
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Evobrutinib is a highly selective, covalent, central nervous system-penetrant Bruton’s tyrosine kinase (BTK) inhibitor, currently in Phase III trials for the treatment of relapsing multiple sclerosis. One major circulating metabolite of evobrutinib has been previously identified as the racemic dihydro-diol M463-2 (MSC2430422) in a Phase I human mass balance study.
Phenotyping experiments were conducted to confirm the metabolic pathway of evobrutinib to M463-2. Ratio of the enantiomers was determined by enantioselective liquid chromatography with tandem mass spectrometry analysis of plasma samples from humans and preclinical species. Drug-drug interaction (DDI) characterisation, evaluation of pharmacological activity on BTK, and off-target screening experiments followed assessing safety of the metabolite.
The biotransformation of evobrutinib to M463-2 was determined to be a two-step process with a CYP-mediated oxidation acting to form an epoxide intermediate, which was further hydrolysed by soluble and mitochondrial epoxide hydrolase. Only the (S)-enantiomer was determined to be a major metabolite, the (R)-enantiomer was minor. In vitro studies demonstrated the (S)-enantiomer lacked clinically relevant pharmacological activity, off-target effects and DDIs.
The biotransformation of evobrutinib to its major metabolite has been elucidated, with the major (S)-enantiomer being shown to pose no on/off target or DDI risks.
Introduction
Bruton’s tyrosine kinase (BTK) is a non-receptor tyrosine kinase that is expressed in multiple immune cells and plays a role in multiple signalling pathways, including that of B cell receptors (López-Herrera et al. Citation2014; Bender et al. Citation2017). BTK inhibitors are a promising therapy in multiple sclerosis (MS), likely acting to limit B cell activation and B and T cell interactions (Cencioni et al. Citation2021; Li et al. Citation2022). BTK inhibitors may also impact MS through other types of cells such as myeloid cells, mast cells, basophils and neutrophils (Carnero Contentti and Correale Citation2022).
Evobrutinib is a highly selective, small-molecule, covalent, central nervous system-penetrant BTK inhibitor currently being investigated in two Phase III trials in patients with relapsing MS (NCT04338022 and NCT04338061). Evobrutinib has high kinase selectivity and low potential for off-target related adverse events due to its binding to a poorly conserved cysteine residue in BTK and interaction with a unique threonine gatekeeper-mediated selectivity pocket (Caldwell et al. Citation2019; Haselmayer et al. Citation2019).
In a Phase I, mass balance and metabolite profiling study involving 6 healthy male participants dosed with 75 mg of 14C-evobrutinib, the structures of 40 Phase I and 18 Phase II metabolites of evobrutinib were identified (Scheible et al. Citation2021). Of these metabolites only one, M463-2 (the diol metabolite MSC2430422), was identified as a major circulating metabolite due to its total plasma exposure being above the 10% of the total drug-related material threshold set by the US Food and Drug Administration and the European Medicines Agency-International Conference on Harmonisation (EMA-ICH Citation2013; FDA Citation2020) guidelines. After reaching Cmax at 1 h, the metabolite was observed in plasma for up to 8 h. M463-2 accounted for 14.9% of the mean plasma exposure (area under the curve [AUC]0–24 h) of total drug-related material and the mean metabolite-to-parent AUC ratio was 202.5%. Biotransformation of evobrutinib to form M463-2 was proposed to occur via an epoxide intermediate through epoxidation and hydrolysis of the acrylamide moiety, as is the case for many xenobiotics in the literature (Bertilsson and Tomson Citation1986; Shah et al. Citation2019). M463-2 was assumed to be a racemic mix of enantiomers, since the structure of the metabolite contains a chiral carbon in what was the acrylamide moiety of the parent molecule, although the ratio of enantiomers in vivo has not been previously investigated.
We carried out this study to understand the mechanism of biotransformation of evobrutinib to M463-2, to identify the ratio of the two enantiomers of the racemic diol metabolite M463-2 in humans and relevant toxicological species (Coelho et al. Citation2021), and to then further evaluate any relevant enantiomer in vitro for potential drug-drug interactions (DDIs), pharmacological activity on BTK inhibition or off-target adverse effects.
Materials and methods
Chemicals
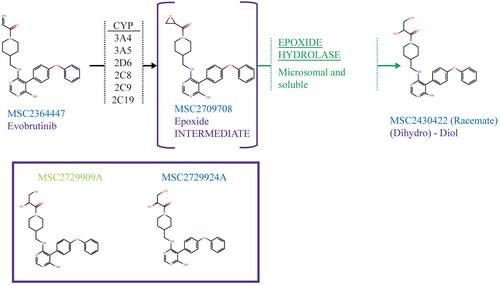
The structure of evobrutinib (MSC2364447 or M2951) can be seen in . M463-2 (the (dihydro)-diol major metabolite of evobrutinib) can also be seen in either as a racemate (MSC2430422) or as (S)- or (R)-enantiomers (MSC2729909A and MSC2729924A, respectively). The epoxide intermediate (MSC2709708A) formed during the proposed two-step metabolism pathway of evobrutinib to the major metabolite can also be seen in .
Figure 1. Proposed 2-step metabolism of evobrutinib to its major metabolite (top) and the two enantiomers of the previously identified major metabolite (bottom). MSC2729909A is the (S)-enantiomer and MSC2729924A the (R)-enantiomer, MSC2430422 the racemate of the major metabolite. CYP2C8, CYP2C9 and CYP2C19 catalysed the formation of the epoxide intermediate in incubations with recombinantly expressed CYP enzymes but were not quantitatively relevant in incubations of suspended hepatocytes.
Standards of the enantiomers were synthesised by Enamine (Ukraine). Evobrutinib and MSC2709708A were synthesised in house at Merck KGaA. Radiolabelled evobrutinib (14C-evobrutinib) was synthesised by Nuvisan GmbH, Grafing, Germany and by Pharmaron UK LTD, Rushden, UK.
Materials
For enzyme phenotyping, human hepatocytes were obtained from BioIVT, USA and modified Krebs-Henseleit, pH 7.4 was used as the assay buffer. For the qualitative assessment of enzyme involvement, E. coli Bactosomes were obtained from Cypex Ltd, UK. Epoxide hydrolase enzymes were obtained from the University of California Davis, USA.
Samples taken from absorption, distribution, metabolism, and excretion (ADME) and toxicology studies in mice were generated using Crl:NMRI(Han) mice from Charles Rivers Laboratories, USA. Samples from rat toxicology studies were generated from Crl:WI(Han) rats obtained from Charles Rivers, Germany. Samples from dog ADME studies were generated using Marshall Beagle® dogs (Marshall Bio Resources, USA).
For mRNA induction experiments, mRNA levels were determined by bDNA-based QuantiGene Plex assay (Thermo Fisher Scientific, USA). UDP-glucuronosyltransferase (UGT) and CYP inhibition potential was evaluated using human liver microsomes supplied by Sekisui XenoTech (USA). P-glycoprotein (P-gp) inhibition data were collected using membrane vesicles prepared from insect Sf9 cells expressing the transporter (baculovirus system; Solvo Biotechnology, Szeged, Hungary, Lot number ME47AD581).
For all off-target screening, excluding hERG channel inhibition, experiments were performed by Eurofins Cerep (France).
For hERG activity experiments the following equipment was used (HEKA Electronics, Germany): computerised system, EPC 11/LAN-43; amplifier, EPC 10 USB; headstage, preamplifier EPC 10; software, PatchMaster Version v2x73.2.
For enantioselective quantification in samples, the following equipment and conditions were used: pump, Agilent 1290 Infinity II High Speed Pump G7120A; multisampler, Agilent 1290 Infinity II Multisampler G7167B with integrated cooler; autosampler temperature, 10 °C; column manager, Agilent 1290 Infinity II MCT G7M6B with integrated column oven; column, Daicel Chiralpak Zwix(−), 250 × 3.00 mm, 3 µm; pre-column/filter, Daicel Porous Glass Guard filter 2.0 mm ID × 2.00 mm L; column oven temperature, 25 °C; detector, Sciex Triple Quadrupole MS-Detector API6500.
Qualitative investigation of the enzymes involved in the formation of the epoxide intermediate MSC2709708A and the diol M463-2
14C-evobrutinib (1 µM) was incubated with E. coli Bactosomes expressing recombinant human CYPs (rhCYPs; 200 pmol/mL) (CYP2C8, 2C9, 2C19, 2D6, 3A4 or 3A5) with an NADPH regenerating system for 2 or 60 min, followed by acetonitrile precipitation. Metabolic profiling was completed using UPLC-QTof with fraction collection and TopCount™ microplate scintillation counting, and radio-chromatograms generated by Laura software (integration limit set at 1.0% ROI).
For quantification of depletion of the epoxide, MSC2709708A (0.1 and 1.0 µM) was incubated with soluble or mitochondrial epoxide hydrolase (sEH or mEH, respectively; 1–50 µg/mL) or buffer for 10 min. Concentrations of the epoxide (MSC2709708A) and the diol (MSC2430422) were determined by LC-MS/MS peak area ratio relative to internal standard (IS) and values were expressed as a percentage of concentration at time 0 or as mean analyte/IS area ratios, respectively.
To determine whether mEH or sEH are capable of catalysing the formation of the diol metabolite from the epoxide intermediate, 1, 5, 10 and 50 µg/mL recombinant enzyme were incubated with 1 µM of the epoxide for up to 10 min. Negative control incubations contained only buffer at pH 9.0 for mEH and buffer at pH 7.5 for sEH. Mixtures of 1 µM epoxide and 0, 1, 5, 10 and 50 µg/mL heat inactivated recombinant mEH (3 h at 65 °C) were used as timepoint t = 0. The buffer for enzyme incubations consisted of 100 mM Tris HCl at pH 9.0 for mEH and at pH 7.5 for sEH and 100 µg/mL fatty acid free bovine serum albumin. The compound was dissolved in dimethyl sulfoxide (DMSO) and further diluted with buffer. Final DMSO concentrations were below 0.05%. Incubations were performed in triplicate at 37 °C and stopped after 2, 4, 6, 8 and 10 min by adding equal volumes of ice-cold acetonitrile including the IS. Samples were centrifuged and supernatant was combined with 4 times the volume of acetonitrile/water (15:85%).
Enzyme phenotyping in human hepatocytes to identify metabolic pathways
Evobrutinib (0.5 µM) was incubated with human hepatocyte suspensions from 50 donors at 0.5 × 106 cells/mL for 2 to 120 min with or without various inhibitors with evobrutinib loss detected by UPLC-MS/MS using a previously qualified method including a calibration curve consisting of 7 concentrations. In brief, hepatocytes frozen in liquid nitrogen were thawed, resuspended in assay buffer, dispensed in 96-well plates, preincubated at 37 °C for 30 min, with the reaction started by the addition of substrate and stopped by the addition of ice-cold methanol. Inhibitors used: CYP2C19: (-)-N-3-benzylphenobarbital (1.0 µM), CYP2D6, quinidine (2.0 µM); CYP3A, troleandomycin (10 µM); pan-CYP, Aminobenzotriazole (1000 µM) + tienilic acid (10 µM); aldehyde oxidase (AO), hydralazine (50 µM). Positive controls: CYP2C19: S-mephenytoin (20 µM), CYP2D6, dextromethorphan (7.5 µM); CYP3A, midazolam (4 µM); AO, zaleplon (20 µM). Individual fm CYP values are based on in vitro intrinsic clearance values with or without specific inhibitors corrected for cross-inhibition according to Njuguna et al. (Citation2016) and normalised to overall CYP fm determined using a pan-CYP inhibitor cocktail. fm,AO was calculated by fm,Pan-CYP+AO – fm,Pan-CYP.
Absorption, metabolism and excretion studies in humans, mice and dogs
Human mass balance, metabolite profiling, and identification data after an oral administration of 14C-evobrutinib were previously published from a Phase I human mass balance study (Scheible et al. Citation2021). In brief, 6 healthy male participants (eligibility: age, 18–55 years; body weight, 50–120 kg; body mass index, 19.0–30.0 kg/m2) were given a single oral 75 mg dose of 14C-evobrutinib (∼3.6 MBq (∼100 μCi)) as a 30 mL solution. Whole blood, plasma, urine and faeces samples were taken and analysed by liquid scintillation counting (LSC). Radioactivity was determined by LSC using a Tri-Carb counter (Tri-Carb 2300 TR and Tri-Carb 2910, TR PerkinElmer, Rodgau, Germany) equipped for computing quench-corrected dpm values using the Transformed Spectral Index of the External Standard Spectrum (tSIE) method, with a tSIE cut-off of 100. Background values were subtracted from each measurement.
In a mouse ADME study, mice aged 8 weeks were given a single oral dose of 500 mg/kg 14C-evobrutinib by gastric intubation with a nominal radioactive dose of 10 MBq/kg and a nominal specific activity of 0.02 MBq/mg. The radioactivity in plasma, urine, cage wash and chamber wash samples were directly analysed by LSC as above, whereas radioactivity in faeces and blood samples was determined after dissolving using a tissue solubiliser (Solvable™, PerkinElmer, USA) followed by LSC.
Dog ADME data were generated using female Marshall Beagle® dogs aged 2 years at the time of dosing. Dogs were given a single oral dose 80 mg/kg 14C-evobrutinib by oral gavage with a radioactive dose of 4.2 MBq/kg specific activity of 0.05 MBq/mg and administration volume of 2 mL/kg body weight. The radioactivity was determined as stated above for mice. More experimental details for the mouse and dog ADME studies are displayed in supplementary methods.
Enantiomer quantification in human, dog, mouse and rat plasma
Samples from the human ADME study and the dog ADME study that were conducted with 14C-evobrutinib were used for quantification of the diol enantiomers. Samples from mouse and rat toxicology studies conducted with unlabelled evobrutinib were also used for the quantification of the diol enantiomers. Plasma samples were stored at −80 °C −15/+10 °C.
A qualified and later validated enantioselective LC-MS/MS method was established for human plasma, and a qualified enantioselective LC-MS/MS was established for dog, mouse, and rat plasma to determine concentrations of enantiomers MSC2729909A and MSC2729924A ().
The potential of interconversion between the two metabolites stereoisomers was tested during the development of the bioanalytical method for enantiomer quantification. No relevant amounts pointing to an interconversion were detected in this experiment. Additionally, stability experiments were performed on samples containing both enantiomers. There was no sign for interconversion and the spiked ratios were always stable.
50 µL (human) or 10 µL (animal) of plasma samples were aliquoted in 96-well PCR plates, followed by 220 µL (human) or 150 µL (animal) of stable-labeled internal standard MSC2430422 in methanol. After shaking and centrifugation, the supernatant was transferred and directly injected (10 µL for human, 5.0 µL for animals). An isocratic LC method on a 1290 Infinity II (Agilent, Germany) with 0.1% formic acid + 25 mM ammonium formate in methanol:water (98 + 2, v/v) was run at a flow rate of 500 µL/min for 10 min using a chiral column Chiralpak Zwix(-), 250 × 3.00 mm, 3 μm (Daicel Chiral Technologies, Japan), giving retention times of approximately 6.8 and 5.6 min for MSC2729909A and MSC2729924A respectively. MS detection was performed on a Triple Quadrupole MS-Detector API6500 (Sciex, US) in positive mode with a mass transition of 464.3–434.2 Da (for human) and to 279.1 (for animals) for both enantiomers.
In vitro DDI assessment of MSC2729909A
The ability of MSC2729909A at different concentrations (0.1–115 µM) to directly inhibit CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4/5 was measured in incubations of human liver microsomes. Experiments were performed using a low microsomal protein concentration (≤0.1 mg/mL) and a short incubation time of 5 min in order to minimise binding to microsomes.
The following substrates were used at concentrations equivalent to their Km to assess CYP activity: CYP1A2, Phenacetin O-dealkylation; CYP2B6, Efavirenz 8-hydroxylation; CYP2C8, Amodiaquine N-dealkylation; CYP2C9, Diclofenac 4′-hydroxylation; CYP2C19, S-Mephenytoin 4′-hydroxylation; CYP2D6, Dextromethorphan O-demethylation; CYP3A4/5, Midazolam 1′-hydroxylation; CYP3A4/5, Nifedipine oxidation; CYP3A4/5, Testosterone 6β-hydroxylation. To examine its ability to act as a metabolism-dependent inhibitor, MSC2729909A was preincubated at 37 ± 2 °C with human liver microsomes and an NADPH-regenerating system for approximately 30 min. This preincubation allowed for the generation of intermediates that could inhibit human CYP enzymes. To examine its ability to act as a time-dependent inhibitor, MSC2729909A was preincubated for 30 min in the presence of pooled human liver microsomes but in the absence of NADPH. This preincubation allowed assessment of whether any potential increase in inhibition was dependent upon NADPH.
Induction by MSC2729909A was studied for CYP1A2, CYP2B6 and CYP3A4. The mRNA expression in human hepatocytes were evaluated. Cryopreserved hepatocytes from 3 human donors were incubated at 37 °C in a humidified atmosphere containing 5% (v/v) CO2 and treated with 0.1–100 µM evobrutinib once daily for 2 consecutive days. mRNA levels were determined 48 h after first treatment. Cytotoxicity and exposure to MSC2729909A was monitored over the incubation time and assessed as sufficient.
UDP-glucuronosyltransferase (UGT) inhibition potential was evaluated using human liver microsomes (pool of 200, mixed gender). MSC2729909A 0.1–100 µM was incubated with the pools of liver microsomes and appropriate substrates at 37 °C, pH 7.7 in 96-well plates. After 5–10 min, reactions were stopped by the addition of acetonitrile, plate spun, and the supernatant analysed by LC-MS/MS for the substrate. The following reactions were monitored for inhibition by MSC2729909A: UGT1A1, 17β-Estradiol 3-O-glucuronidation; UGT1A3, Chenodeoxycholic acid 24-O-glucuronidation; UGT1A4, Trifluoperazine N-glucuronidation; UGT1A6, 1-Naphthol O-glucuronidation; UGT1A9, Propofol O-glucuronidation; UGT2B7, Morphine 3-O-glucuronidation; UGT2B15, S-Oxazepam O-glucuronidation; UGT2B17, Testosterone 17-O-glucuronidation. Inhibition at various concentrations was completed. Troglitazone, a known direct inhibitor of UGT enzyme activity was included as a positive control in each assay.
P-gp inhibition data were collected using membrane vesicles prepared from insect Sf9 cells expressing the transporter. To evaluate MSC2729909A, as an inhibitor of human P-gp, the accumulation of the P-gp substrate N-methylquinidine (NMQ) at 0.5 µM in human P-gp expressing inside-out vesicles was measured in the presence of multiple concentrations of MSC2729909A (0.04, 0.13, 0.39, 1.25, 3.68, 12.8, 72.4 and 250 µM, actual values) for the purpose of determining inhibition. Known reference inhibitors of human P-gp (verapamil, diltiazem, and valspodar) were included as positive controls. The accumulation of NMQ in the vesicles was measured by LC-MS/MS.
Breast Cancer Resistance Protein (BCRP) inhibition data were collected using colon adenocarcinoma (Caco-2)/TC7 cell monolayers cultured in Transwell™ plates for 8 days. 3H-2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (3H-PhIP) was used as the substrate to assess BCRP uptake. The inhibition by MSC2729909A was investigated using a concentration range from 0.5–150 µM and an incubation time of 2 h. The amount of probe substrate was measured by LSC.
Hepatic uptake transporters human organic anion transporting polypeptide (hOATP) 1B1 and 1B3, human organic cation transporter (hOCT) 1 and 2, human organic anion transporters (hOAT) 1 and 3 and renal efflux transporters human multidrug and toxin extrusion transporter (hMATE) 1 and 2-K inhibition data were collected using stably transfected human embryonic kidney (HEK)-cell lines. The uptake of the following radiolabelled compounds was used to assess uptake transporter activity: hOATP1B1, 3H-estrone sulphate; hOATP1B3, 3H-sulfobromophthalein; hOCT1, 3H-1-methyl-4-phenylpyridinium iodide; hOAT1, 3H-p-aminohippuric acid; hOAT3, 3H-estrone sulphate; hOCT2, 3H-1-methyl-4-phenylpyridinium iodide; and hMATEs, 14C-1,1 dimethylbiguanide hydrochloride (14C-metformin). The inhibition by MSC2729909A was investigated using a concentration range from 0.1 to 200 µM. The amount of probe substrate was measured by LSC.
To determine half maximal inhibitory concentration (IC50) values in all cases above, the Levenberg-Marquardt algorithm was used to perform non-linear regression fitting of the data to the 4-parameter sigmoidal-logistic IC50 equation:
DDI risk via direct inhibition was assessed for CYPs (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4/5) and UGTs (1A1, 1A3, 1A4, 1A6, 1A9, 2B7, 2B15, 2B17) using the following equation:
where the maximum unbound plasma concentration (Imax,ss,u) is equal 0.063 µM as calculated from the human mass balance study (146 ng/mL), and Ki was calculated as half of the IC50 values (Cheng and Prusoff Citation1973).
DDI risk via induction was assessed for CYP1A2, CYP2B6 and CYP3A4 using the following equation:
where Emax is the maximal inhibition, EC50 is the concentration resulting half Emax and d is equal to a scaling factor of 1 (ICH Citation2022). For donors, where for the studied concentration range, Emax and EC50 values were not estimated, the mRNA fold change method was used (ICH Citation2022) i.e. DDI liability is excluded if the fold-change of mRNA i) is <2-fold compared to the vehicle control, and ii) <20% compared to the positive control up to the expected hepatic concentration of 15·Cmax,u (∼1 µM).
DDI risk of MSC2729909A as the perpetrator was evaluated for transporters BCRP, P-gp; hMATE1, hMATE2K, hOCT2, hOAT1, and hOAT3 using appropriate cut-offs for Imax,u/IC50 (ICH Citation2022).
Pharmacological activity of MSC2729909A on B cells and BTK
Inhibition of CD69 expression on B cells after cell stimulation was used as a pharmacodynamic marker for BTK inhibition. Peripheral blood mononuclear cells were isolated from healthy volunteers, pre-incubated for 1 h with increasing amounts of compound (0.0001–10 µM), and stimulated by the addition of anti-immunoglobulin M. After culturing overnight, the cells were stained for CD3, CD19, and CD69. The percentage of CD69-positive B cells was measured by flow cytometry and used to calculate the IC50 values for B cell inhibition. Tamatinib was used as a positive control to measure B cell inhibition using a non-BTK related mechanism.
The effect of MSC2729909A (1 µM) on BTK was also evaluated in a standard assay format (Reaction Biology Corp., USA).
Off-target screening of MSC2729909A
MSC2729909A was studied in a wide-range off-target binding and enzyme assay settings (Eurofins Cerep, France) as outlined in Table S1. For binding assays, compound binding was calculated as a % inhibition of the binding of a radiolabelled cognate ligand for a receptor, ion channel transporter or other enzyme. For enzyme and uptake assays, compound inhibitory/stimulatory effect was calculated as a percentage change of activity by measuring substrate depletion/product formation/tracer uptake. MSC2729909A was tested at 10 µM in all cases. An inhibitory or stimulatory effect of ≥50% (corresponding to an IC50 or EC50 of ≤10 µM, respectively) was set as a cut-off point to initiate further investigation. The validity of assays were tested with positive controls of known affinity/potency. Values are calculated as the mean value of two experiments.
In addition to the effect of MSC2729909A on BTK, the effect of MSC2729909A (1 µM) on 370 other different protein kinases was evaluated in a standard assay format (Reaction Biology Corp., USA).
hERG inactivating tail currents were recorded from stably transfected CHO cells (hERG DUO) and the effect of four test item concentrations (2 μM, 6 μM, 20 μM and 60 μM) of MSC2729909A on these currents were investigated at near physiological temperature (36 °C ± 1 °C) with three replicates.
Ethics
All studies involving humans and animals were performed after obtaining appropriate ethical or animal welfare approvals. All in vivo animal studies were performed at Nuvisan GmbH (Grafing, Germany), which has full accreditation by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Human studies were conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice Guidelines. Written informed consent was obtained from each patient before any trial-related activities were performed.
Results
Phenotyping experiments to identify metabolic pathways
After the identification of the major metabolite of evobrutinib, M463-2, it was hypothesised that this metabolite was formed via a 2-step process involving the formation of an epoxide intermediate (Scheible et al. Citation2021). Following the incubation of 14C-evobrutinib with rhCYPs, no M463-2 was formed by the rhCYPs, however, an epoxide intermediate of evobrutinib MSC2709708A, was detected for all CYP isoforms tested (2C8, 2C9, 2C19, 2D6, 3A4, 3A5) (). This confirmation of the epoxide formation adds confidence to the previously stated hypothesis of the metabolic route for the formation of M463-2, as has been seen previously described in the literature (Shah et al. Citation2019).
Table 1. Formation of selected metabolites of 14C-evobrutinib incubated with rhCYPs expressed in E. coli Bactosomes.
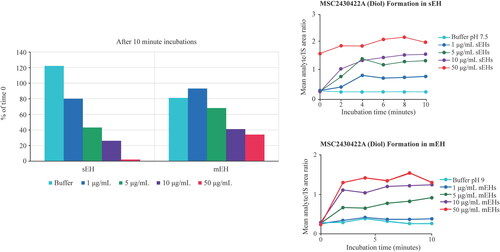
The subsequent conversion of the epoxide MSC2709708A to M463-2 requires a hydrolytic step from the epoxide moiety to form the diol product (). Exploratory experiments suggested that both sEH and mEH were capable of metabolising the epoxide intermediate to form the diol (). In particular, sEH depleted >95% of MSC2709708A after 10 min at a concentration of 50 µg/mL enzyme. Further experiments would be required to fully confirm the contributions and understand the kinetics of the conversion. Non-enzymatic hydrolysis was ruled out, as M463-2 was not formed by incubation of the epoxide in the buffer.
Figure 2. Exploratory experiments looking at M445-10 (MSC2709708A) depletion and diol formation by sEH and mEH. IS: internal standard; mEH: mitochondrial epoxide hydrolase; sEH: soluble epoxide hydrolase. 1–50 µg/mL of recombinant enzyme were incubated with 1µM of the epoxide for up to 10 minutes. Negative control incubations contained only buffer at pH 9.0 for mEH and buffer at pH 7.5 for sEH. Concentrations of the epoxide (MSC2709708) and the diol (MSC2430422) were determined by LC-MS/MS peak area ratio relative to IS. Left, the values for the epoxide were expressed as a percentage of concentration at timepoint t = 0. Right, the values for diol formation were expressed as mean analyte over IS area ratios.
Further experiments in human hepatocytes were conducted to assess the quantitative contribution of individual pathways to the overall metabolism of evobrutinib. In pre-experiments in recombinant enzymes and hepatocytes significant contribution of several CYPs (including 1A2, 2A6, 2B6, 2C8, 2C9) to the metabolism of evobrutinib could be excluded (data on file), therefore a specific set of targeted enzyme inhibitors was used. Incubation of evobrutinib plus targeted enzyme inhibitors, and normalisation to overall CYP contribution to metabolism as well as correction for cross-inhibition, revealed the hepatic fractions metabolised (fm) (). The overall CYP fm after correction for cross-inhibition and normalisation was 0.82, and therefore the non-CYP fm was calculated to be 0.18. Of the CYP contribution, the major contribution was from CYP3A (fm = 0.62), including enzymes CYP3A4 and CYP3A5, followed by CYP2D6 (fm = 0.20). Of the non-CYP fraction metabolised, AO was determined to have a very small contribution with overall AO fm of 0.05.
Table 2. Phenotyping of evobrutinib hepatic metabolism by inhibition of CYP isoenzymes and AO in cryopreserved human hepatocytes.
Mouse, dog and human ADME studies
Human ADME data from a Phase I trial where 6 healthy males were given a single 75 mg dose of evobrutinib have been previously reported (Scheible et al. Citation2021). Key results from this study are summarised in along with data from experiments in mice (n = 46, female) and dogs (n = 6, female) ADME studies. In humans, the dose was chosen to be in the anticipated therapeutic dose range; in mice and dog, the dose was based on the assessment in the 4-week toxicological studies and within the respective No-Observed-Adverse-Effect Level (NOAEL) established. The majority of the administered drug-related material was recovered in excreta (88.8–91.6%), with faeces being the main contributor in all cases. In humans, no unchanged evobrutinib was detected in excreta, however, in mice and dogs unchanged evobrutinib was detected in faeces (27.5% and 66.7% of total radioactivity, respectively) as well as a minor component in dog urine (0.0067% of total radioactivity). The presence of evobrutinib in the faeces of mice is mainly due to non-absorption of the parent compound as can be observed in the difference of evobrutinib concentrations in the faeces of mice who received oral versus intravenous routes of administration (data on file). Similarly, this explanation fits for the dog data as well based on intravenous and oral pharmacokinetic studies in dogs at lower doses, the bioavailability was low (∼12%) (data on file). In human and the two animal species, M463-2 was the major circulating metabolite with AUC0-24h ≥10% AUC total radioactivity (AUCTR) (human, 14.9%; mice, 23.1%; dog, 13.5%) and in all cases AUC0–24 h for M463-2 was approximately 2-fold of the AUC0-24h for evobrutinib. The mean Cmax for evobrutinib versus M463-2 for mice was 8860 ng/mL versus 3980 ng eq/mL, for the dog was 361 versus 72.2 ng/mL, and for human was 321 ng/mL versus 131 ng eq/mL.
Table 3. Summary of ADME data for mouse, dog and human after a single oral dose of 14C-evobrutinib.
Similar to the human situation, evobrutinib was extensively metabolised in mouse and dog to several additional minor metabolites. Metabolism proceeds in all species via the same pathways, including CYP-mediated epoxidation and further hydrolysis to M463-2, further oxidations, oxidative cleavage of either the acrylamide or the acrylamide-piperidine linkage and several conjugation reactions.
Enantiomer contributions
The previously identified major metabolite M463-2 contains a single chiral carbon and, therefore, two enantiomers could exist in vivo. The structure of the enantiomers and their respective names can be seen in . The concentrations of the enantiomers were determined in samples derived from human, dog, mouse, and rat using an enantioselective (chiral chromatography) LC-MS/MS method (). In the human mass balance study, the contribution of (S)-enantiomer MSC2729909A to total radioactivity geometric mean AUC0–24 h was approximately 10%, and hence MSC2729909A was classified as a major metabolite. The contribution of (R)-enantiomer MSC2729924A to the total radioactivity AUC0–24 h was below 10% (<3%) and hence this was not classified as a major metabolite. Similarly, for all three preclinical species tested, AUC was higher for MSC2729909A compared with MSC2729924A (4.7-fold higher in dogs [AUC0–8 h, 80 mg/kg dose: 138 versus 29.2 h*ng/mL], 35.1-fold higher in mice [AUC0–24 h, 1500 mg/kg dose: 40,000 versus 1140 h*ng/mL], and 13.5-fold higher in rats [AUC0–24 h 500 mg/kg dose: 1280 versus 94.6 h*ng/mL]). For dogs it should be noted that the determination was performed at a dose level below the NOAEL, therefore the absolute AUC for the metabolite in that species is lower.
Table 4. Enantiomer contributions in samples from human ADME experiments and preclinical ADME and toxicology studies.
MSC2729909A was used instead of racemate MSC2430422 in studies investigating potential DDIs according to recent DDI guidance (FDA Citation2020), and both on- and off-target effects in order to precisely investigate potential additional risks from the relevant enantiomer as a major metabolite of evobrutinib.
DDIs
The IC50 values for MSC2729909A at CYP enzymes obtained relating to direct inhibition (no preincubation), time-dependent inhibition (30 min preincubation without NADPH) and metabolism-dependent inhibition (30 min preincubation with NADPH) were ≥60 µM for all CYP enzymes tested (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4/5) (). The R1 values for direct inhibition of CYP enzymes (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4/5) were below the limit (1.02) specified in ICH M12 draft guidance and hence no DDI risk via hepatic direct inhibition was concluded. Furthermore, MSC2729909A was not a time- or metabolism-dependent inhibitor of any of the tested CYP enzymes.
Table 5. In vitro inhibition of CYP enzymes by MSC2729909A.
The mRNA induction for CYP1A2, CYP2B6, and CYP3A4 in all three donors tested are shown in . For CYP1A2 (donors ACZ, VKB), CYP2B6 (all donors) and CYP3A4 (all donors), at the studied concentration range (0.1–100 µM), Emax and EC50 were not estimated because CYP induction was too low. Therefore, the R3 values were not estimated. For these CYP donors, the mRNA fold-change method was applied. Based on the mRNA fold-change method, no DDI liability for induction was concluded at clinically relevant concentration. Emax and EC50 could only be determined and the R3 value was calculated in a single donor for CYP1A2 (donor HJK, R3 = 0.95): the R3 value was above the liability threshold of 0.8 and therefore a risk for clinically relevant CYP1A2 induction was also excluded.
Table 6. Induction of mRNA expression of CYPs in cultured human hepatocytes from 3 donors after treatment with MSC2729909A.
The R1 values for direct inhibition of UGTs (1A1, 1A3, 1A4, 1A6, 1A9, 2B7, 1B15, 2B17) were below the limit (1.02) specified in ICH M12 draft guidance and hence the DDI liability risk via direct inhibition could be excluded ().
Table 7. In vitro inhibition of UGTs by MSC2729909A.
No DDI risks due to the inhibition of transporters (BCRP, P-gp, hMATE1, hMATE2K, hOAT3) by MSC2729909A were identified (). A minimal (<50%) inhibition of hOAT1 and hOCT2 in the studied concentration range (0.1–200 µM) resulted in no IC50 values for these transporters and, therefore, suggested no strong DDI risk via inhibition. For all the CYP enzymes, UGT enzymes, and various transporters tested, the IC50 values of MSC2729909A were higher than those of the parent compound (data not shown).
Table 8. In vitro inhibition of transporters by MSC2729909A.
DDIs relating to intestinal enzymes or transporters were not investigated as the compound is a metabolite.
Pharmacological activity on B cells and BTK
At the concentrations tested (up to 10 µM), the enantiomer MSC2729909A had no inhibitory effect on CD69 expression on B cells and the percentage of CD69 was comparable to stimulated control. The racemate MSC2430422 showed a slight inhibition at 10 µM with a mean % effect of −16%. Therefore, in both cases the IC50 value must be higher than 10 µM, confirming that the major metabolite at clinical human concentrations does not contribute to on-target pharmacological effect.
In the kinase panel assay, the enantiomer MSC2729909A showed no activity on BTK at a 1 µM concentration. No other kinases in the panel were inhibited by greater than 50%, except for FLT3 and CK1 epsilon (IC50 625 nM and 1725 nM, respectively) (Table S2).
Off-target effects
In addition to potential DDIs, in general it is important to consider and assess potential stimulatory or inhibitory effects of major metabolites on common off-targets in addition to those of the parent compound. MSC2729909A was screened against a panel of known off-targets in a selective binding profiling in vitro assay against 124 cell receptors, enzymes, transporters, and ion channels at a concentration of 10 µM (Table S1). An inhibitory or stimulatory effect of ≥50% (corresponding to an IC50 or EC50 of ≤10 µM, respectively) was set as a cut-off point to initiate further investigation. MSC2729909A did not reach this cut-off point for any target tested and therefore no further investigation was initiated. Similarly, no inhibitory effect was seen for the panel of off-target kinases, tested at 1 µM (Table S2).
In addition, a whole-cell patch-clamp experiment was conducted to assess the risk of MSC2729909A to human Kv11.1 (hERG) ion channel activity in stably transfected CHO cells expressing the hERG gene. Actual concentrations tested were determined to be 2.02, 6.26, 20.5, 60.0 µM. The highest concentration corresponded to a percentage inhibition ± SD of 64.2 ± 5.7% (n = 3). An IC50 value was determined at the high concentration of 31.9 µM, Hill coefficient 0.92, based on nominal concentrations, and 32.4 µM, Hill coefficient 0.94, based on actual concentrations.
Discussion
The previously proposed biotransformation of evobrutinib to its only major metabolite, the diol M463-2, via an epoxide intermediate in a two-step process (Scheible et al. Citation2021) has now been supported with data. It was found that the major metabolite could be more precisely defined as a single enantiomer, MSC2729909A, as only this enantiomer reached the threshold for classification as a major metabolite whereas the other enantiomer, MSC2729924A, only had a minor contribution. MSC2729909A did not appear to show any pharmacological activity on BTK inhibition or risk of in vivo DDI or off-target inhibition effects.
In vitro, evobrutinib is metabolised primarily by CYP3A4/5 in human hepatocytes (fm 0.62), followed by CYP 2D6 (fm 0.20). The first of the two metabolic steps to the major metabolite were shown to be mediated by several CYP isoforms (including CYP3A and 2D6), which form the epoxide intermediate MSC2709708A. Preliminary data suggest both sEH and mEH enzymes were capable of converting the epoxide intermediate to M463-2 in the second step of this metabolism. The epoxide intermediate has not been seen in any in vivo ADME studies nor in in vitro systems with epoxide hydrolase activity (e.g. microsomes, hepatocytes), confirming that the second step is faster than the first step. Previous investigations by Shah et al. (Citation2019) of the metabolism of the urate transporter inhibitor lesinurad have shown lesinurad forms an epoxide intermediate (referred to as M3c) that was efficiently depleted by mEH. The authors concluded that no clinically relevant effects of M3c were anticipated due to this rapid elimination and lack of detectable M3c in the plasma. This conclusion was accepted by the Committee for Medicinal Products for Human Use (Coppola et al. Citation2019).
The previously identified racemic metabolite of evobrutinib, M463-2, contains a single chiral carbon and, therefore, two enantiomers of this metabolite are theoretically present in vivo. Before conducting DDI and off-target studies, it was pertinent to assess the relevant contributions of each enantiomer rather than using a racemic mixture for these experiments that may not be reflective of physiological conditions. By retesting samples from the previously published mass balance study of evobrutinib (Scheible et al. Citation2021) with a specifically developed enantioselective LC-MS/MS method, it was determined that only one enantiomer, MSC2729909A, met the definition of a major metabolite of ≥10% of drug related material plasma exposure. This was corroborated by data from preclinical species showing the presence of MSC2729909A in plasma (), adding validity to only consider MSC2729909A as the major metabolite, and confirming that preclinical toxicology studies provide sufficient toxicology coverage for the major metabolite with >20-fold higher Cmax concentrations and >100-fold higher AUC observed in mice.
With the previously identified major metabolite of evobrutinib now more precisely identified as a single enantiomer rather than a racemic mixture, DDI, on-target and off-target studies were conducted to assess if any safety risks were associated with the metabolite. No notable potency was observed for the metabolite for inhibition of key transporters and enzymes tested commonly associated with DDIs, or inhibition or stimulation of any of the tested common off-target proteins. A Ki value for inhibition of hERG by evobrutinib has been previously reported of 3.1 µM, approximately 10-fold more potent than data obtained in this study for MSC2729909A (32.4 µM) (Caldwell et al. Citation2019). In clinical studies, MSC2729909A showed a lower mean Cmax compared with unchanged evobrutinib of 146 ng/mL versus 334 ng/mL and a lower ratio of AUC0–24 h of the total radioactivity of 10% versus 13.5%. Notably, in human plasma MSC2729909A has approximately 3-fold higher unbound fraction compared with evobrutinib. However, as MSC2729909A did not show any off-target effects up to 10 µM, which is about 1000-fold higher than the maximum unbound plasma human concentration, there would be no clinically meaningful alteration of efficacy or safety of the drug unaccounted for by differences in unbound fraction between metabolite and parent.
The DDI risk assessment for MSC2729909A showed no risk via direct inhibition for all evaluated CYPs (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4/5) and UGTs (1A1, 1A3, 1A4, 1A6, 1A9, 2B7, 2B15, 2B17). The DDI risk assessment excluded any DDI liability via induction for CYP1A2, CYP2B6 and CYP3A4. No DDI risk via inhibition was concluded for BCRP, P-gp, hMATE1, hMATE2-K, and hOAT3 transporter.
It should be noted that the exposure (geometric mean Cmax) used for DDI liabilities evaluation was determined from the human mass balance study. The DDI risk assessment may be confirmed as more clinical data are collected in the development of evobrutinib.
The limitations of this study include the exploratory nature of the experiments investigating the conversion of the epoxide intermediate to M463-2. As the second step of the transformation is more rapid than the first, similar to the case of lesinurad, there is no safety risk. Given that the contribution of CYP3A to the overall metabolism of evobrutinib was much larger than any other enzyme, CYP3A can be considered as the major contributor to the formation of the epoxide intermediate in vivo.
In summary, the biotransformation of evobrutinib to its previously identified major metabolite has been confirmed to be a two-step process: formation of an epoxide intermediate, primarily via CYP3A, followed by rapid hydrolysis to form a diol metabolite by sEH and mEH. The single chiral carbon containing major metabolite, M463-2, was found to exist primarily in one of two enantiomeric forms, MSC2729909A, such that only this enantiomer should be considered a major metabolite. MSC2729909A did not appear to exhibit any in vivo DDI liability or off-target modulation risks in addition to its parent compound. Therefore, no additional safety issues would be expected to be associated with evobrutinib due to its major metabolite.
Supplemental Material
Download PDF (285.5 KB)Acknowledgments
Merck Healthcare KGaA, Darmstadt, Germany was involved in the experimental design, collection, analysis, and interpretation of the data, and the development of this manuscript. Medical writing and editorial support were provided by Jack Lochray of Bioscript Group Ltd, Macclesfield, UK, and supported by Merck Healthcare KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945). The authors would like to thank Karthik Venkatakrishnan for reviewing the manuscript and providing valuable input, Birikiti Kidane for conducting some of the phenotyping experiments, Hammock Lab from University of California Davis for supporting with the epoxide hydrolase enzymes, Ursula Boschert and Yasemin Begum Alankus for the support and monitoring of the kinase profiling study, and Maximillian Kranz and Matthias Bader for supporting with the metabolite profiling and identification studies.
Disclosure statement
H Scheible, H Schieferstein, K Pusecker, U Gradhand, S Gopalakrishnan, K Iqbal and K Georgi are or were employees of Merck Healthcare KGaA, Darmstadt, Germany.
R Schmidt, J Dong, R Jones, J Bolleddula and M Dyroff are or were employees of EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA. C Meli is an employee of Merck Ltd., Piedmont, Italy, an affiliate of Merck KGaA.
Data availability statement
Data are available upon reasonable request. Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck Healthcare KGaA, Darmstadt, Germany’s Data Sharing Policy. All requests should be submitted in writing to Merck Healthcare KGaA, Darmstadt, Germany’s data sharing portal https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. When Merck Healthcare KGaA, Darmstadt, Germany has a co-research, co-development, or co-marketing or co-promotion agreement, or when the product has been out-licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck Healthcare KGaA, Darmstadt, Germany will endeavour to gain agreement to share data in response to requests.
Additional information
Funding
References
- Bender AT, Gardberg A, Pereira A, Johnson T, Wu Y, Grenningloh R, Head J, Morandi F, Haselmayer P, Liu-Bujalski L. 2017. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Mol Pharmacol. 91(3):208–219. doi: 10.1124/mol.116.107037.
- Bertilsson L, Tomson T. 1986. Clinical pharmacokinetics and pharmacological effects of carbamazepine and carbamazepine-10,11-epoxide. An update. Clin Pharmacokinet. 11(3):177–198. doi: 10.2165/00003088-198611030-00001.
- Caldwell RD, Qiu H, Askew BC, Bender AT, Brugger N, Camps M, Dhanabal M, Dutt V, Eichhorn T, Gardberg AS, et al. 2019. Discovery of Evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem. 62(17):7643–7655. doi: 10.1021/acs.jmedchem.9b00794.
- Carnero Contentti E, Correale J. 2022. Current perspectives: evidence to date on BTK inhibitors in the management of multiple sclerosis. Drug Des Devel Ther. 16:3473–3490. doi: 10.2147/DDDT.S348129.
- Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA. 2021. B cells in multiple sclerosis – from targeted depletion to immune reconstitution therapies. Nat Rev Neurol. 17(7):399–414. doi: 10.1038/s41582-021-00498-5.
- Cheng Y, Prusoff WH. 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2.
- Coelho MM, Fernandes C, Remião F, Tiritan ME. 2021. Enantioselectivity in drug pharmacokinetics and toxicity: pharmacological relevance and analytical methods. Molecules. 26(11):3113. doi: 10.3390/molecules26113113.
- Coppola P, Andersson A, Cole S. 2019. The importance of the human mass balance study in regulatory submissions. CPT Pharmacometrics Syst Pharmacol. 8(11):792–804. doi: 10.1002/psp4.12466.
- EMA-ICH. 2013. ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. [accessed 2023 January 24]. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m3r2-non-clinical-safety-studies-conduct-human-clinical-trials-marketing-authorisation_en.pdf.
- FDA. 2020. Safety testing of drug metabolites guidance for industry. [accessed 2023 January 24]. https://www.fda.gov/media/72279/download.
- Haselmayer P, Camps M, Liu-Bujalski L, Nguyen N, Morandi F, Head J, O'Mahony A, Zimmerli SC, Bruns L, Bender AT, et al. 2019. Efficacy and pharmacodynamic modeling of the BTK inhibitor Evobrutinib in autoimmune disease models. J Immunol. 202(10):2888–2906. doi: 10.4049/jimmunol.1800583.
- ICH. 2022. ICH harmonised guideline – drug interaction studies M12. https://database.ich.org/sites/default/files/M12_Step1_draft_Guideline_2022_0524.pdf.
- Li R, Tang H, Burns JC, Hopkins BT, Le Coz C, Zhang B, de Barcelos IP, Romberg N, Goldstein AC, Banwell BL, et al. 2022. BTK inhibition limits B-cell-T-cell interaction through modulation of B-cell metabolism: implications for multiple sclerosis therapy. Acta Neuropathol. 143(4):505–521. doi: 10.1007/s00401-022-02411-w.
- López-Herrera G, Vargas-Hernández A, González-Serrano ME, Berrón-Ruiz L, Rodríguez-Alba JC, Espinosa-Rosales F, Santos-Argumedo L. 2014. Bruton’s tyrosine kinase–an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol. 95(2):243–250. doi: 10.1189/jlb.0513307.
- Njuguna NM, Umehara KI, Huth F, Schiller H, Chibale K, Camenisch G. 2016. Improvement of the chemical inhibition phenotyping assay by cross-reactivity correction. Drug Metab Pers Ther. 31(4):221–228.
- Scheible H, Dyroff M, Seithel-Keuth A, Harrison-Moench E, Mammasse N, Port A, Bachmann A, Dong J, van Lier JJ, Tracewell W, et al. 2021. Evobrutinib, a covalent Bruton’s tyrosine kinase inhibitor: mass balance, elimination route, and metabolism in healthy participants. Clin Transl Sci. 14(6):2420–2430. doi: 10.1111/cts.13108.
- Shah V, Yang C, Shen Z, Kerr BM, Tieu K, Wilson DM, Hall J, Gillen M, Lee CA. 2019. Metabolism and disposition of Lesinurad, a uric acid reabsorption inhibitor, in humans. Xenobiotica. 49(7):811–822. doi: 10.1080/00498254.2018.1504257.