
Abstract
Bictegravir (BIC) is a potent small-molecule integrase strand-transfer inhibitor (INSTI) and a component of Biktarvy®, a single-tablet combination regimen that is currently approved for the treatment of human immunodeficiency virus type 1 (HIV-1) infection. The absorption, metabolism, distribution, and elimination (ADME) characteristics of BIC were determined through in vivo nonclinical and clinical studies (IND 121318).
[14C]BIC was rapidly absorbed orally in mice, rats, monkeys and human. The cumulative dose recovery was high in nonclinical species (>80%) and humans (95.3%), with most of the excreted dose recovered in faeces. Quantifiable radioactivity with declining concentration was observed in rat tissues suggesting reversible binding. Unchanged BIC was the most abundant circulating component in all species along with two notable metabolites M20 (a sulphate conjugate of hydroxylated BIC) and M15 (a glucuronide conjugate of BIC). BIC was primarily eliminated by hepatic metabolism followed by excretion of the biotransformed products into faeces. In vitro drug-drug interaction (DDI) studies with M15 and M20 demonstrated that no clinically relevant interactions were expected.
Overall, BIC is a novel and potent INSTI with a favourable resistance, PK, and ADME profile that provides important improvements over other currently available INSTIs for the treatment of HIV-1.
Introduction
Bictegravir (BIC; ) is a potent small-molecule human immunodeficiency virus type 1 (HIV-1) integrase strand-transfer inhibitor (INSTI) with a high barrier to resistance in vitro (Tsiang et al. Citation2016; Santoro et al. Citation2020). Biktarvy, the fixed-dose combination (FDC) tablet comprising BIC (50 mg), emtricitabine (FTC; 200 mg), and tenofovir alafenamide (TAF; 25 mg) is approved for the treatment of HIV-1 infection. Biktarvy is indicated as a complete regimen for the treatment of HIV-1 infection in patients who have no antiretroviral treatment history or to replace the current antiretroviral regimen in those who are virologically suppressed (HIV-1 RNA <50 copies/ml) and on a stable antiretroviral regimen, with no history of treatment failure and no known substitutions associated with resistance to the individual components of Biktarvy (EMA (European Medicines Agency) Citation2018).
Figure 1. Chemical structure of BIC (*denotes position of the [14C] label).
![Figure 1. Chemical structure of BIC (*denotes position of the [14C] label).](/cms/asset/3e91da06-f243-47f5-95ca-6808a275a0ab/ixen_a_2159569_f0001_b.jpg)
Radiolabeled pharmacokinetic (PK) and absorption, metabolism, distribution, and excretion (ADME) studies in animal species and human serve as the foundation for the understanding the drug disposition in the body. These studies provide understanding of the fraction absorbed, quantitative understanding of clearance mechanisms, structure elucidation of metabolites and define excretion routes for parent drug and metabolites. The results from radiolabeled ADME and in vitro studies are important in identifying the susceptibility to drug interactions as a victim. Finally, understanding of the plasma exposure of humans to the parent drug and metabolites enable confirmation of the suitability of the animal species used to evaluate the safety of the drug. These aspects of drug development appear in several guidance documents issued by regulatory agencies. The radiolabeled ADME studies reported herein for [14C]BIC were conducted in RasH2 transgenic mouse, Long Evans and Wistar-Han rat, cynomolgus monkey and healthy human subjects. The results described in this report fulfil the above described objectives to fully characterise the ADME of BIC. In addition, these studies informed the suitability of coadministration of BIC with FTC and TAF in Biktarvy.
Materials and methods
Reagents and compounds
BIC, and internal standards for mass spectrometry-based analyses were synthesised by Gilead Sciences (Foster City, CA, USA). Metabolite M15 was synthesised by Kalexsyn, Inc. (Kalamazoo, MI); M20 and M23 were synthesised by Gilead Sciences (Jin et al. Citation2019). [14C]BIC (≥55.9 μCi/μmol; ≥2.07 MBq/μmol; ≥97.3% purity) was synthesised by Moravek Biochemicals (Brea, CA, USA) or ViTrax (Placentia, CA, USA). The [14C] in [14C]BIC was incorporated at the carbonyl group of the trifluorobenzyl acetamide moiety of the molecule ().
All other chemicals or reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA; Gillingham, United Kingdom), VWR (West Chester, PA, USA), Toronto Research Chemicals (North York, ON, Canada), Thermo Fisher Scientific (Carlsbad, CA, USA), or BD Biosciences (Woburn, MA, USA) unless otherwise specified.
Nonclinical in vivo ADME studies
The PK and disposition of [14C]BIC were assessed at Covance Laboratories (Madison, WI, USA) following a single oral dose in male intact mice (n = 34); nonpigmented (Wistar-Han), intact and bile duct-cannulated (BDC) rats (n = 27 and 3, respectively); pigmented (Long Evans), intact rats (n = 9); and intact and BDC monkeys (each n = 3). An overview of the designs including the animal strain of these nonclinical ADME studies is provided in . All nonclinical studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health and were approved by the Institution’s Animal Care and Use Committee or local equivalent.
Table 1. Summary of nonclinical and clinical in vivo study designs.
Mice aged 7 weeks (Taconic Biosciences, Germantown, NY, USA), rats aged 7 to 12 weeks (Charles Laboratories, Wilmington, MA, USA), and monkeys aged 3 to 5 years (Covance Research Products, Alice, TX, USA) were assigned into groups and administered [14C]BIC via oral gavage at a dose of 2 mg/kg for mice and rats (300 and 100 μCi/kg, respectively) and 1 mg/kg for monkeys (25 μCi/kg). For mice and rats, the dosing vehicle was (by volume) 5% ethanol, 55% polyethylene glycol 300, and 40% water; for monkeys, the dosing vehicle was 30% Captisol in reverse osmosis water. Rats and monkeys were fasted overnight and through approximately 4 h postdose; mice were not fasted.
Blood was collected for PK analyses at specified time points through 168 h postdose. At designated times after dosing, urine, faeces, bile, residual carcasses, or nonbiological samples (cage rinses, cage wash, cage wipe, cage debris, bile cannula rinse, and jacket rinse, as applicable) were collected for analysis of excretion of radioactivity and mass balance. At designated time points, nonpigmented and pigmented rats were sacrificed, exsanguinated and frozen prior to embedding and sectioning for analysis by quantitative whole-body autoradiography (QWBA).
Clinical ADME study
Study design
The PK, metabolism, and excretion of BIC following administration of a single oral dose of [14C]BIC was evaluated in a phase 1, open-label, single-centre, mass-balance clinical study (GS-US-141-1481, IND 121318) conducted in eight healthy male subjects. An overview of the design of this study is presented in . The study was approved by an institutional review board before initiation and was conducted in accordance with recognised international scientific and ethical standards, including, but not limited to, the International Conference on Harmonisation guideline for Good Clinical Practice and the principles embodied in the Declaration of Helsinki. Written informed consent was obtained from all subjects.
Prospective subjects were screened no more than 28 days prior to dosing. Eligible subjects were confined at the study centre starting on Day −1, received a single oral dose of [14C]BIC on the morning of Day 1, and remained confined for a minimum of 6 days and a maximum of 22 days following dosing. The overall duration of the confinement period was based on real-time monitoring of the amount of [14C] radioactivity present in the whole blood, plasma, urine, and stool samples using liquid scintillation counting (LSC) radio analysis. A safety follow-up telephone call was conducted 7 (± 1) days after subjects were discharged from the study centre.
Subjects
Eight male subjects were enrolled into the study and completed the study. The median age was 37 years (range: 20 − 43), the median body mass index was 27.5 kg/m2 (range: 19.7 − 30.0), and the median estimated glomerular filtration rate (Cockcroft-Gault method; Cockcroft and Gault Citation1976) was 117.48 ml/min (range: 96.53 − 141.11) and no significant medical history. Subjects were negative for hepatitis B/C and HIV and had refrained from blood donation or other activities that could have affected ADME assessments.
Subjects were ineligible if they engaged in substance or alcohol abuse that could have potentially interfered with subject compliance or safety; had taken any over-the-counter or prescription medications (except for vitamins, acetaminophen, or ibuprofen) within 28 days prior to dosing; or had taken systemic steroids, immunosuppressant therapies, or chemotherapeutic agents within 3 months prior to screening or expected to receive these agents during the study.
Treatment and sampling
Radiation dosimetry results from the rat tissue distribution study informed the BIC radioactive dose in humans. Administration of a single 100-μCi (3.7 MBq) oral dose of [14C]BIC was not expected to represent a significant radiation exposure risk to adult male subjects. Each subject received a single 100-mg dose of [14C]BIC (99 mg of nonradiolabeled BIC sodium salt and approximately 100 μCi of [14C]BIC [equivalent to approximately 1 mg of BIC]) and was administered as an 40-ml water/ethanol solution (4:6, v/v). The dosing container was then rinsed twice, each time with 50 ml of cranberry juice, and administered to the subject. BIC was administered on the morning of Day 1 within 5 min of completing a standard moderate-fat breakfast and following an overnight fast. The entire study drug solution and cranberry juice rinse (total of approximately 140 ml) was administered within a 10-minute window.
Whole blood and plasma samples were collected for total radioactivity, BIC bioanalysis, and metabolite profiling and identification at prespecified time points. After the 120-hour postdose time point, additional whole blood or plasma samples were collected at 24-hour intervals up to the 504-hour postdose time point (morning of Day 22) or until one of the following two criteria was met: LSC indicated that the radioactivity in two consecutive whole blood or plasma samples had decreased to levels below the limit of quantitation, or both urine and faeces sample collections were discontinued. Urine and faeces were collected for total radioactivity at prespecified time points. After the 120-hour postdose time point, urine and faeces continued to be collected over successive 24-hour intervals up to the 504-hour postdose time point (morning of Day 22), or until LSC showed that the radioactivity in samples from two consecutive 24-hour collection intervals were ≤1% of the administered dose and the cumulative [14C] radioactivity recovered in urine and faeces was ≥90% of the administered dose.
Urine samples for PK analysis were taken from the pooled urine collected for radioactive monitoring at each of the collection intervals, beginning predose (within 12 h prior to dosing) until 144 h postdose.
Analytical methods
Quantification of BIC in human plasma
Concentrations of non-radiolabeled BIC in human plasma were determined using liquid chromatography-tandem mass spectrometry (LC-MS/MS) bioanalytical methods, which were performed and validated by QPS, LLC (Newark, DE, USA). The method involved protein precipitation of BIC and its internal standard from human plasma. The quantification range for BIC in human plasma was 20 − 20 000 ng/ml. The interassay precision (percentage coefficient of variation) and accuracy (percentage relative error) for BIC ranged from 3.9 to 5.7% and 2.4 to 5.8%, respectively.
Quantification of 14C radioactivity
The amount of radioactivity present in samples from mice, rats, monkeys, and humans was quantified by LSC using Model 2900TR or 2910TR liquid scintillation counters (PerkinElmer, Waltham, MA, USA). All sample combustions were performed using a Model 307 Sample Oxidiser (PerkinElmer); the resulting 14CO2 was absorbed by Carbo-Sorb and mixed with Permafluor (PerkinElmer). Plasma, urine, or bile samples from animals and human subjects were mixed with either EcoLite(+) scintillation cocktail (MP Biomedicals, Irvine, CA, USA) or Ultima Gold XR scintillation cocktail (PerkinElmer) and analysed directly by LSC. Blood samples from humans were mixed, combusted, and analysed by LSC. Blood samples from animals were digested using a commercial solubilising agent, incubated for ≥1 h at approximately 60 °C, and mixed with disodium EDTA (0.1 M) and 30% hydrogen peroxide. Foaming was allowed to dissipate, and the blood samples were mixed with scintillation cocktail and analysed by LSC. Faeces samples from human subjects were combined for each subject at 12- or 24-hour intervals. Each sample was mixed with ethanol/water (20:80, v/v), homogenised using a probe-type homogeniser, then combusted and analysed by LSC. Faeces samples from animals were homogenised in acetonitrile/water (20:80, v/v), combusted, and analysed by LSC. Residual mouse carcasses were homogenised in a precooled food grinder, digested in sodium hydroxide, and mixed with 30% hydrogen peroxide; foaming was allowed to dissipate, and the samples were mixed with scintillation cocktail and analysed by LSC. Residual rat carcasses were digested in sodium hydroxide, homogenised in ethanol, and analysed by LSC.
The amount of radioactivity in 52 tissues was quantified, where possible, in nonpigmented and pigmented rats by QWBA. Sagittal sections (40-μm thickness) were collected on adhesive tape in a CM3600 cryomacrotome (Leica Biosystems, Buffalo Grove, IL) and dried at −20 °C. The mounted sections were exposed to phosphorimaging screens along with blood standards for 4 days, and the screens were scanned using a Storm scanner. MCID analysis software (InterFocus Imaging, Cambridge, UK) was used to generate calibrated standard curves from the autoradiographic standard image data, and 14C concentrations in tissues were quantified from each standard curve.
Metabolite profiling and identification
Details on sample preparation and analytical methods for metabolite profiling and identification, as well as representative LC-MS/MS data and proposed fragmentation patterns for BIC and the main metabolites M15 and M20, are provided in the Supplemental Material. Briefly, plasma samples were pooled by time point across intact mice (0.5, 1, 4, 12, 24, 48, and 72 h), intact rats (0.5, 1, 4, 12, 24, 72, and 168 h), intact monkeys (0.5, 2, 8, 12, 24, 48, and 72 h), and human subjects (1, 4, 8, 12, 24, 48, and 72 h). For mice and rats, these samples were then further pooled to prepare a single area under the curve (AUC)-pooled sample using a time-weighted pooling method (Hop et al. Citation1998), while for monkeys and human subjects the AUC was calculated from concentrations of each pooled time point sample using the trapezoidal method. Urine, bile, and faeces samples were prepared by weight-proportional pooling across time intervals, and the prepared plasma and excreta samples were analysed by liquid chromatography-high-resolution mass spectrometry (HRMS) analysis with radioactivity detection.
Quantification of [14C]BIC and metabolites
The radioactivity associated with the parent drug and each identifiable metabolite was determined. Quantification of [14C]BIC and its metabolites in pooled plasma, urine, bile, and faeces samples in the nonclinical and clinical ADME studies was based on integration of the respective metabolite profile peaks in the liquid chromatography-radiochromatograms and the radioactive concentration or dose recovered in the corresponding sample.
The limit of quantitation for radioactivity in plasma and urine from mice, rats, and monkeys; bile from rats and monkeys; and faeces from mice and rats was established at 1% of the injected radioactivity and 10 cpm peak height in a chromatogram. The limit of quantitation for radioactivity in faeces from monkeys was established at 1% of each chromatogram. For a given metabolite and matrix, values below the limit of quantitation were only reported if at least one time point or interval had a value above the limit of quantitation. For samples from human subjects, the limit of quantitation for radioactivity was established at 15 dpm (up to 1.5, 0.78, and 0.84% of the run for the pooled plasma, urine, and faeces samples injected). Peaks with a peak height ≥15 dpm on the radiochromatogram were integrated for quantitation.
In vitro drug-drug interaction studies with BIC metabolites
The in vitro methods used to assess the DDI potential of M15 and M20 were comparable to the methods applied previously for BIC (Subramanian et al. submitted manuscript). Binding of M20 (2 μM) to human plasma and liver microsomal proteins (0.5 mg/ml) was measured using the in vitro methods applied for BIC. M20 was also assessed for its potential to inhibit human cytochrome P450 (CYP) enzymes (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3 A), uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1), and select human transporter proteins including P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), organic anion transporter (OAT) 1, OAT3, organic cation transporter (OCT) 2, multidrug and toxin extrusion (MATE) 1, MATE2-K, organic anion transporting polypeptide (OATP) 1B1, OATP1B3, and OATP2B1. The inhibition of UGT1A1, OATP1B1, OATP1B3, and OATP2B1 by M15 was also assessed. For the present analysis, the potential for M15 and M20 to inhibit human multidrug-resistance protein (MRP) 2-mediated transport was also assessed. Briefly, M15 (0 − 300 μM), M20 (0 − 300 μM), or positive control (200 μM benzbromarone) was incubated with membrane vesicle preparations (total protein: 50 μg/well) and probe substrate estradiol-17β-glucuronide in the absence or presence of ATP, and the amount of substrate in the filtered vesicles was determined by liquid scintillation. Fractional transport activities were calculated, and half-maximal inhibitory concentration (IC50) values were determined using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA).
Pharmacokinetic and human dosimetry analyses
Blood and plasma PK parameters for total radioactivity in mice, rats, monkeys, and human subjects, as well as plasma PK parameters for non-radiolabeled BIC in humans were estimated using standard non-compartmental analysis by Phoenix WinNonlin software, versions 6.2.1 or 6.3 (Pharsight Corporation, Mountain View, CA, USA), including maximum concentration (Cmax), time to Cmax (Tmax), t1/2, AUC from time 0 to the last measurable time point (AUClast), and AUC from time 0 to infinity (AUCinf). Select plasma PK parameters were also calculated for [14C]BIC and its metabolites, and the relative exposure of each component in plasma was expressed as percentage of the total radioactivity AUClast. For excreta, the percentage of [14C] dose recovered as parent drug and metabolites was determined.
Human dosimetry parameters were calculated for a radioactive dose administration to adult male subjects of approximately 100 μCi (∼1.43 μCi/kg or ∼0.53 MBq/kg for a 70-kg male) using the rat QWBA data. Tissue PK parameters for radioactivity were estimated for non-pigmented and pigmented rats (Groups 3 and 4, respectively, ), and the overall whole-body effective dose was determined by summing the effective doses of selected tissues (bone marrow, uveal tract of eye, testes, epididymis, and skin).
Results
Nonclinical in vivo ADME studies
Pharmacokinetics of total radioactivity in blood and plasma
The blood and plasma PK parameters for total radioactivity following a single oral administration of [14C]BIC in intact mice, intact rats, and intact and BDC monkeys are summarised in .
Table 2. Blood and plasma PK parameters for total radioactivity following a single oral administration of [14C]BIC in mice, rats, and monkeys.
[14C]BIC was rapidly absorbed following oral administration to intact mice. The mean Cmax values of total radioactivity were 4830 and 9900 ng Eq/g in blood and plasma, respectively, and were observed at the first sampling time point (0.5 h postdose). Mean radioactivity concentrations then decreased through 144 and 120 h in blood and plasma, respectively, and were below the limit of quantitation thereafter through 168 h postdose. The mean AUClast values for total radioactivity were 51,726 and 107,605 ng Eq•h/g in blood and plasma, respectively, and mean t1/2 values were 21.0 and 15.8 h, respectively.
[14C]BIC was also rapidly absorbed following oral administration in intact rats, reaching mean Cmax values of 14,400 and 30,600 ng Eq/g in blood and plasma, respectively, by 1 h postdose. Total radioactivity in blood and plasma then generally decreased, with quantifiable radioactivity observed through 168 h postdose. The mean AUCinf values for total radioactivity were 541 634 and 1 021 223 ng Eq•h/g in blood and plasma, respectively, and mean t1/2 values were 46.9 and 50.3 h, respectively.
Following oral administration of [14C]BIC in intact monkeys, the mean Cmax values for total radioactivity were 3200 and 5410 ng Eq/g in blood and plasma, respectively, and were observed by 1.67 h postdose. Concentrations of total radioactivity in blood and plasma then decreased through 48 and 96 h postdose, respectively, and were below the limit of quantitation thereafter through 168 h postdose. The mean t1/2 values for total radioactivity were 9.05 and 14.4 h in blood and plasma, respectively.
The mean blood-to-plasma ratio of total radioactivity following oral dosing of [14C]BIC was approximately 0.46–0.53 for mice, 0.46–0.57 for rats, and 0.50–0.84 for monkeys over the course of each study, suggesting a limited association of radioactivity (BIC + metabolites) with blood cells.
Tissue distribution
Concentrations of total radioactivity in blood and representative tissues determined by QWBA following a single 2-mg/kg dose of [14C]BIC (100 μCi/kg) in non-pigmented and pigmented rats are presented in . The [14C]BIC-derived radioactivity was rapidly (0.25 h postdose) and widely distributed to most tissues and was similar in both non-pigmented and pigmented rats. The Cmax for total radioactivity was reached by 1 to 4 h postdose in most tissues. Concentrations of radioactivity in tissues were lower compared with blood and decreased throughout the course of the study (168 h postdose); however, due to the long BIC PK half-life (Wang et al. Citation2017), radioactivity was not eliminated from most tissues by 168 h postdose.
Table 3. Concentrations of total radioactivity (ng Eq/g) in blood and selected tissues determined by QWBA following a single 2-mg/kg oral dose of [14C]BIC in non-pigmented and pigmented rats.
The tissue distribution study also informed the radiation dosimetry for the clinical ADME study with [14C]BIC. For a 100-μCi human radioactive dose, the overall whole-body exposure was calculated to be approximately 2.5% of the allowable 3000-mrem exposure limit established by the Food and Drug Administration.
Excretion
The cumulative excretion of radioactivity over a 1-week collection period following a single oral administration of [14C]BIC in intact mice, intact and BDC rats, and intact and BDC monkeys is summarised in . The mean cumulative overall recovery of dosed radioactivity was >80% in all species. The excretion routes in intact animals were consistent across species, with the majority of the excreted dose recovered in faeces (>40% of [14C] dose) and minor amounts recovered in urine (<21% of [14C] dose). The excretion of radioactivity into bile in BDC rats and monkeys was approximately 34 and 40% of the [14C] dose, respectively. The mean recovery of radioactivity in rat carcasses was 13.7% of the [14C] dose. This residual radioactivity was attributed to the long PK half-life of BIC.
Table 4. Cumulative dose recovery following a single oral administration of [14C]BIC in mice, rats, monkeys, and human subjects (mean ± S.D.).
Clinical ADME study
Pharmacokinetics of total radioactivity and BIC in blood and/or plasma
Concentration-time profiles for total radioactivity in blood and plasma by LSC and for non-radiolabeled BIC in plasma by LC-MS/MS following a single oral dose of [14C]BIC are shown in , and a summary of the respective PK parameters are presented in . The median Tmax values for total radioactivity in blood and plasma were 3.0 and 3.5 h postdose, respectively, with median t1/2 values of 22.9 and 21.3 h, respectively. The mean blood-to-plasma concentration ratio of total radioactivity was approximately 0.50 to 0.55 through 120 h postdose, indicating negligible association of radioactivity with blood cells.
Figure 2. Semilogarithmic concentration-time profiles for total radioactivity in whole blood and plasma by LSC and for nonradiolabeled BIC in plasma by LC-MS/MS following a single 100-mg (100 μCi) dose in healthy subjects (n = 8, mean ± S.D.).
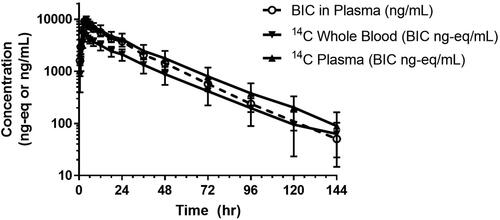
Table 5. Blood and plasma PK parameters for total radioactivity by LSC and plasma PK parameters for non-radiolabeled BIC by LC-MS/MS following a single 100-mg (100 μCi) dose in healthy human subjects (n = 8, mean ± S.D.).
The median plasma Tmax of non-radiolabeled BIC was 3.5 h, with a median t1/2 of 17.3 h. The plasma PK parameters of BIC observed in this study (e.g. apparent oral clearance, apparent volume of distribution, and t1/2) were comparable to those observed following administration of single-agent BIC 75 or 100 mg after a moderate- or high-fat meal, respectively (Zhang et al. Citation2017).
Excretion
The mean cumulative recovery of total radioactivity was 95.3% (range: 92.5 − 96.9%), with 60.3% (range: 51.2 − 68.0%) recovered in faeces and 35.0% (range: 29.0 − 44.0%) recovered in urine. Most (90.9%) of the administered total radioactivity was recovered in the first 120 h postdose. Renal clearance of non-radiolabeled BIC was minimal, with approximately 1.3% of the [14C] dose recovered in urine through 144 h postdose.
Safety
One subject had two treatment-emergent adverse events (AEs) considered related to treatment with BIC (feeling hot and in a euphoric mood). Both AEs were of low severity (Grade 1). No other adverse events, serious adverse events, or deaths were reported, and no subject discontinued study drug or the study due to an adverse event.
Metabolite profiling and quantitation
Summaries of BIC and its metabolites observed in AUC-pooled plasma and excreta from mice, rats, monkeys, and human subjects are provided in and and (pooling method details are provided in the Supplemental Information). The proposed in vivo biotransformation scheme is shown in . The criteria for the metabolic scheme was to include human metabolites that were present at >1% of the [14C] dose in excreta or >1% of the plasma AUClast of total radioactivity. The structures of metabolites M15, M20, and M23 were also confirmed with synthetic standards. Nonclinical metabolites that were present at >1% of the [14C] dose in excreta or >1% of the plasma AUClast of total radioactivity were listed in the footnotes.
Figure 3. Radiochromatograms of (A) plasma (8 h postdose; pool of all subjects), (B) urine (0 − 24 h postdose; Subject #3002), and (C) faeces (0 − 48 h post dose; pool of all subjects) following a single 100-mg (100 μCi) oral dose of [14C]BIC in human subjects. Only select metabolites are labelled in the radiochromatograms. A more complete listing is provided in .
![Figure 3. Radiochromatograms of (A) plasma (8 h postdose; pool of all subjects), (B) urine (0 − 24 h postdose; Subject #3002), and (C) faeces (0 − 48 h post dose; pool of all subjects) following a single 100-mg (100 μCi) oral dose of [14C]BIC in human subjects. Only select metabolites are labelled in the radiochromatograms. A more complete listing is provided in Table 7.](/cms/asset/f5b5d009-7e9a-41d4-8944-fcd2e90d6060/ixen_a_2159569_f0003_b.jpg)
Figure 4. Proposed biotransformation scheme of BIC. Identities were matched based on HRMS. In vitro metabolites M465a and M465b were both hydroxylated BIC metabolites and positional isomers; HRMS data were consistent with in vivo metabolites M21 or M25 (exact match unknown). In vitro metabolite M465c was also a hydroxylated BIC metabolite; HRMS data were consistent with in vivo metabolite M23. In vitro metabolite M625 was a glucuronide conjugate of BIC; HRMS data were consistent with in vivo metabolite M15. Additional in vitro and in vivo matched metabolites included M611 (BIC-glucoside) with M39 observed in monkey excreta and M641 (hydroxy-BIC-glucuronide) with M35 and M45 observed in monkey and human excreta, respectively. Additional metabolites present at >1% of the [14C] dose in excreta or >1% of the plasma AUClast of total radioactivity in mouse, rat, or monkey included the following: M7 (desfluoro-hydroxy-BIC-cysteine conjugate-1), M10 (desfluoro-hydroxy-BIC-cysteine-glycine conjugate), M12 (hydroxy-BIC-glucuronide), M16 (desfluoro-hydroxy-BIC-glucuronide), M18 (hydroxy-BIC-1), M19 (desfluoro-hydroxy-BIC-1), M26 (desfluoro-hydroxy-BIC-3), M28 (desfluoro-hydroxy-BIC-4), M30 (desfluoro-dihydroxy-BIC-cysteine conjugate), M32 (desfluoro-hydroxy-BIC-cysteine-glycine conjugate-2), M35 (hydroxy-BIC-glucuronide-2), M37 (desfluoro-hydroxy-BIC-cysteine conjugate-3), M38 (dihydroxy-BIC), M39 (BIC-glucoside), and M42 (hydroxy-BIC-5).
![Figure 4. Proposed biotransformation scheme of BIC. Identities were matched based on HRMS. In vitro metabolites M465a and M465b were both hydroxylated BIC metabolites and positional isomers; HRMS data were consistent with in vivo metabolites M21 or M25 (exact match unknown). In vitro metabolite M465c was also a hydroxylated BIC metabolite; HRMS data were consistent with in vivo metabolite M23. In vitro metabolite M625 was a glucuronide conjugate of BIC; HRMS data were consistent with in vivo metabolite M15. Additional in vitro and in vivo matched metabolites included M611 (BIC-glucoside) with M39 observed in monkey excreta and M641 (hydroxy-BIC-glucuronide) with M35 and M45 observed in monkey and human excreta, respectively. Additional metabolites present at >1% of the [14C] dose in excreta or >1% of the plasma AUClast of total radioactivity in mouse, rat, or monkey included the following: M7 (desfluoro-hydroxy-BIC-cysteine conjugate-1), M10 (desfluoro-hydroxy-BIC-cysteine-glycine conjugate), M12 (hydroxy-BIC-glucuronide), M16 (desfluoro-hydroxy-BIC-glucuronide), M18 (hydroxy-BIC-1), M19 (desfluoro-hydroxy-BIC-1), M26 (desfluoro-hydroxy-BIC-3), M28 (desfluoro-hydroxy-BIC-4), M30 (desfluoro-dihydroxy-BIC-cysteine conjugate), M32 (desfluoro-hydroxy-BIC-cysteine-glycine conjugate-2), M35 (hydroxy-BIC-glucuronide-2), M37 (desfluoro-hydroxy-BIC-cysteine conjugate-3), M38 (dihydroxy-BIC), M39 (BIC-glucoside), and M42 (hydroxy-BIC-5).](/cms/asset/99061fbc-d4df-47b7-8033-4dbd165d20db/ixen_a_2159569_f0004_b.jpg)
Table 6. Metabolite profile observed in AUC-pooled plasma following a single oral administration of [14C]BIC in mice, rats, monkeys, and human subjects.
Table 7. Metabolite profile observed in pooled urine, bile, and faeces following a single oral administration of [14C]BIC in mice, rats, monkeys, and human subjects.
Unchanged BIC was the most abundant circulating component in all nonclinical species (76.5% to 96.5%) and in human (67.9%) of the plasma AUClast of total radioactivity (). Unchanged BIC in human plasma (i.e. the ratio of LC-MS/MS bioanalysis AUC to total radioactivity AUC) determined from intensive PK timepoint sampling () showed a value of 86.1% ± 25.6%. Human plasma metabolite profiles were generated from sparsely sampled time-points () and resulted in a single point estimate for the unchanged BIC to total radioactivity AUC ratio (67.9%); this value is within the range from the PK analysis estimate (86.1% ± 25.6%).
Metabolite M20 (hydroxy-BIC-sulphate) was the most abundant circulating metabolite in humans (20.1% of the total radioactivity AUClast) and rats (11.3% of the total radioactivity AUClast) and was also present in monkeys but at a much lower abundance (0.77% of the total radioactivity AUClast). Metabolite M15 (BIC-glucuronide) was the next most abundant circulating metabolite in humans (8.6% of the total radioactivity AUClast) and was also observed in monkeys at low levels (0.55% of the total radioactivity AUClast). The major circulating metabolite in monkeys was the M42 metabolite (hydroxy-BIC [isomer of M21]; 12.2% of the total radioactivity AUClast), which was not detected in mouse, rat, or human plasma.
Unchanged BIC was the most abundant component identified in faeces from humans (33.7% of [14C] dose), intact mice (64.4% of [14C] dose), intact and bile-duct cannulated (BDC) rats (23.9 and 13.4% of [14C] dose, respectively), and intact and BDC monkeys (each 10.7% of [14C] dose). M9 (cysteine conjugate of desfluoro-hydroxy-BIC) was the major metabolite excreted in faeces in human subjects (9.50% of [14C] dose), followed by coeluted M21/M22 (hydroxy-BIC/desfluoro-hydroxy-BIC; 7.05% of [14C] dose), and M23 (hydroxy-BIC; 6.75% of [14C] dose). Coeluted M21/M22 were also the most abundant metabolites in faeces from intact mice (4.21% of [14C] dose), intact and BDC rats (8.07 and 1.32% of [14C] dose, respectively), and BDC monkeys (3.02% of [14C] dose). Coeluted M23/M42 were the most abundant metabolites identified in faeces from intact monkeys (10.5% of [14C] dose).
M15 was the main component detected in bile from BDC rats (12.7% of [14C] dose) and was also a major component in bile from BDC monkeys (6.14% of [14C] dose). The most abundant metabolite identified in bile from BDC monkeys was the M9 metabolite, accounting for 10.9% of the [14C] dose. Low levels of unchanged BIC were recovered in bile from BDC rats and monkeys.
Coeluted M15/M58 (both BIC-glucuronides) were the major components recovered in human urine (21.4% of [14C] dose). M15 was also the main component in urine from intact mice (0.88% of [14C] dose) and an abundant component in urine from intact and BDC monkeys (3.84 and 3.94% of [14C] dose, respectively). Each component observed in urine from intact and BDC rats accounted for <1% of the [14C] dose with the exception of M21, which accounted for 1.01 and 1.10% of the [14C] dose, respectively. The M42 metabolite was the most abundant component detected in urine from intact and BDC monkeys (6.15 and 2.61% of [14C] dose, respectively) and was not present in mouse, rat, or human urine. Low levels of unchanged BIC were recovered in urine from mice, rats, and human subjects, and no unchanged BIC was detected in urine from monkeys.
In vitro drug-drug interaction studies with BIC metabolites
M20, the sulphate conjugate of hydroxylated BIC, was the major circulating metabolite detected in human plasma, accounting for approximately 20% of the plasma AUClast of total radioactivity. M15, a glucuronide conjugate of BIC, accounted for close to 9% of the plasma AUClast of total radioactivity. Additional in vitro studies were performed to evaluate the DDI potential of these two metabolites.
M20 had little or no inhibitory effect on the activities of human hepatic microsomal enzymes CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A up to concentrations of 25 μM (IC50 > 22.4 μM) and was a very weak inhibitor of UGT1A1 activity at concentrations up to 300 μM (IC50 = 153 μM) (). M20 showed no inhibition of P-gp or BCRP-mediated transport at up to the highest tested concentration (25 µM) (). At concentrations up to 100 μM, M20 showed no inhibition of OATP1B1 and was a weak inhibitor of OATP1B3 and OATP2B1 (IC50 >100 μM). M20 showed concentration-dependent inhibition of MRP2, with an IC50 value of 45 μM at the highest tested concentration of 300 µM. M20 also showed concentration-dependent inhibition of renal transporters in vitro. For OAT1 and OAT3, the IC50 values were determined to be 0.91 and 0.88 μM, respectively. For OCT2, MATE1, and MATE2-K, the IC50 values were determined to be >5.9, >7.4, and >7.1 μM, respectively. M20 was highly bound to human plasma proteins (mean fraction unbound of 0.13%) and was negligibly bound to human microsomal proteins (mean fraction unbound of 86%).
Table 8. Inhibition of major human CYP enzymes (mean, n = 7) and UGT1A1 (mean, n = 2) by metabolites M15 and M20.
Table 9. Inhibition of transporters by metabolites M15 and M20.
M15 showed no detectable inhibitory effect on UGT1A1 activity at up to the highest tested concentration (300 μM), with an IC50 value >300 μM (<2% inhibition). At concentrations up to 100 μM, M15 showed no inhibition of OATP1B3 but was a weak inhibitor of OATP1B1 (IC50 > 100 μM). M15 showed concentration-dependent inhibition of MRP2, with a calculated IC50 value of 256 μM at the highest tested concentration of 300 μM ().
Discussion
The disposition of [14C]BIC was extensively characterised following single-dose oral administration in nonclinical species and human. [14C]BIC was rapidly absorbed in all species studied, with maximum mean concentrations of drug-derived radioactivity in blood and plasma observed within a few hours after dosing. The blood-to-plasma concentration ratio of BIC is <0.65 across species and was <1 for total radioactivity suggesting that BIC and the circulating metabolites are excluded from the cellular components of blood as expected with the high plasma protein binding of BIC (percent unbound of <0.5% in all species tested except for dogs (1.24%), including 0.25% unbound in humans; Wang et al. Citation2017) and M20.
Following oral administration of [14C]BIC in male rats, all tissue/blood concentrations of radioactivity were <1 consistent with the low volume of distribution of BIC (Wang et al. Citation2017) and likely the circulating metabolites. Low levels of radioactivity were detected in the brain (<4% relative to blood [14C]BIC AUC), suggesting that unbound [14C]BIC-derived radioactivity crossed the blood-brain barrier. Following Biktarvy administration in virologically suppressed patients, the total and unbound BIC cerebrospinal fluid concentrations (CSF) were above the half-maximal effective concentration value in all patients, and all subjects had HIV viral suppression in plasma and CSF; thus, BIC may contribute to inhibition of viral replication in this compartment (Tiraboschi et al. Citation2020). Quantifiable radioactivity was observed in rat tissues at 168 h postdose, but concentrations were declining, suggesting reversible binding. Distribution trends of radioactivity in pigmented skin and uveal tract of the eye indicated that [14C]BIC-related radioactivity was not selectively associated with melanin-containing tissues. These data supported the safe administration of a single 100-μCi dose of [14C]BIC to male subjects in the clinical ADME study.
The cumulative overall recovery of dosed radioactivity was high (>80%) in all species studied. Following a single 100-mg dose of [14C]BIC in human subjects, the mean cumulative combined urinary and faecal recovery of [14C] radioactivity was 95.3%, with 90.9% of the administered [14C] radioactivity being recovered in the first 120 h postdose; this result was consistent with the observed t1/2 of total radioactivity. The high recovery of radioactivity suggests no notable sequestration binding of BIC or its metabolites.
The metabolism and elimination profiles of BIC in human subjects were consistent with those observed in nonclinical species. Following oral administration of [14C]BIC, unchanged parent drug was the predominant circulating component in mice (95.5%), rats (76.5%), monkeys (80.2%), and humans (67.9%). Renal clearance of unchanged BIC was minimal (0–1.3% of dose) in all species. Additionally, biliary excretion of unchanged BIC was found to be negligible in BDC rats and monkeys (0.58 and 0.46% of dose, respectively).
[14C]BIC was extensively metabolised in all species through direct glucuronidation, oxidation, and oxidation followed by phase II conjugation. In monkey, [14C]BIC was metabolised through the oxidative pathways to a greater extent compared with rat and human. All human circulating metabolites were also found in nonclinical species and in toxicology species. Overall, clinical and nonclinical data collectively indicate that BIC was primarily eliminated by hepatic metabolism followed by excretion of the biotransformed products into faeces.
In rat and monkey, absorption was high (>86% of dose) based on the cumulative radiodose recovery in excreta and the amount of intact BIC in faeces. In human, following a single oral dose of 100-mg [14C]BIC under fed conditions, the fractions of radioactive dose recovered in urine (∼35%) and excreted in the faeces as metabolites (∼27%) indicate that at least 65% of the recovered dose (∼95%) was absorbed. Additionally, M15, a direct glucuronide conjugate, was observed in rat and monkey bile with none observed in human faeces. The nonclinical data suggest that a portion of the unchanged BIC recovered in human faeces (∼34% of dose) is from M15 in bile that undergoes biliary secretion to gastrointestinal tract, is deconjugated (presumably by bacteria) and recovered as BIC in faeces. The cumulative data shows absorption of BIC was high in human (65–88% of dose). As BIC was a substrate for intestinal efflux transporters P-gp and BCRP in vitro (EMA (European Medicines Agency) Citation2018), and the transepithelial permeability of BIC was high across human colon carcinoma-derived cell monolayers (Subramanian et al. submitted manuscript), passive transcellular diffusion is expected to be the primary mechanism for BIC intestinal absorption. BIC was not a substrate or inhibitor of uptake transporters organic anion transporting polypeptide (OATP) 1B1 or OATP1B3 (FDA (Food and Drug Administration) Citation2018); hepatic uptake is not expected to contribute to the disposition of BIC.
The plasma PK parameters estimated in the clinical ADME study were comparable to those observed in studies in which BIC 75 or 100 mg was administered following a moderate- or high-fat meal, respectively (Studies GS-US-141-1485 and GS-US-141-1218, respectively); therefore, the results of the present study are applicable to other clinical studies investigating BIC PK (EMA (European Medicines Agency) Citation2018; Zhang et al. Citation2017). Food had been noted to modestly increase BIC exposure during administration as a single agent (Study GS-US-141-1418), but with improved formulation for oral absorption the effect became less significant.
BIC metabolism was predominantly mediated by CYP3A and UGT1A1 (FDA (Food and Drug Administration) Citation2018; EMA (European Medicines Agency) Citation2018). Metabolite profiling in total excreta from the clinical ADME study suggested that approximately 30–31% of the dose was oxidised metabolites of BIC which may be further metabolised to conjugated products, and approximately 3–5% of the dose was unidentified metabolites, which were likely oxidative metabolites. Radioactivity in urine consisted primarily of BIC glucuronide(s) (≥21% of dose).
M20, the sulphate conjugate of hydroxylated BIC, was the only metabolite in human plasma that was present at >10% of the circulating drug-related material. Based upon recovery of M23 and M20, conversion to M20 represents only a very minor fraction of the overall metabolic clearance of BIC despite being a significant fraction of radioactivity in plasma. Therefore, further studies were not conducted to understand the enzymology of M23 and M20 formation since they represent insignificant clearance pathways for BIC. While not evaluated, the presence of this conjugate in plasma suggests transport from the hepatocyte to blood, potentially by MRP3/4. The very high plasma protein binding and anionic nature of M20 restricts this metabolite to the plasma compartment and explains why M20 exposure is high in plasma while being a very minor metabolite in excreta. Since M20 is a phase II metabolite and is pharmacologically inactive, no additional nonclinical safety testing of this metabolite was required (CDER (Center for Drug Evaluation and Research)), Citation2016). However, given the results from multiple methods identifying M20 as a major circulating metabolite in humans, additional in vitro studies were performed to evaluate the DDI potential of this metabolite. Results from these studies demonstrate that clinical interactions between M20 with substrates of the major CYP enzymes or UGT1A1 were unlikely. While BIC is a weak inhibitor of P-gp and BCRP transporters, M20 shows no inhibition of transport by P-gp or BCRP. Like BIC, M20 does not inhibit OATP1B1. BIC does not inhibit OATP1B3, while M20 is a weak inhibitor of OATP1B3 and OATP2B1. While M20 shows concentration-dependent inhibition of MRP2, the IC50 value (45 μM) is approximately 20-fold higher than the estimated mean plasma concentration of M20 (estimated average total plasma concentration is ∼2.3 μM), and thus, no clinically relevant interaction is expected. As M20 is excreted via urine and multiple interactions have been reported between BIC/FTC/TAF and drugs that are renally excreted, the inhibition potential of M20 on renal transporters OAT1, OAT3, OCT2, MATE1, and MATE2-K was evaluated. While M20 also showed concentration-dependent inhibition of the renal transporters in vitro, the inhibition constant (Ki=IC50/2 assuming competitive inhibition; lowest value 0.44 μM; ) of each evaluated renal transporter was >142-fold higher compared with the unbound M20 mean plasma concentration (∼3.2 nM); therefore, M20 was not an inhibitor of these renal transporters at the observed clinical concentration, and no in vivo DDI assessments were warranted.
BIC, which was shown to have a low potential to perpetrate clinically meaningful DDIs via known drug metabolising enzymes or transporters when dosed alone (EMA (European Medicines Agency) Citation2018; FDA (Food and Drug Administration) Citation2018). In the FDC product Biktarvy® (EMA (European Medicines Agency) Citation2018), BIC is coadministered with FTC and TAF. BIC, FTC, and TAF have distinct metabolic and excretion pathways for elimination: BIC is metabolised by CYP3A-mediated oxidation and conjugation by UGT enzymes and then eliminated into faeces; FTC is eliminated primarily intact by renal excretion; and the initial step in TAF elimination is intracellular hydrolysis by CatA and CES1 and subsequently to TFV which is eliminated by renal excretion (Birkus et al. Citation2016). Thus, coadministration of BIC, FTC, and TAF was not anticipated to change the excretion of the individual compounds or result in clinically relevant DDIs (EMA (European Medicines Agency) Citation2018). This was confirmed in a clinical DDI study, wherein concomitant administration of BIC and FTC/TAF showed no significant PK DDIs, and no dose adjustment was necessary when BIC was administered or coformulated together with FTC/TAF (Zhang et al. Citation2017).
Overall, BIC has favourable resistance, PK, and ADME profiles.
Supplemental Material
Download PDF (279.7 KB)Acknowledgements
The authors thank all the volunteers who participated in the human ADME study. We also thank Peter Pyun for synthesis of BIC metabolite standards. Authors thank Sibylle Wilbert for editorial assistance of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s). All authors are current employees except that BS, HJ, HZ, and JW are former employees.
Additional information
Funding
References
- Birkus G, Bam RA, Willkom M, Frey CR, Tsai L, Stray KM, Yant SR, Cihlar T. 2016. Intracellular activation of tenofovir alafenamide and the effect of viral and host protease inhibitors. Antimicrob Agents Chemother. 60(1):316–322.
- CDER (Center for Drug Evaluation and Research). 2016. Guidance for industry: safety testing of drug metabolites. Silver Spring, MD: U.S. Department of Health and Human Services, Food and Drug Administration. [accessed 2022 Dec 7]. https://www.fda.gov/media/72279/download.
- Cockcroft DW, Gault MH. 1976. Prediction of creatinine clearance from serum creatinine. Nephron. 16(1):31–41.
- EMA (European Medicines Agency). 2018. Public assessment report Biktarvy®. [accessed 2022 Dec 7]. https://www.ema.europa.eu/documents/assessment-report/biktarvy-epar-public-assessment-report_en.pdf. Last.
- FDA (Food and Drug Administration). 2018. Biktarvy® (bictegravir, emtricitabine, and tenofovir alafenamide) tablets, for oral use. [accessed 2022 Dec 10]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210251s000lbl.pdf.
- Hop CE, Wang Z, Chen Q, Kwei G. 1998. Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J Pharm Sci. 87(7):901–903.
- Jin H, Pyun H, Smith BJ, Subramanian R, Wang J, inventors; Gilead Sciences, Inc., assignee. 2019 Jan 18. Metabolites of bictegravir. International Patent WO/2019/144015 Al.
- Santoro MM, Fornabaio C, Malena M, Galli L, Poli A, Menozzi M, Zazzi M, White KL, Castagna A, for the PRESTIGIO Study Group. 2020. Susceptibility to HIV-1 integrase strand transfer inhibitors (INSTIs) in highly treatment-experienced patients who failed an INSTI-based regimen. Int J Antimicrob Agents. 56(1):106027.
- Tiraboschi J, Imaz A, Khoo S, Niubo J, Prieto P, Saumoy M, Penchala SD, Garcia B, Padilla C, Videla S, et al. 2020. Total and unbound bictegravir concentrations and viral suppression in cerebrospinal fluid of human immunodeficiency virus-infected patients (Spanish HIV/AIDS Research Network, PreEC/RIS 56). J Infect Dis. 221(9):1425–1428.
- Tsiang M, Jones GS, Goldsmith J, Mulato A, Hansen D, Kan E, Tsai L, Bam RA, Stepan G, Stray KM, et al. 2016. Antiviral activity of bictegravir (GS-9883), a novel potent HIV-1 integrase strand transfer inhibitor with an improved resistance profile. Antimicrob Agents Chemother. 60(12):7086–7097.
- Wang J, Lazerwith S, Morganelli P, Pyun H, Jin H, Tang J, Matles M, Mwangi J, Wang K, Eisenberg G, et al. 2017. P16. Prediction of bictegravir human pharmacokinetics from protein binding and in vitro-in vivo correlation. Poster Session Presented at: 21st North American ISSX Meeting; Sep 24 − 28; Providence, RI.
- Zhang H, Custodio J, Wei X, Wang H, Wu A, Ling J, Martin H, Quirk E, Elliot C, Kearney B. 2017. Clinical pharmacology of the HIV integrase strand transfer inhibitor bictegravir. Sex Transm Infect. 93(Suppl 1):A74.