

Abstract
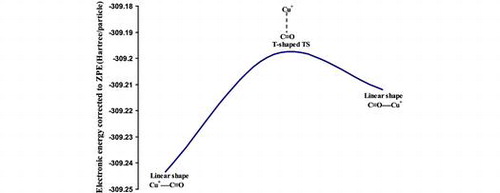
Density functional theory calculations, with an effective core potential for the copper ion, and large polarized basis set functions have been used to construct the potential energy surface of the Cu+·(CO)n (n = 1–3) complexes. A linear configuration is obtained for the global minimum of the Cu+·CO and Cu+·(CO)2 complexes with a bond dissociation energy (BDE) of 35.9 and 40.0 kcal mol-1, respectively. For the Cu+·(CO)3 complex, a trigonal planar geometry is obtained for the global minimum with a BDE of 16.5 kcal mol−1. C-coordinated copper ion complexes exhibit stronger binding energy than O-coordinated complexes as a result of Clp → 4s σ-donation. The computed sequential BDEs of Cu+·(CO)n (n = 1–4) complexes agree well with experimental findings, in which the electrostatic energy and σ-donation play an important role in the observed trend.
Acknowledgement
JND would like to thank the Deanship of the Graduate Studies of the Hashemite University (Jordan) for the financial support.