screw-cap vials and immediately frozen and stored at −60 °C. Urine was collected from and pooled for each subject by time period post-dose (0–6, 6–12, 12–24, 24–48, 48–72, and 72–96 h). Feces were collected as produced from time of dosing at pre-dose and at 24 h intervals until at least 96 h post-dose. Plasma urine and feces samples were stored at −20 °C and whole blood was stored at 4 °C until analysis. Subsamples of urine and feces were stored at −80 °C prior to metabolite profiling and identification analysis.
Determination of total radioactivity
Radioactivity measurements in plasma and urine were carried out using direct LSC (TriCarb Scintillation Counter; PerkinElmer, USA). Feces (homogenized to a paste with water) and whole-blood replicate samples were combusted in oxygen (Automatic Sample Oxidizer Model 307; PerkinElmer, USA) prior to measurement of radioactivity. Processing for metabolite profiling is described in following paragraphs.
Sample preparation for characterization of viloxazine and related components
Plasma samples were pooled to prepare a single “AUC pool” for each subject using the method of Hamilton et al. (Citation1981). Pooled plasma samples (1 mL) were extracted with 3 mL acetonitrile, vortexed, and centrifuged. The supernatant was retained and the pellet resuspended and extracted with an additional 3 mL acetonitrile. The supernatants were combined and the radioactivity measured. Extraction recovery was shown to have a mean recovery of 94%. The supernatant was evaporated to dryness and reconstituted in 500 µL of methanol:water (20:80 v/v). The recovery on reconstitution was determined to be 92%. Pooled urine samples were analyzed directly without prior extraction.
Metabolite profiling by HPLC
The metabolite profiles were generated using the HPLC with in-line fraction collection using Dionex Ultimate 3000 chromatography system (Thermofisher, UK). Separation was achieved using a Phenomenex Kinetex Luna, 3 µm, 150 × 4.6 mm column (Phenomenex, UK) maintained at 30 °C by using two mobile phases: A (0.1% formic acid in water) and B (0.1% formic acid in ACN:isopropanol (95:5)). A linear gradient at a flow rate of 1 mL/min was set as follows: 1% B (0 min) → 1% B (5 min) → 40% B (40 min) → 100% B (45 min) → 1% B (45.1 min) → 1% B (50 min). The analysis of plasma and urine was performed by collecting 8-second fractions using a PAL-HTX-xt (Presearch, UK) fraction collector and measurement by TopCount (PE Life Sciences, UK) to determine the radioactive metabolite profile.
Structural elucidation by LC-MS/MS
Selected plasma and urine samples were taken for structural elucidation of key metabolites using Thermo Fisher Scientific Q Exactive mass spectrometer under the following conditions: (1) atmospheric pressure ionization interface, electrospray ionization; (2) acquisition, full scans in the positive ion mode; (3) scan range, 50–750 m/z (MSn data dependent); (4) source voltage, 4.2 kV; (5) and capillary temperature, 250 °C.
Deconjugation of urine samples
1.8 mg β-glucuronidase from Helix Pomatia (Sigma-Aldrich, UK) was added to a pooled urine sample and allowed to incubate at 37 °C overnight. The deconjugation reaction was halted by addition of acetonitrile and the sample was subsequently concentrated under nitrogen to remove the acetonitrile. Subsamples of each sample were analyzed by LC-MS/MS using Dionex Ultimate 3000 chromatography system (Thermofisher, UK). Separation was achieved using Restek Allure BiPhenyl, 5 µm, 150 × 4.6 mm column (Thames Restek, UK) maintained at 30 °C by using two mobile phases: A (0.1% formic acid in water) and B (0.1% formic acid in methanol). A linear gradient at a flow rate of 1 mL/min was set as follows: 1% B (0 min) → 1% B (5 min) → 40% B (40 min) → 100% B (50 min) → 100% B (55 min) → 8% B (33 min) → 1% (55.1 min) → 1% (60 min). The column elute was split post-column in a ratio of 3:2 between a mass spectrometer and Β-RAM Model 5 radiodetector (Lablogic Systems Ltd., UK). The mass spectral analysis was performed using a Q-Exactive mass spectrometer (Thermo Fisher Scientific, UK). The analytical conditions of the mass spectrometer were as follows: (1) atmospheric pressure ionization interface, electrospray ionization; (2) acquisition, full scans in the positive ion mode; (3) scan range, 50–600 m/z (MSn data dependent); (4) source voltage, 3.0 kV; and (5) capillary temperature, 320 °C.
Results
In vitro species comparison with rat, dog, and human hepatocytes
The extent of metabolism of [14C]-viloxazine by hepatocytes over a 120 min incubation (0.3 µg/mL viloxazine) varied from 10% in humans, to approximately 50% in dogs, and exceeded 90% in rat. The degree of metabolism in hepatocytes was rat > dog≫human. The metabolite profiles are outlined in . A summary of the detected metabolites in shown in .
Figure 2. Representative radiochromatograms from 120 min incubations of 3 µg/mL of [14C]-viloxazine with rat, dog, and human hepatocyte incubations.
![Figure 2. Representative radiochromatograms from 120 min incubations of 3 µg/mL of [14C]-viloxazine with rat, dog, and human hepatocyte incubations.](/cms/asset/08cd5666-2678-4ed1-ad14-a64d42a83f76/ixen_a_1767319_f0002_b.jpg)
Table 1. Metabolites of [14C]-viloxazine formed in rat, dog, and human hepatocytes following a 120 min incubation. Data expressed as the average percentage of total sample radioactivity.
The major metabolite of viloxazine in the rat was desethyl viloxazine sulfate (M5), while the major metabolite in the dog resulted from hydroxylation of the morpholine group (M11). Of the four metabolites formed in human hepatocytes incubations, three resulted from hydroxylation on the phenyl ring (M2, M6, M8) and one from hydroxylation on the morpholine group (M11). Based on relative retention time and previous data suggesting that the major route of metabolism in humans is via 5-hydroxylation with subsequent glucuronidation (Case & Reeves, Citation1975), M2 is likely to be 5-hydroxyviloxazine glucuronide. There were no metabolites detected in the human hepatocyte incubations that were not detected in rat or dog incubations.
Rat ADME
Mass balance
Following oral administration of [14C]-viloxazine to male and female rats, radioactivity was rapidly excreted with approximately 88.6 to 88.8% of the radioactive dose eliminated in the initial 24 h post-dose. Urine was the major route of elimination representing approximately 84.2% and 82.0% of the administered radioactive dose in male and female rats, respectively, at the end of the 168 h post-dose collection period. A lesser amount of the radioactive dose was found in feces, 5.39% and 6.37% in male and female rats, respectively. The extent and route of elimination were similar in male and female rats with total recovery of 92.0% and 93.3% of administered dose in male and female rats, respectively, suggesting near complete elimination of radioactivity at 168 h post-dose.
Rat in vivo metabolism
Viloxazine is extensively metabolized by rat with no obvious difference between male and female rats. Male rat plasma showed up to 14 quantifiable metabolites, whereas female plasma showed 13 metabolites with no unique gender specific metabolites observed. Typical metabolite profiles for plasma, urine, and feces are shown in .
Figure 3. Metabolite profiles in the plasma (8 h), urine (8 to 24 h), and feces (8 to 24 h) following administration of a single 100-mg/kg oral dose of [14C]-viloxazine to male rats.
![Figure 3. Metabolite profiles in the plasma (8 h), urine (8 to 24 h), and feces (8 to 24 h) following administration of a single 100-mg/kg oral dose of [14C]-viloxazine to male rats.](/cms/asset/316e1f7b-7eb3-4dca-bb75-f024595cc5bd/ixen_a_1767319_f0003_b.jpg)
The metabolite identified as desethyl viloxazine sulfate (M5) represented the highest proportion of the circulating radioactivity in the plasma, accounting for up to 60.2% and 45.6% of the radioactivity in a 1 h plasma sample from male and female rats, respectively. Unchanged viloxazine was seen at a very low level, representing a minor component of the total radioactivity in rat plasma.
Analysis of urine, which represents the major route of excretion of drug-related radioactivity, revealed the presence of small amounts of [14C]-viloxazine and as many as 15 metabolites. The same major metabolite present in plasma, desethyl viloxazine sulfate (M5), is a major metabolite in urine. Viloxazine is also metabolized by hydroxylation of the phenyl ring and hydroxylation and oxidation of the morpholine ring. Glucuronidation of numerous primary metabolites also occurred.
A summary of the radioactive components that represent greater than 5% of the radioactivity in plasma or greater than 5% of the dose in urine is provided in . No single metabolite of greater than 5% of the dose was observed in feces; therefore, these data are not included in the table.
Table 2. Metabolites of [14C]-viloxazine formed in male (A) and female rats (B) following an oral dose of 100 mg/kg.
Radiolabeled human metabolism
Blood and plasma concentrations of radioactivity
Following a single 100 mg/kg dose of [14C]-viloxazine, the greatest concentrations of radioactivity measured in plasma were noted between 0.75 and 2.5 h post-dose and ranged from 1635–2880 ng equivalents/mL. Radioactivity was quantifiable in all samples up to 36 h. In whole blood, the greatest concentrations of radioactivity were noted between 1 and 2 h and ranged from 884–1745 ng equivalents/g). Radioactivity was quantifiable in all samples up to 24 h.
Excretion and mass balance in urine and feces
Elimination of radioactivity was primarily in urine (mean 99.4% dose; Supplementary Table S1). The majority of the radioactivity was recovered in urine within the first 24 h following dose administration. Total radioactivity eliminated in feces was minimal (mean 0.67 % dose). Overall dose recovery was judged to be complete (mean of 100.5 %) within the 96 h collection period.
Viloxazine metabolism in human
Metabolite profiling and identification was performed on human plasma and urine samples obtained from the radiolabeled clinical study. The amount of radiolabeled material in feces was minimal and less than 1% of the radioactive dose; therefore, metabolite analysis was not performed on fecal samples.
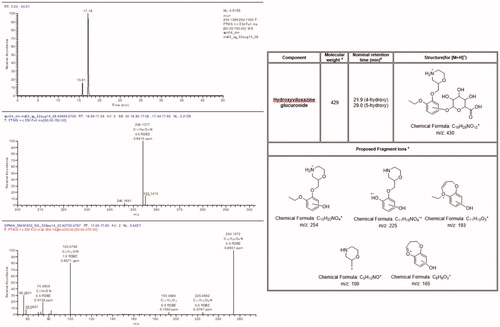
Analysis of the plasma indicated that there were 2 major radioactive components that accounted for 10% or more of the radioactivity present (). Using LC-MS/MS these components were identified as viloxazine and a hydroxyglucuronide metabolite. The latter metabolite was subsequently identified as the 5-hydroxyglucuronide metabolite ().
Figure 4. Representive chromatogram from human plasma and urine from a single subject following a single oral administration of [14C]-viloxazine (nominal 100 mg; 8.7 MBq) to seven male human subjects. Metabolites P1 and U1 are considered to be the same metabolite and are indicated as such in the plasma chromatogram.
![Figure 4. Representive chromatogram from human plasma and urine from a single subject following a single oral administration of [14C]-viloxazine (nominal 100 mg; 8.7 MBq) to seven male human subjects. Metabolites P1 and U1 are considered to be the same metabolite and are indicated as such in the plasma chromatogram.](/cms/asset/95a86f80-a5c4-48a8-b69a-9e0ed19c7809/ixen_a_1767319_f0004_c.jpg)
Figure 5. Accurate mass extracted ion chromatogram, positive ion full scan, and product ion spectra with a summary of theoretical accurate mass values, characteristic fragment ions, and proposed assignments for the metabolite P1 / U1.
Analysis of urine indicated that as many as four components exceeded 10% of the total radioactivity in at least one sample with a number of minor metabolites comprising the remaining 20–30% of the total radioactivity. These radioactive components were identified as the hydroxyglucuronide metabolite, hydroxyviloxazine, viloxazine, and an N-carbamoyl glucuronide metabolite.
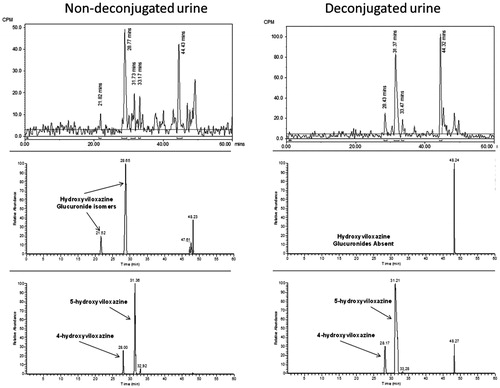
Several positional isomers could account for the position of hydroxylation on the phenyl ring. Therefore, 3-hydroxyviloxazine, 4-hydroxyviloxazine, 5-hydroxyviloxazine, and 6-hydroxyviloxazine were synthesized. The HPLC method was modified to allow the separation of the isomer reference standards. The position of the site of hydroxylation was determined to be the 5-hydroxy position by analyzing urine samples following deconjugation with β-glucuronidase. shows the transformation of the glucuronide metabolite back to the unconjugated 5-hydroxyviloxazine metabolite.
Figure 6. Radiochromatograms and extracted ion chromatograms for selected components from human urine before and after deconjugation with β-glucuronidase to show position of glucuronidation.
The relative proportions of metabolites are described for pooled plasma and urine samples (). Viloxazine and 5-hydroxyglucuronide metabolite (P1) are the major components in plasma (>10% of the total sample radioactivity), while 5-hydroxyviloxazine metabolite (U2) and N-carbamoyl glucuronide metabolite (U4) are detected as minor plasma metabolites.
Table 3. Summary of the relative proportions of the human plasma and urinary metabolites following a single oral administration of [14C]-viloxazine (nominal 100 mg; 8.7 MBq) to seven male human subjects.
The previous study by Case & Reeves (Citation1975) had identified N-methylviloxazine as a metabolite in human. This metabolite was also seen in urine in this study but was not quantified since the levels were less than 10% of the sample radioactivity.
In vitro CYP phenotyping
Identification of the metabolites of [14C]-viloxazine from microsomal incubations
Several metabolites of [14C]-viloxazine were detected in incubations with human hepatic microsomes (data not shown). Samples were analyzed by LC-MS/MS to identify the metabolites, and the major metabolite detected corresponded to M8 previously observed in human hepatocytes. Thus, M8 was chosen as the metabolite for which all subsequent incubation conditions were optimized. Subsequent experiments using an authentic standard confirmed M8 to be 5-hydroxyviloxazine.
Kinetic analysis with pooled human hepatic microsomes
After incubating [14C]-viloxazine (10 µM) with human liver microsomes at different time and protein concentrations, analysis showed that the rate of 5-hydroxyviloxazine formation was linear for 30 min at 0.5 mg/mL of microsomal protein, which were chosen as conditions for the subsequent experiments.
Following the incubation of [14C]-viloxazine over a concentration range of 10 to 500 µM with human liver microsomes, the rate of formation of 5-hydroxyviloxazine was determined. The enzyme kinetics of formation of 5-hydroxyviloxazine were determined by Michaelis–Menten analysis. The Km and Vmax values for 5-hydroxyviloxazine were determined to be 35.2 ± 9.4 µM and 64.3 + 4.7 pmol/min/mg (data not shown). A value close to the Km, 40 µM, was chosen as the concentration of [14C]-viloxazine to be used in all subsequent experiments.
Chemical inhibition analysis using pooled human hepatic microsomes
[14C]-Viloxazine (40 µM) was incubated with pooled human hepatic microsomes (0.5 mg protein/mL) at 37 °C for 30 min in the presence and absence of CYP-specific inhibitory antibodies or sulfaphenazole, a selective inhibitor of CYP2C9. The formation of 5-hydroxyviloxazine was markedly inhibited by anti-CYP2D6 (79% inhibition), suggesting that CYP2D6 is primarily involved with its formation. Less than 20% inhibition was seen with sulfaphenazole and the other antibodies.
Incubations with cDNA-expressed human cytochromes P450 (SupersomesTM)
The capacity of selected cDNA-expressed human CYPs to metabolize [14C]-viloxazine (40 µM) to 5-hydroxyviloxazine in vitro was examined in SupersomesTM containing CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, or 3A4 (). The results indicate that the formation of 5-hydroxyviloxazine is mediated primarily by CYP2D6 at a rate of 12.9 pmol/min/pmol CYP2D6.
Figure 7. Formation of 5-hydroxyviloxazine from the incubations of [14C]-viloxazine (40 µM) with a panel of recombinant cytochrome P450s.
![Figure 7. Formation of 5-hydroxyviloxazine from the incubations of [14C]-viloxazine (40 µM) with a panel of recombinant cytochrome P450s.](/cms/asset/eb49e7ae-0204-4fa0-a10a-a920c052cd18/ixen_a_1767319_f0007_b.jpg)
In vitro CYP inhibition
The ability of viloxazine to inhibit CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5 was investigated in human liver microsomes up to a final viloxazine concentration of 1010 µM, and the data are summarised in . Under these conditions, viloxazine did not inhibit CYP2C8, 2C9, or 2C19 activities (IC50 values >1010 µM); but it was a reversible inhibitor of CYP1A2, 2B6, 2D6, and 3A4/5 activities, with IC50 values of 0.269, 184, 141, 221 (midazolam), and 352 (testosterone) µM, respectively.
Table 4. Viloxazine in vitro reversible and time dependent CYP inhibition.
None of the enzymes investigated showed time-dependent inhibition (TDI) by viloxazine, with the exception of CYP1A2 (). In this case, viloxazine was potentially a time-dependent inhibitor of CYP1A2, with a 6.2-fold shift in IC50 upon pre-incubation. However, a shift of 3.7-fold was also obtained following a pre-incubation in the absence of cofactor, suggesting that the TDI could be due to slow-binding inhibition or non-NADPH-dependent conversion to an inhibitory product, rather than time-dependent inactivation of the enzyme.
In vitro CYP induction
The results for the induction of CYP1A2, 2B6, and 3A4 mRNA (donors 1, 2, and 3) and CYP1A2, 2B6, 2C9, 2C19, and 3 A enzyme activities (donors A, B, and C) by viloxazine are shown in Supplementary Table S2.
Concentration dependent increases in CYP1A2 mRNA levels were observed following the treatment of hepatocytes from all three donors with viloxazine. The increase in expression of CYP1A2 mRNA from donors 1 and 2 was sufficient to generate Emax values of 44.8-fold and 2040-fold, and EC50 values were 70.7 µM and 2930 µM, respectively. For donor 3, no increases of >2-fold were observed at concentrations below 70 µM, so Emax and EC50 values could not be determined. Following the exposure to viloxazine, CYP1A enzyme activity was increased in all three donors in a dose-responsive manner (Supplementary Table S2). This induction was 125% and 212% of the extent of induction of CYP1A2 activity seen with 25 μM omeprazole in hepatocytes from donors A and B, while it was 36.2% in hepatocytes from donor C.
Concentration dependent increases in CYP2B6 mRNA levels were observed following treatment of hepatocytes from donor 1, with viloxazine up to maximum fold increase of 11.7-fold at 140 µM. These data generated an Emax value of 16-fold and an EC50 value of 64.9 µM. Increases in CYP2B6 mRNA expression was also seen in donor 2 at 70 µM and 140 µM, but no increases were observed at the lower concentrations. For donor 3, no increases of >2-fold were observed at concentrations below 70 µM. Therefore, Emax and EC50 values could not be determined for these donors. Similarly, CYP2B6 activity was increased in all three donors in a dose-dependent manner following treatment with viloxazine. At 73 µM viloxazine, CYP2B6 was increased 6.59-, 4.26-, and 9.07-fold in hepatocytes from donors A, B, and C, respectively. This equates to 67.8%, 103%, and 82.0% of the extent of induction CYP2B6 enzyme activity seen with of 1 mM phenobarbital in hepatocytes from these donors.
Following the exposure to 73 µM viloxazine, CYP2C9 enzyme activity was increased 1.60- and 1.65-fold in hepatocytes from donors A and C, respectively. This equates to 41.6% and 16.3% of that of 50 μM rifampicin in donors A and C, respectively. Thus, there appears to be little or no viloxazine-mediated induction of CYP2C9. There was no measurable induction by rifampicin or viloxazine in donor B.
A similar pattern was seen with CYP2C19 enzyme activity with only donors A and C showing induction with rifampicin (50 μM). Following exposure to viloxazine at 73 μM, CYP2C19 activity was minimally affected in hepatocytes from donors A and C (1.54- and 1.69-fold in hepatocytes from donors A and C, respectively). Thus, there appears to be little or no viloxazine-mediated induction of CYP2C19.
Minimal increases in CYP3A4 mRNA expression were seen in donor 1 (maximum of 4.40-fold at 140 µM) and donor 2 (8.13-fold at 140 µM). However, no increases were observed at lower concentrations. No increases of greater than 2-fold in CYP3A4 mRNA expression were observed in donor 3. Therefore, Emax and EC50 values could not be determined for CYP3A4 induction. Following the exposure to viloxazine, CYP3A activity was minimally affected in hepatocytes from all three donors. At 73 µM, CYP3A4/3A5 activity was increased 1.67-, 0.954-, and 1.21-fold in hepatocytes from donors A, B, and C, respectively. Thus, there appears to be little or no viloxazine-mediated induction of CYP3A enzyme activity.
Viloxazine as an inhibitor of efflux (P-gp and BCRP), hepatic uptake (OATP1B1 and OATP1B3), and renal (MATE1 and MATE-2K) transporters
Viloxazine produced concentration-dependent inhibition of the net efflux ratio of the P-gp substrate (digoxin) and BCRP substrate (prazosin) resulting in IC50 values of 1.50 mM and 1.68 mM, respectively. Viloxazine also appeared to be an inhibitor of the OATP1B1 and OATP1B3 transporters, with IC50 values of 1.84 and 2.70 mM, respectively. Viloxazine was a weak inhibitor of the MATE1 transporter, with a reported IC50 value of 140 µM. In contrast, there was little evidence for a concentration-dependent inhibition of [14C]-metformin MATE-2K transport by viloxazine and so, by inference, the IC50 was >3300 µM.
Viloxazine as a substrate for OATP1B1 and 1B3
There was evidence of concentration-dependent uptake of viloxazine into OATP1B1, OATP1B3, and non-transfected (wild type) TransportoCells™, with the rate of uptake increasing with increasing viloxazine concentration. The uptake rates in OATP1B1 and OATP1B3, compared to the non-transfected cells, were similar, resulting in uptake ratios <2.
The uptake ratio of 0.659 μM viloxazine, relative to control in both OATP1B1 and OATP1B3 TransportoCells™, was reduced by approximately 50% by 10 μM cyclosporine A. However, this reduction was caused by the increased rate of viloxazine transport into the wild-type cells. There was no effect of cyclosporine A on the rate of uptake of viloxazine into transfected cells. Therefore, inhibition of viloxazine in the presence of cyclosporine A was considered not to be significant.
Discussion
Viloxazine metabolism has previously been studied in rat, dog, and humans (Case et al., Citation1975; Case & Reeves, Citation1975). The current studies were conducted using modern techniques, including in vitro metabolism in hepatocytes and human microsomes and mass spectroscopy, to expand our understanding of viloxazine metabolism. The summary of the metabolism of viloxazine in rat and human is detailed in and , respectively.
Figure 8. Summary metabolic scheme for viloxazine in rat.
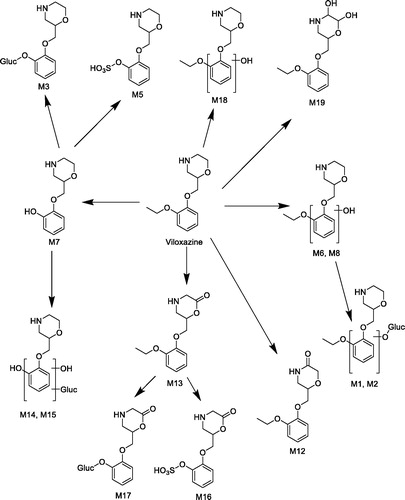
Figure 9. Summary metabolic scheme for viloxazine in human.
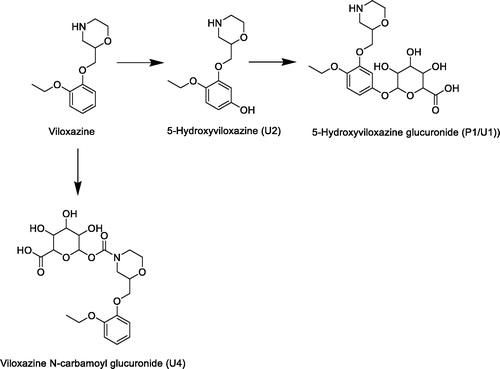
In this study, the major route of viloxazine metabolism in the rat, both in vitro and in vivo, was oxidative deethylation followed by sulfation to M5. Although consistent with the previously reported metabolism in the rat (Case et al., Citation1975), the current study also shows the presence of other metabolic routes, including oxidation and hydroxylation with subsequent conjugation of the morpholine ring and hydroxylation with glucuronidation of the phenyl ring (). Furthermore, this study identified the major route of metabolism in human as hydroxylation followed by glucuronidation which is in line with that seen previously (Case et al., Citation1975). The exception was the carbomyl glucuronide metabolite which was identified in this study but not in the previous one.
In the rat, two notable circulating metabolites (M1 and M2) have a chemical structure indicated to be equivalent by mass spectrometry to human P1, which was confirmed as 5-hydroxyviloxazine glucuronide. Although the identity of the rat metabolite was not confirmed as 5-hydroxyviloxazine glucuronide, M2 is likely to be the same metabolite and would provide sufficient exposure cover in the rat for 5-hydroxyviloxazine glucuronide. Hence, M2 circulates in male and female rat plasma at a higher exposure (0.632 µg equivalent/g and 0.681 µg equivalent/g, respectively, at 8 h) than 5-hydroxyviloxazine glucuronide in humans (0.118 µg equivalent/g at 8 h).This meets the ICH M3 (R2) guidance (ICH, Citation2010) which states that “characterization of metabolite toxicity would generally be considered adequate when animal exposure is at least 50 percent the exposure seen in humans,” and the FDA requirements in the Safety Testing of Drug Metabolites Guidance for Industry (FDA, Citation2016). Furthermore, 5-hydroxyviloxazine, which was seen in human microsomes and hepatocytes, was also observed in dog hepatocytes, and was also seen as a circulating metabolite in dog plasma (data not shown), supporting the view that it is not a unique human metabolite.
Based on the in vitro data presented here, the major enzyme contributing to the formation of 5-hydroxyviloxazine is CYP2D6, with minor involvement of CYP1A2, CYP2B6, CYP2C9, CYP2C19, and CYP3A4. Subsequent glucuronidation to 5-hydroyviloxazine glucuronide is mediated by UGT1A9 and 2B15 (data not shown). Insofar as viloxazine is a CYP2D6 substrate, its pharmacokinetic profile can vary with CYP2D6 genetic polymorphisms. However, Gibbs et al. (Citation2006) demonstrated that the overall fraction of the drug metabolized by CYP2D6 is key to predicting whether there are differences in area under the curve (AUC) between CYP2D6 poor metabolizers (PMs) and extensive metabolizers (EMs). This analysis compared data from human in vivo studies for a series of CYP2D6 substrates, and it showed that for relatively poor substrates (fraction metabolized by CYP2D6 ≤ 60%), the difference in AUC between CYP2D6 EMs and PMs was very modest (≤2.5-fold). For good CYP2D6 substrates where the fraction metabolized by CYP2D6 is ≥60% the AUC change was more significant (≥ 2.5-fold).
In the case of viloxazine, the presence of other minor metabolites, such as the carbamoyl glucuronide, will modulate the impact of CYP2D6. The majority of the radioactivity is excreted as the parent molecule and metabolites in the urine after 48 h. The 0 to 48 h pooled urine data in shows that for the excreted radioactivity, approximately 22% is viloxazine; that is, total fraction metabolized equals 78%. Since only one of the subjects had 5-hydroxyviloxazine levels over the 10% cutoff, the metabolites from the 5-hydroxylation pathway (5-hydroxyviloxazine [U2] + 5-hydroxyviloxazine glucuronide [U1]) account for less than 39% of excreted radioactivity. Thus, the fraction metabolized via the CYP2D6-mediated pathway (the sum of the 5-hydroxylation pathway/total fraction metabolized) is less than 50% (i.e., 39/78). Based on this fraction metabolized and using the analysis presented by Gibbs et al. (Citation2006), the AUC for viloxazine would show less than a 2-fold difference between CYP2D6 EMs and PMs — a minor difference in the clinical setting.
Viloxazine also forms an N-carbamoyl glucuronide conjugate in humans that is detected in plasma and urine. The metabolite constitutes an average of 6.4% and 4.4% of the circulating drug-related material at 1 and 8 h, respectively, after administration of viloxazine. This minor metabolite is not detected in either rat or, from the earlier study, dog plasma. Although the metabolite is disproportionate in humans and is not well represented in the toxicology species, it falls into a class of metabolites that are generally judged as low risk.
N-carbamoyl glucuronide conjugates form via a two-step process and is observed as a metabolite for many primary and secondary amine containing drugs (Schaefer, Citation2006). The first step involves the reversible formation of a carbamic acid adduct of the amine via reaction with CO2. The carbamic acid product, in some cases, then serves as a substrate for UDP-glucuronosyl transferase enzymes to form the N-carbamoyl glucuronide conjugate. There does not seem to be any general species selectivity in the formation of these conjugates. Drugs such as lorcaserin, sertraline and sitagliptin all form N-carbamoyl glucuronide conjugates in humans (Obach et al., Citation2005; Sadeque et al., Citation2012; Vincent et al., Citation2007).
Distinct from reactive acyl glucuronides, N-carbamoyl glucuronide conjugates have not been reported to be associated with any adverse events. This is almost certainly a consequence of the relative stability of the conjugate. The stability of the conjugate is inherent in the carbamate moiety, which allows delocalization of electron density through the ester and amide portion of the carbamate (Ghosh & Brindisi, Citation2015). The carbamate group is a key structural motif in many approved drugs and prodrugs because of its stability and resistance to hydrolytic and proteolytic cleavage as well as other desirable properties (Ghosh & Brindisi, Citation2015). Although the use as a prodrug does require carbamate cleavage, this reaction is almost always carried out by a specific esterase and not mediated by endogenous nucleophiles. An example of the stability of a typical N-carbamoyl glucuronide can be seen in a study by Gunduz et al. (Citation2010) where β-glucuronidase efficiently cleaved the conjugate to yield the parent; however, the conjugate was completely stable in the control incubation (buffer at 37 °C for 4 h). Problematic acyl glucuronides typically have buffer half-lives of <2 h (Gunduz et al., Citation2018).
From a toxicity standpoint, the two most important reactions for acyl glucuronides are: 1) transacylation reactions with various endogenous nucleophiles to yield drug-labelled compounds/macromolecules, and 2) acyl migration that allows opening of the hexose ring and glycation and glycosylation of proteins by the resulting aldehyde (Gunduz et al., Citation2018). Both of these reactions lead to covalent binding with intracellular proteins that is thought to mediate the toxicity of the conjugates. The reactions require a reactive moiety, such as the acyl glucuronide bond. The general stability of the N-carbamoyl glucuronide structure distinguishes it from acyl glucuronides and disfavors either of the reactions listed above. For this reason, when evaluating the safety of the metabolites of new chemical entities, N-carbamoyl glucuronide conjugates should be treated as stable PhaseII conjugates.
Of the CYP enzymes investigated, viloxazine was only a significant inhibitor of CYP1A2. In addition, viloxazine induced the mRNA levels of CYP1A2 and CYP2B6, as well as CYP1A2, CYP2B6, and CYP2C9 enzyme activity levels, albeit with a considerable amount of between donor variability. This suggests that the interplay of induction and inhibition of CYP1A2 is important for potential clinical drug-drug interaction.
This CYP1A2 drug-drug interaction is demonstrated by the reported clinical interaction of viloxazine with carbamazepine in epileptic patients (Pisani et al., Citation1986). Following 3 weeks of viloxazine at 300 mg/day, there was an increase (∼55%) in steady-state carbamazepine concentrations. This was accompanied by a slight (16%) increase in the CYP1A2 metabolite carbamazepine-10, 11-epoxide. Once viloxazine dosing was stopped, the carbamazepine and carbamazepine-10, 11-epoxide levels returned to steady state within 1 week, suggesting that the observed potential CYP1A2 time-dependent inhibition would not be observed clinically. These data suggest that although viloxazine is both an inhibitor and an inducer of CYP1A2 in vitro, the dominant in vivo effect is inhibition rather than induction.
A similar interaction was seen when viloxazine was administered with theophylline, another CYP1A2 substrate, where a modest change in its plasma concentration (approximately a doubling) resulted in a clinical effect; that is, theophylline intoxication (Laaban et al., Citation1986). However, theophylline has a narrow therapeutic range, and viloxazine is one of many drugs that, while only modestly changing plasma concentrations, may produce such clinical effect (Laaban et al., Citation1986). Thus, it can be concluded that impact of viloxazine on the pharmacokinetics of CYP1A2 metabolized drugs is modest (>2-fold), and so will be only clinically relevant for the small number of drugs with a narrow therapeutic range.
Viloxazine has been extensively studied as an inhibitor for the efflux transporters (P-gp and BCRP), hepatic uptake (OATP1B1 and OATP1B3), and renal (MATE1 and MATE2-K) transporters. Based on these in vitro data, it can be reasonably excluded as an inhibitor of these transporters, although viloxazine appears to be a weak inhibitor of the MATE1 transporter. Based on these in vitro data, it can be reasonably excluded as an inhibitor of these transporters, although viloxazine appears to be a weak inhibitor of the MATE1 transporter. The extensive oral absorption (based on high degree of urinary excretion from human mass balance study) of viloxazine makes it unlikely to be a substrate of efflux transporters. In addition, it was investigated as a substrate of OATP1B1 and OATP1B3, but the data suggest that it was not a substrate for either transporter.
Conclusion
In summary, the ADME studies presented here demonstrated safe metabolic profile of viloxazine. In vitro drug-drug interaction (DDI) testing suggest that viloxazine is not a significant inhibitor or inducer of CYPs and transporters with the exception of CYP1A2. Future clinical DDI studies will be incorporated into clinical development plan to further assess the significance of these findings in humans.
Supplemental Material
Download PDF (390.4 KB)Disclosure statement
Chungping Yu is an employee of Supernus Pharmaceuticals, Inc. The study was funded and conducted by Supernus Pharmaceuticals, Inc.
Additional information
Funding
References
- Case DE, Reeves PR. (1975). The disposition and metabolism of I.C.I. 58,834 (Viloxazine) in humans. Xenobiotica 5:113–29.
- Case DE, Illston H, Reeves PR, et al. (1975). The disposition and metabolism of LCI. 58,834 (ViloxazJne) in animals. Xenobiotica 5:83–111.
- FDA. (2016). Safety Testing of Drug Metabolites Guidance for Industry. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/safety-testing-drug-metabolites
- Garcia-Olivares J, Schwabe S, Yu C. 2020. Effects of viloxazine on central neurotransmitter systems: a microdialysis study in freely moving rats. Presented at the American Society for Experimental Neurotherapeutics Annual Meeting; March 2–5; Bethesda, MD.
- Ghosh AK, Brindisi M. (2015). Organic carbamates in drug design and medicinal chemistry. J Med Chem 58:2895–940.
- Gibbs JP, Hyland R, Youdim K. (2006). Minimizing polymorphic metabolism in drug discovery: evaluation of the utility of in vitro methods for predicting pharmacokinetic consequences associated with CYP2D6 metabolism. Drug Metab Dispos 34:1516–22.
- Gunduz M, Argikar UA, Baeschlin D, et al. (2010). Identification of a novel N-carbamoyl glucuronide: in vitro, in vivo, and mechanistic studies. Drug Metab Dispos 38:361–7.
- Gunduz M, Argikar UA, Cirello AL, Dumouchel JL. (2018). New perspectives on acyl glucuronide risk assessment in drug discovery: investigation of in vitro stability, in situ reactivity, and bioactivation. Drug Metab Lett 12:84–92.
- Hamilton RA, Garnett WR, Kline BJ. (1981). Determination of mean valproic acid serum level by assay of a single pooled sample. Clin Pharmacol Ther 29:408–13.
- ICH. (2010). Nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m3r2-nonclinical-safety-studies-conduct-human-clinical-trials-and-marketing-authorization
- INTUNIV®. (2019). Prescribing information. Lexington, MA. Shire US Inc. Available from: https://www.shirecontent.com/PI/PDFS/Intuniv_USA_ENG.pdf
- KAPVAY®. (2017). Prescribing information. St. Michael, Barbados. Concordia Pharmaceuticals Inc. Available from: https://www.lupin.com/US/pdf/18/02/clonidine-hcl-er-tabs-kapvay-pi-11-2017.pdf
- Laaban JP, Dupeyron JP, Lafay M, et al. (1986). Theophylline intoxication following viloxazine induced decrease in clearance. Eur J Clin Pharmacol 30:351–3.
- Obach RS, Cox LM, Tremaine LM. (2005). Sertraline is metabolized by multiple cytochrome P450 enzymes, monoamine oxidases, and glucuronyl transferases in human: an in vitro study. Drug Metab Dispos 33:262–70.
- Pinder RM, Brogden RN, Speight TM, Avery GS. (1977). Viloxazine: a review of its pharmacological properties and therapeutic efficacy in depressive illness. Drugs 13:401–21.
- Pisani F, Fazio A, Oteri G, et al. (1986). Carbamazepine-viloxazine interaction in patients with epilepsy. J Neurol Neurosurg Psychiat 49:1142–5.
- Sadeque AJM, Usmani KA, Palamar S, et al. (2012). Identification of human UDP-glucuronosyltransferases involved in N-carbamoyl glucuronidation of lorcaserin. Drug Metab Dispos 40:772–8.
- Schaefer WH. (2006). Reaction of primary and secondary amines to form carbamic acid glucuronides. Curr Drug Metab 7:873–81.
- STRATTERA®. (2017). Prescribing information. Indianapolis, IN: Lilly USA, LLC.
- Vincent SH, Reed JR, Bergman AJ, et al. (2007). Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [14C]sitagliptin in humans. Drug Metab Dispos 35:533–8.