
Abstract
In order to develop new, low toxicity and readily accessible supramolecular simulants for V-series organophosphorus chemical warfare agents (OP CWAs) a series of organothiophosphate compounds, that are structurally analogous to the V-series OP CWAs, were designed and synthesized. Solution spectroscopic studies (luminescence and UV-vis) into the binding behaviors of these compounds and some organothiophosphate pesticides with the simple model trivalent lanthanide complex [Eu(phen)2(NO3)3]·3H2O were performed and association constants (Kassoc) determined. Binding behaviors of these compounds with other OP CWAs and simulants investigated previously in analogous studies are presented and the implications of this in the context of OP CWA and related chemical sensing are discussed.
1. Introduction
The V-series chemical warfare agents (CWAs) are a sub-class of the traditional organophosphorus (OP) CWAs that are comprised of a characteristic phosphonothiolate (VX) or phosphorothiolate (VG) core (P(O)-S moiety) with an appended tertiary amine () [Citation1]. They are extremely potent and fast acting acetylcholinesterase inhibitors that can result in incapacitation and death within minutes of exposure to milligram quantities [Citation2] and are scheduled chemicals under the Chemical Weapons Convention (CWC) [Citation2]. Due to this, the V-series OP CWAs are handled only in specialist facilities and therefore research, development, and test and evaluation of concepts, systems and instrumentation outside of these controlled environments often relies upon the use of low toxicity simulants (also commonly termed surrogates, analogues or mimics). Selection or design of new simulants is based upon the properties that the proposed technology exploits such as reactivity, supramolecular interactions or molecular structure and also on understanding the correlation between simulant and agent behavior [Citation3]. Many commonly used, simple, simulants for OP CWAs include monodentate dialkyl methylphosphonates, trialkyl phosphates, and dialkyl phosphorohalidates [Citation4]. However, when investigating supramolecular and coordinative interactions (particularly with trivalent lanthanides) these simulants have been shown to be more closely correlated to G-series OP CWA behavior as the V-series possess greater potential for bidentate/multivalent supramolecular interactions [Citation5].
Figure 1. (a) Structure of the V-series chemical warfare agents VX and VG (Amiton) and the V-series supramolecular simulant VO. (b) The core structures of the organothiophosphate compounds of interest where (i) phosphonothiolate, (ii) phosphorothiolate, (iii) phosphorothionate, and (iv) phosphorothiolothionate.
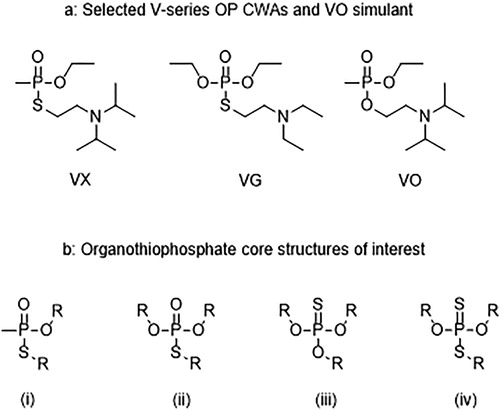
In our previous work we elucidated the mechanisms of trivalent lanthanide luminescence quenching upon exposure to the V-series OP CWAs VX and VG and the V-series simulant VO () [Citation3b, Citation5a]. This was performed by spectroscopically monitored (luminescence and UV-vis) titrations of the V-series agents into solutions of simple model trivalent lanthanide complexes of the type [Ln(phen)2(NO3)3]·xH2O] (where phen = 1,10-phenanthroline, Ln = Eu and x = 3 or Ln = Tb and x = 2). VX and VG were shown to rapidly quench the lanthanide luminescence at low molecular equivalents via a competitive binding process (with some significant dynamic effects) displacing the sensitizing phen ligand and disrupting the antenna effect. This was proposed to occur via the formation of a seven-membered bidentate chelate ring through the P = O and N moieties (Scheme 1). Additional investigations using NMR and IR techniques and related La-based complexes were used to further elucidate the coordination mechanism. In particular, the use of 31P and 1H NMR to monitor changes in P-center and aliphatic-amine resonances in the simulant VO () upon addition of La(NO3)3(H2O)6 were highly indicative of the proposed [OP,N] coordination mode, although quantitative association and kinetic parameters could not be derived [Citation3b].
Scheme 1. General mechanism of the displacement of the 1,10-phenanthroline ligand from complexes of the type Ln(phen)2(NO3)3·xH2O by the addition of ligands capable of forming bidentate [OP,N] chelates, such as VX, VG, and VO. Additional remaining phen and/or second [OP,N] ligands, nitrates and H2O omitted from product species for clarity. X = O, S; x = 1, 2.
![Scheme 1. General mechanism of the displacement of the 1,10-phenanthroline ligand from complexes of the type Ln(phen)2(NO3)3·xH2O by the addition of ligands capable of forming bidentate [OP,N] chelates, such as VX, VG, and VO. Additional remaining phen and/or second [OP,N] ligands, nitrates and H2O omitted from product species for clarity. X = O, S; x = 1, 2.](/cms/asset/e0191a34-972b-468f-ad5b-3e5ec5dc0c0d/gcoo_a_1624729_c0001_b.jpg)
In related work by Martinez-Manez the bidentate coordination ability of V-type ligands was also exploited. In this work, a BODIPY-based ligand system was coordinated to Eu3+ or Au3+ metal-centers via a five-membered chelate ring, which was displaced effectively by the putative V-series simulants Demeton-S via the formation of a [OP, S] seven-membered chelate ring [Citation6]. Both this work and our own series of studies indicate the ability of V-type ligands to coordinate effectively to lanthanides, and that this could be a route to new sensors or, as is addressed here, to the design of new simulants.
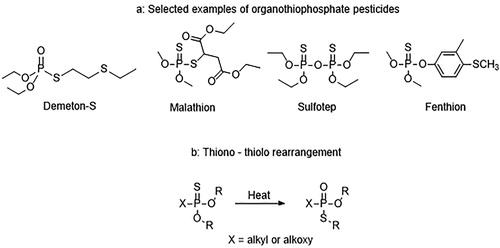
Herein, we extend our own studies of potential simulant candidates to consider organothiophosphate analogues such as the phosphorothionates and phosphorothiolothionates depicted in , which contain a P = S moiety instead of a P = O moiety, and sterically hindered phosphorothiolates (identical phosphorus core to VG). These compounds are closely related to, but generally display significantly lower toxicities, than the V-series CWAs which make them excellent targets for the design of V-series simulants. Detection of P = S containing compounds would also be beneficial as there are numerous examples of toxic pesticides within this class of compounds such as Malathion, Sulfotep, and Fenthion () [Citation7]. Finally, known synthetic routes to V-series OP CWAs can proceed via thionate precursors followed by heat-induced irreversible thiono-thiolo rearrangement to produce the thiolate () [Citation8].
Figure 2. (a) Structures of Demeton-S, Malathion, Sulfotep and Fenthion organophosphorus pesticides. (b) General scheme for the thiono-thiolo rearrangement.
2. Experimental
2.1. Chemicals and instrumentation
Unless otherwise indicated, all reagents and solvents were obtained from commercial suppliers (Sigma-Aldrich, Alfa Aesar, Merck, VWR) and were used without purification. [Eu(phen)2(NO3)3]·3H2O was prepared as previously described [Citation5].
1H, 13C, and 31P NMR spectra were recorded with a Bruker AV400TR FT-NMR spectrometer, using TMS for 1H (400.1 MHz) and 13C (100.6 MHz), and 85% H3PO4 for 31P (161.9 MHz) as external standards. Chemical shifts are expressed in parts per million (δ) using TMS or residual solvent protons as internal standards. Coupling constants (J) are reported in Hz. Splitting patterns are designated as s (singlet), d (doublet), t (triplet), q (quartet), qui (quintet), sex (sextet), sept (septet), oct (octet), and m (multiplet). A Thermo ScientificTM Trace GC 1310 with PTV injector and an Agilent J&W GC-column (CP-Sil 8 CB Low Bleed/MS, 30 m 0.25 µm), Triplus RSHTM auto sampler and TSQ Duo triple quadrupole mass spectrometer was used in single quad mode. ChromeleonTM 7.2 Chromatography Management Software was used for system control and data processing. HPLC-Separations were performed on a modular DIONEX UltiMate™ 3000HPLC System (Thermo ScientificTM) equipped with a SRD-3400 (4-channel degasser) Solvent Racks, HPG-3400SD gradient pump, WPS-3000TSL (Analytical) auto sampler, TCC-3000SD column oven, DAD-3000 Photometer, MSQ-Plus mass detector. An Accucore RP-MS column (3.0 × 150.0 mm, particle size 2.6 µm Thermo ScientificTM Part No. 17626-153030) was used for separation. ChromeleonTM 7.2 Chromatography Management Software was used for system control and data processing. The IR-spectra were recorded with a Spectrum One FT-IR spectrometer from Perkin Elmer, equipped with a Golden Gate ATRTM unit from Specac. The spectra were recorded from 600 to 4000 cm−1. As a rule, before and after each measurement a blank was taken and four spectra were accumulated to give a good average. For IR and Raman data processing, the OMNIC 8 Software from Thermo ScientificTM was used. The Refractive Index measurements were performed on an Abbe-refractometer with a temperature controlled water bath. Luminescence experiments were performed on an Ocean Optics portable USB 4000 fluorimeter with 1000 nm fibre optic cables and a PX-2 pulsed xenon light source [Integration time (υ/s) 100000, Spectra Averaged: 10 - 20, Boxcar smoothing: 0]. UV-vis experiments were performed on a Thermo ScientificTM Evolution 201 spectrophotometer. Absorbance, band width: 2 nm, data interval: 1.00 nm, scan speed: 1200 nm/min, integration time: 0.05, smoothing: low.
2.2. Synthesis of ligands 1–6
The appropriate amino alcohol analogue (1 molar equivalent) was added dropwise to a suspension of sodium hydride (1 molar equivalent) in anhydrous benzene heated under reflux. Upon formation of the amino ethanolate anion the solution became almost transparent. The reaction mixture was cooled on an ice bath and O,O′-diethyl chlorothiophosphate (1 equivalent) was added dropwise. The reaction mixture was allowed to warm to room temperature and was extracted three-times with distilled water at pH 2. Ammonia solution (8% v/v) was added to the aqueous extracts until a pH value of >10 was reached. This solution was washed three times with diethyl ether, the organic layer dried over anhydrous sodium sulfate and the solvent was removed by rotary evaporation. Experimental yield is calculated with respect to the educt used in the smallest quantity.
Full synthesis and characterization details for 1–6 are given in the Supplementary Information.
2.3. Titration experiments
[Eu(phen)2(NO3)3]·3H2O (7.5 mg, 0.01 mol) was dissolved in 1 mL of anhydrous DMF. This solution was made up to 10 mL with anhydrous MeCN. 0.25 mL of this solution was further diluted in anhydrous MeCN (25 mL) to generate a 1 × 10−5 mol dm−3 solution. 0.2, 0.02, and 0.002 mol dm−3 solutions of the analytes were prepared in anhydrous MeCN. 2 mL of the 1 × 10−5 M complex solution was placed into a screw cap fluorescence cuvette with a PTFE septum and a small stir bar and reference fluorescence and UV-vis spectra were obtained for the complex. Additions of the appropriate equivalents of simulant solutions were made via a 10 μL syringe through the septum and the solution was allowed to stir at room temperature for 1 min in-between additions before spectra were recorded.
3. Results and discussion
3.1. Ligand design and synthesis
A series of phosphorothionates (1–4), a phosphorothiolothionate (5) and a sterically hindered phosphorothiolate (6) with appended amine binding sites were designed as potential ligands for complexes of the type [Ln(phen)2(NO3)3·xH2O] (). These putative ligands also incorporated varied substitution at the N- and P-centers in order to further elucidate the effect on coordination properties. In addition to these ligands, the commercially available pesticides Malathion (containing a single P = S group) and Sulfotep (containing two P = S groups) were also selected for incorporation into the study ().
Figure 3. Structures of O,O-diethyl O-[1-dimethylamino)propan-2-yl] phosphorothionate (1), O,O-diethyl O-[2-(dimethylamino)ethyl] phosphorothionate (2), O,O-diethyl O-[2-(pyrrolidyl)ethyl] phosphorothionate (3), O,O-diethyl O-[3-(dimethylamino)propyl] phosphorothionate (4), O,O-diethyl S-[2-(diethylamino)ethyl] phosphorothionothiolate (5) and O,O-diethyl S-[2-(diisopropylamino)ethyl] phosphorothiolate (6).
![Figure 3. Structures of O,O-diethyl O-[1-dimethylamino)propan-2-yl] phosphorothionate (1), O,O-diethyl O-[2-(dimethylamino)ethyl] phosphorothionate (2), O,O-diethyl O-[2-(pyrrolidyl)ethyl] phosphorothionate (3), O,O-diethyl O-[3-(dimethylamino)propyl] phosphorothionate (4), O,O-diethyl S-[2-(diethylamino)ethyl] phosphorothionothiolate (5) and O,O-diethyl S-[2-(diisopropylamino)ethyl] phosphorothiolate (6).](/cms/asset/c64fe06c-64a7-49ce-844f-e0935783ac5b/gcoo_a_1624729_f0003_b.jpg)
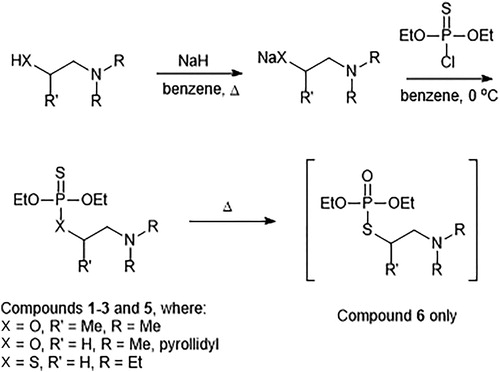
The general reaction pathway for the preparation of the analogues follows the methodologies described by Timperley [Citation8] or Gupalo [Citation9] (Scheme 2). This is the first synthesis of 3 reported in the literature; we reported the synthesis of 4 previously [Citation10–12].
Scheme 2. General methodology for the synthesis of the organothiophosphonates 1–6. Compound 4 is not represented by the scheme shown, but its synthesis follows the same overall strategy.
All products were characterized by 1H, 13C, and 31P NMR and IR spectroscopy, GC-MS, high resolution mass spectrometry and elemental analysis. The preparation and characterization of [Eu(phen)2(NO3)3]·3H2O follows standard procedures, as previously reported [Citation5].
3.2. Spectroscopic studies of complexation behavior
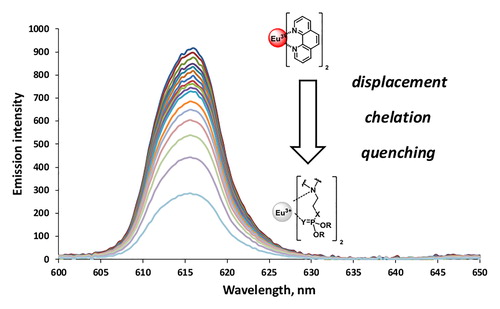
A solution of the lanthanide complex [Eu(phen)2(NO3)3]·3H2O (1 × 10−3 mol dm−3) was prepared in 1:9 DMF:MeCN. This solution was further diluted in MeCN ([complex] = 1 × 10−5 mol dm−3) and the emission and absorbance spectra were recorded. In the luminescence experiments the strong, narrow emission band characteristic for the 5D0-7F2 transition in Eu3+ complexes was observed, centered at ca. 615–617 nm () [Citation13]. Solutions of the synthesized organothiophosphate compounds 1–6 and the pesticides Malathion and Sulfotep in MeCN were titrated incrementally into the prepared solution of the [Eu(phen)2(NO3)3]·3H2O complex ([complex] = 1 × 10−5 mol dm−3) and the emission and absorption spectra recorded after each addition.
Figure 4. Luminescence emission spectra of [Eu(phen)2(NO3)3]·3H2O upon incremental addition of 1 up to 20 molar equivalents, where λem = 615.97 nm; [complex]initial = 1 × 10−5 mol dm−3, MeCN, 293 K.
![Figure 4. Luminescence emission spectra of [Eu(phen)2(NO3)3]·3H2O upon incremental addition of 1 up to 20 molar equivalents, where λem = 615.97 nm; [complex]initial = 1 × 10−5 mol dm−3, MeCN, 293 K.](/cms/asset/64c26f5f-d667-4ce1-945e-0592b4fffa47/gcoo_a_1624729_f0004_c.jpg)
Addition of the synthesized organothiophosphate simulants 1–6 to the solution of [Eu(phen)2(NO3)3]·3H2O resulted in an immediate decrease in the luminescence emission intensity with all analytes ().
The molar equivalents at which complete luminescence quenching was achieved varied between the analogues (see Supplementary Information) indicating that the structural, steric, and electronic differences affected spectral and binding behaviors. In general, the molar equivalents required for complete, or near-complete, luminescence quenching with the organothiophosphate analogues (20–100 equivalents) were significantly higher than those recorded for the V-series CWAs (approximately 7 equivalents) in our previous studies [Citation5].
Stern-Volmer (SV) analysis of the luminescence data was performed in which I0/I (where I0 is the initial emission intensity and I is the intensity in the presence of a known concentration of quenching ligand) was plotted against the concentration of the added ligand (see Supplementary Information) [Citation14]. Analysis at low concentrations indicated a single quenching mechanism, postulated to be static quenching through displacement of the phen ligand as in agreement with previous studies and through UV-vis data (vide infra). At higher concentrations of ligand an upward curvature of the SV plot indicated the presence of a secondary quenching mechanism, postulated to be either excited-state dynamic quenching or quenching through the PET-properties of the amine functionality present in ligands 1–6. Unlike our previous studies, we were unable to satisfactorily separate the KSV and KD components of the quenching behavior, and thus luminescence measurements were unable to furnish quantitative binding affinity data.
Although the luminescence data are qualitatively in agreement with our previous reports and the postulated ligand displacement mechanism, further confirmation of ligand displacement and quantitative analysis of binding affinity was required and therefore analysis of ligand titration behavior using UV-vis spectral measurements was also conducted. In agreement with our previous studies [Citation5], UV-vis titrations resulted in significant changes in the absorption spectra upon addition of the organothiophosphate analogues (1–6). The loss of absorption intensity related to the coordinated phen ligand at 271 nm with a simultaneous absorbance increase and bathochromic shift towards the free phen absorbance at 263 nm was observed over the entire titration range with an isosbestic point observed at 266 nm (). This displacement of coordinated phen with concurrent loss of luminescence was indicative of competitive binding (static quenching) of the organothiophosphate analogues to the Eu3+ ion.
Figure 5. UV–vis absorbance data of [Eu(phen)2(NO3)3]·3H2O upon addition of 1 up to 50 molar equivalents; [complex]initial = 1 × 10−5 mol dm−3, MeCN, 293 K.
![Figure 5. UV–vis absorbance data of [Eu(phen)2(NO3)3]·3H2O upon addition of 1 up to 50 molar equivalents; [complex]initial = 1 × 10−5 mol dm−3, MeCN, 293 K.](/cms/asset/5551775b-a9a2-4586-acbc-cb00209a3156/gcoo_a_1624729_f0005_c.jpg)
Using the parameters determined in previous work [Citation5], the UV-vis titration data of the organothiophosphate analogues (at low molecular equivalents, <2) were fitted to a 1:1 binding mode using the software package HypSpec [Citation15]. The analysis of the UV–vis data displayed no evidence of 1:2 speciation, which indicates that the europium binding sites act independently of one another (no positive or negative cooperativity). This result is consistent with the results seen with VX, VG, and VO [Citation3b, Citation5]. The association constants (log Kassoc) for these compounds are presented in .
Table 1. Association constants for ligands 1–6 with [Eu(phen)2(NO3)3]·3H2O determined by analysis of UV–vis titration data up to 10 molar equivalents ligand in MeCN at 293 K.
Titration experiments using Malathion and Sulfotep did not result in changes to UV-vis spectral data, indicating the lack of ligand displacement through competitive binding. This is in agreement with qualitative luminescence data on which only 11 and 25% emission quenching was observed at 10 molar equivalents of Malathion and Sulfotep, respectively, through dynamic quenching processes.
3.3. Agent-simulant correlations
Evaluation of the current study alongside our previous studies [Citation3b, Citation5] of both CWAs and simulant ligands allows for some comparisons to be made. For example, ligands presenting a single donor group for coordination, such as diethyl chlorophosphate, dimethyl methylphosphonate (DMMP) and the G-series agent GB, do not lead to ligand displacement in the systems studied. Within this study the pesticide Malathion, possessing a single P = S donor group, also failed to displace the phen ligand from the Eu-complex, in agreement with those previous observations. Furthermore, it is suggestive that the two ester moieties in Malathion are not effective in driving competitive coordination in this system, an observation also consistent with previous studies [Citation3b, Citation5].
Clearly, this sets a minimum requirement for bidentate chelation in any ligands proposed as simulants for V-series coordination behavior in systems similar to those studied here and previously. This is also in agreement with the study reported by Martinez-Manez who demonstrated that DMMP, diisopropyl fluorophosphate and diethylcyanophosphate, which all present a single coordination site for Ln3+ binding, were unable to displace five-membered chelate coordinated ligands, whereas Demeton-S was able to displace these ligands via formation of a seven-membered chelate [Citation6]. Our results also suggest that for the bidentate ligands studied to date, those compounds that contain a phosphono-core structure (e.g. VX, VO) demonstrate greater affinities than those containing phosphoro-core structures. This general observation is likely to be a result of electronic effects. For example, in the phosphorothionates (1–5) the replacement of the P = O moiety with a P = S group results in a less polarized P = X bond as a result of the decreased electronegativity of the S atom, thus lowering Lewis basicity. Similarly, when comparing phosphono- and phosphoro-compounds, the additional electronegative substituent (O or S) lowers the electron density, and thus coordinative affinity, of the donor group.
Differences in binding affinity between the analogues studied herein are less apparent using these methods. Any subtle differences are likely to be a result of small modulations in both electronic and steric effects. Overall, the phosphorothionate systems (1–4) appeared to demonstrate slightly lower binding affinities than the phosphorothiolate ligand VG, and this may be a result of the stronger Lewis basicity of P = O groups compared to P = S groups. Surprisingly, the increase in chelate size from seven- to eight-membered when comparing ligands 2 and 4 did not result in a marked decrease in complex stability, as might be expected. This is also at odds with theoretical studies of V-series OP CWA complexation behavior with group I and II metal cations reported by Bandyopadhyay et al. [Citation16], although an increase in the number of ligands studied in our work would be required to fully elucidate this. Similarly, the introduction of additional steric bulk, 1, and steric constraints, 3, when compared to 2 also did not appear to have an effect on binding using the methods presented here. However, it should also be noted that in luminescence titration experiments using 1 the system took longer to stabilize, which could be indicative of a change in the kinetics of the coordination process.
Surprisingly, the pesticide Sulfotep does not competitively bind and displace phen ligands, despite the potential for bidentate P = S coordination and previously observed bidentate coordination with the ligand O,O′-tetraethyl ethylphosphonate (TEEP). The reasons for this are unclear at this stage, but may derive from multiple factors such as the reduced basicity of P = S compared to P = O and a possible tight, unfavorable, bite angle [Citation17].
4. Conclusion
Overall, three main factors were found to determine the binding affinity of OP compounds to trivalent lanthanide ions in all of the ligand displacement assays that we have studied. The most dominant effect is the contrast between mono- and bi-dentate ligands to allow for the formation of [OP, N] or [SP, N] chelates, followed by the electronegativity of the coordinating functionalities and, to a far lesser extent, steric effects that can influence the bidentate complex stability. Knowledge of these factors and the underlying trends can be applied into the design of new simulants for the OP CWAs. Additional experimental and computational approaches may need to be explored in order to further elucidate these factors.
The results from this study demonstrate that organothiophosphate compounds with analogous core structures to the V-series agents (i.e. containing a P = O or P = S moiety with an appended N binding site) have the potential to be utilized as less toxic simulants for V-series CWAs in supramolecular and spectroscopic studies, and in particular in sensing systems based upon ligand displacement and disruption of the antenna effect [Citation18, Citation19]. The most appropriate use of these compounds in this context would be as simulants for VG with its phosphoro-core structure and hence similar binding affinity. Whilst the V-series simulant VO [Citation3b] was shown to be the most appropriate simulant for use with the phosphono-based V-series CWAs such as VX, it should be noted that this simulant does fall under Schedule 2B of the CWC controlled chemicals. In light of this the studied organothiophosphate compounds may offer suitable alternatives for the testing of new V-series OP CWA sensors outside of specialized facilities.
Supplementary data associated with this article can be found in the online version.
Download MS Word (4.6 MB)Acknowledgments
G. H. D. is extremely grateful to Assoc. Prof. Martin Johnston of Flinders University, Australia, for the loan of the PX-2 light source and fibre optic cables for the Ocean Optics system and to the Chemical and Biological Defence Branch (Land Division, DST Group) for funding and support to this work. M. R. S. thanks the Operational Sciences Innovation Fund (CBR Division, Dstl) for funding to support this work. We acknowledge the collaboration between the German Armed Forces (Bundeswehr) and the Ludwig-Maximilian University (LMU) according to the official collaborative agreement between the two institutions, and its beneficial effect in facilitating international collaboration with Australia and the UK. The provision of financial support does not constitute an express or implied endorsement of the results or conclusions presented here by DST Group or the Australian Department of Defence. Content includes material subject to © Crown copyright (2018), Dstl. This material is licensed under the terms of the Open Government Licence except where otherwise stated. To view this licence, visit http://www.nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or E-mail: [email protected].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- (a) K. Kim, O.G. Tsay, D.A. Atwood, D.G. Churchill. Chem. Rev., 111, 5345 (2011). (b) R.M. Black, J.M. Harrison, In The Chemistry of Organophosphorus Compounds, F.R. Hartley (Ed.), Vol. 4, Wiley, New York (1996).
- (a) U. Ivarsson, H. Nilsson, J. Santesson (Eds.), A FOA Briefing Book on Chemical Weapons: Threat, Effects, and Protection, National Defence Research Establishment, Umea (1992). (b) Organisation for the Prohibition of Chemical Weapons (OPCW), https://www.opcw.org/our-work/what-chemical-weapon Convention on the prohibition of the development, production, stockpiling and use of chemical weapons and on their destruction, Annex on Chemicals, OPCW, The Hague, The Netherlands (1997).
- (a) J. Lavoie, S. Srinivasan, R. Nagarajan. J. Hazard. Mater., 194, 85 (2011). (b) G.H. Dennison, C.G. Bochet, C. Curty, J. Ducry, D.J. Nielsen, M.R. Sambrook, A. Zaugg, M.R. Johnston. Eur. J. Inorg. Chem., 1348 (2016). (c) M.R. Sambrook, J.C. Vincent, J.A. Ede, I.A. Gass, P.J. Cragg. RSC Adv., 7, 38069 (2017).
- (a) S.L. Bartelt-Hunt, D.R.U. Knappe, M.A. Barlaz. Crit. Rev. Environ. Sci. Technol., 38, 112 (2008). (b) J.D. Kittle, B.P. Fisher, A.J. Esparaza, A.M. Morey, S.T. Iacono. ACS Omega, 2, 8301 (2017). (c) J. McKenna, E.S. Dhummakupt, T. Connell, P.S. Demond, D.B. Miller, J.M. Nilles, N.E. Manicke, T. Glaros. Analyst, 142, 1442 (2017). (d) C.G.P. Taylor, J.R. Piper, M.D. Ward. Chem. Commun., 52, 6225 (2016).
- (a) G.H. Dennison, M.R. Sambrook, M.R. Johnston. Chem. Commun., 50, 195 (2014). (b) G.H. Dennison, M.R. Sambrook, M.R. Johnston. RSC Adv., 4, 55524 (2014).
- A. Barba-Bon, A.M. Costero, S. Gil, F. Sancenón, R. Martínez-Máñez. Chem. Commun., 50, 13289 (2014).
- S.O. Obare, C. De, W. Guo, T.L. Haywood, T.A. Samuels, C.P. Adams, N.O. Masike, D.H. Murray, G.A. Anderson, K. Campbell, K. Fletcher. Sensors (Basel), 10, 7018 (2010).
- C.M. Timperley, Best Synthetic Methods: Organophosphorus(V) Chemistry, Elsevier Ltd., Oxford (2015).
- A.P. Gupalo, M.I. Khmilevskaya, N.E. Tsepukh. Zhurnal Obschchei Khimii, 49, 93 (1979).
- M.A. Althoff, A. Bertsch, M. Metzulat, O. Kalthoff, K. Karaghiosoff. Phosphorus Sulfur Silicon Relat. Elem., 192, 149 (2016).
- M.A. Althoff, K. Grieger, M.A.C. Härtel, K.L. Karaghiosoff, T.M. Klapötke, M. Metzulat. J. Phys. Chem. A, 121, 2603 (2017).
- M.A. Althoff, A. Bertsch, M. Metzulat, T.M. Klapötke, K.L. Karaghiosoff. Talanta, 174, 295 (2017).
- K. Binnemans. Coord. Chem. Rev., 295, 1 (2015).
- C.D. Geddes. Meas. Sci. Technol., 12, R53 (2001).
- P. Gans, A.A. Sabatini, A. Vacca. Talanta, 43, 1739 (1996).
- I. Bandyopadhyay, M.J. Kim, Y.S. Lee, D.G. Churchill. J. Phys. Chem. A, 110, 3655 (2006).
- B. Coupez, C. Boehme, G. Wipff. Phys. Chem. Chem. Phys., 4, 5716 (2002).
- G.H. Dennison, M.R. Johnston. Chem. Eur. J., 21, 6328 (2015).
- M.L. Aulsebrook, B. Graham, M.R. Grace, K.L. Tuck. Coord. Chem. Rev., 375, 191 (2018).