
Abstract
Room temperature reactions of [(η6-arene)Ru(μ-Cl)Cl]2 (arene = p-cymene) with three new bidentate (SS) organochalcogen ligands, 2a–c (2a: 1,1’-ethylenebis(3-ethyl-imidazole-2-thione), 2b: 1,1’-ethylenebis(3-propyl-imidazole-2-thione), 2c: 1,1’-methylenebis(3-ethyl-imidazole-2-thione)), and in situ anionic exchange with KPF6 afforded the corresponding new 18-electron half-sandwich [(η6-arene)RuLCl]PF6 complexes 3a–c (3a: L = 2a, 3b: L = 2b, 3c: L = 2c). The Ru(II) complexes and the organochalcogen ligands were fully characterized by spectroscopic and analytical techniques. Solid state single crystal X-ray diffraction analysis of 3a–c displayed the Ru(II) center in the expected piano stool geometry. The catalytic activities of the half-sandwich ruthenium complexes toward the transfer hydrogenation of ketones to their corresponding alcohols were investigated using 2-propanol as a hydrogen source and solvent. All three Ru(II) complexes exhibited excellent catalytic activity for the transfer hydrogenation of ketones in the order 3b > 3a > 3c at an appreciably low catalyst loading of 0.5 mol% under a minimal alkaline condition of 10 mol% KOH. The catalytic activities of the complexes are influenced by the length of the alkyl spacer and the steric bulk of the N-substituents on the chelating organochalcogen ligand framework.
1. Introduction
There has been significant and long-standing interest in multidentate ligand systems comprising bis(imidazole-2-thione)borates (BmR) and tris(imidazole-2-thione)borates (TmR) [Citation1, Citation2] (). These anionic scorpionate ligands contain a borohydride bridging unit between the heterocycles and soft sulfur donors. The interest in their coordination compounds with transition and main group metals is due to their potential applications in bioinorganic, coordination, and organometallic chemistry [Citation2–12]. Interestingly, the charge-neutral counterparts of these “soft” scorpionates, namely poly(imidazole-2-thione)methane and poly(imidazole-2-thione)ethane, have received relatively little attention [Citation13]. However, 1,1′-methylenebis(1,3-dihydro-3-methyl-2H-imidazole-2-thione) (Mbit), 1,1′-(1,2-ethanediyl)bis(3-methyl-imidazole-2-thione) (Ebit) () and a few of their metal complexes have been reported [Citation13–16].
Figure 1. Reported scorpionate ligands with soft S-donor groups [Citation13–16].
![Figure 1. Reported scorpionate ligands with soft S-donor groups [Citation13–16].](/cms/asset/8c34c34f-9e46-4b63-9d98-5f7c1b4606e5/gcoo_a_2366275_f0001_b.jpg)
Half-sandwich complexes of group 8 metals with characteristic piano-stool architecture are desired structures in organometallic chemistry and catalysis research [Citation17–22], as they have widespread use in bioinorganic and medicinal chemistry [Citation21–27]. Although the η6-arene ligands, which occupy three coordination sites, stabilize and protect the metal center, changing their electronic and steric properties remains challenging to synthetic organometallic chemists [Citation14, Citation28]. Hence, the addition of soft donor atoms (with large size and polarizable Lewis basic properties) [Citation13], such as sulfur, to the facial site opposite the η6-bonded hydrocarbon ligand serves as a valuable stereocontrol element in the corresponding complexes [Citation14, Citation28, Citation29]. Since many of the 2nd series of the d-block metal ions are also soft Lewis acids, they form stable complexes with imidazole thione ligands [Citation30–32]. Therefore, the synthesis and design of neutral organochalcogen compounds with imidazole-2-thione rings are attractive from organometallic and application points of view, and the ability of these functional groups to bind strongly to late transition metals is of interest. Due to their versatile coordination properties , many transition metal complexes with imidazoline-2-chalcogenone moieties have been synthesized and studied for diverse applications [Citation33–39]. Many complexes of Ru(II), ligated with different organochalcogen ligands, have also been explored for catalyzing the oxidation of alcohols and transfer hydrogenation during the last decade [Citation33]. However, those bearing chalcogene-carbonyl ligands remain relatively rare [Citation40]. Initial catalytic investigations of analogous chelating-N-heterocyclic bis(carbene) complexes of ruthenium in reactions such as hydrosilylation of terminal alkynes, hydrogen transfer between alcohols and ketones, and intramolecular hydroamination were not promising, probably due to the high coordination abilities of the non-labile carbene ligand, which prevents the complex from opening a vacant coordination site needed for catalytic activity [Citation41].
Herein, we describe the preparation of three new neutral organochalcogen ligands (2a–c) and their derivatives with half-sandwich ruthenium precursors. The aim is to explore further neutral organochalcogen coordination chemistry. Previous work has shown the efficiency of analogous ruthenium complexes bearing either pyridine or phenylene-bridged chalcogene-carbonyl ligands as green homogeneous catalysts for both the tandem dehydrogenation of ammonia borane and hydrogenation of R–NO2 to R–NH2 at 353 K in water [Citation38]. Inspired by this work, we explored the catalytic activity of the arene Ru(II) complexes [(η6-arene)RuLCl]PF6 3a–c (3a: L = 1,1′-ethylenebis(3-ethyl-imidazole-2-thione), 3b: L = 1,1′-ethylenebis(3-propyl-imidazole-2-thione), 3c: L = 1,1′-methylenebis(3-ethyl-imidazole-2-thione) at a very low catalyst loading of 0.5 mol% in the transfer hydrogenation of ketones to their corresponding alcohols using 2-propanol as a hydrogen source and solvent.
2. Results and discussion
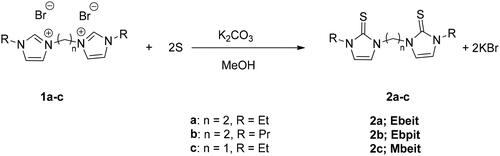
2.1. Synthesis of ligands
Several substituted bis(1-R-imidazole-2-thione)alkanes (where R = Me, tBu, Bn, and iPr) have already been synthesized [Citation15, Citation16, Citation42–45]. However, the related species, i.e. bis(1-R-imidazole-2-thione)ethane, 2a–c (2a: R = Et, Ebeit; 2b: R = Pr, Ebpit) and bis(1-ethane-imidazole-2-thione)methane (2c: Mbeit) presented herein have never been reported to the best of our knowledge. As outlined in Scheme 1, the chelating (SS) imidazole-2-thione ligands were obtained in moderate yields via slight modification of a one-pot literature procedure involving the reaction of respective methanolic solutions of methylene and ethylene-bridged alkylimidazolium dibromide with sulfur powder [Citation16, Citation40]. The modified greener synthetic method described herein has proven to be more cost-effective than the previously reported method involving potassium tertiary butoxide as the auxiliary catalyst/base [Citation16, Citation46, Citation47]. The afforded imidazole-2-thione ligands are obtained as air and moisture-stable solids that are soluble in polar organic solvents like CH2Cl2, CHCl3, and THF. They could be stored at room temperature for several months under ambient conditions. The ligands were characterized by nuclear magnetic resonance (NMR) and Infrared (IR) spectroscopy as well as HR-MS.
Scheme 1. Synthesis of imidazole-2-thione ligands 2a–c.
The formation of 2a (Ebeit) was confirmed by the appearance of the 1H NMR resonance signals at 1.34, 4.06, 4.48, and 6.48 ppm, which correspond to the CH3, CH2, the ethylene linker, and two olefinic protons in the imidazolyl moiety on the ligand framework, respectively. Similarly, the resonance signals at 0.92, 1.77, 3.96, 4.49, and 6.53 ppm in the 1H NMR spectrum of 2b (Ebpit), assigned to the CH3, βCH2, αCH2, the ethylene linker, and olefinic imidazolyl backbone protons, respectively, confirmed its formation. The 1H NMR spectrum of 2c (Mbeit) showed resonance signals at δ 1.35, 4.05, 6.33, 6.62, and 7.64 ppm, which are assigned to the CH3, CH2, the methylene linker, and imidazolyl backbone olefinic protons, respectively. The 13C NMR spectra of Ebeit, Ebpit, and Mbeit confirm the formation of each ligand via resonance signals at δ 161.6, 161.8, and 162.9 ppm, respectively, assigned to the formation of the C = S group.
There has been uncertainty concerning the assignment of the C = S stretching frequency in compounds containing one or two nitrogen atoms [Citation48]. The strong vibrational coupling effect of (CH), (CN), and (CS) makes the assignment range from 850 to 1570 cm−1, which is divided into four thioamide bands [Citation48–52]. The bands of interest are ʋ(C = C)+ʋ(C = N) and thioamide IV ʋs(C = S)+ʋas(C = S). The presence of strong ʋ(C = C)+ʋ(C = N) absorptions at 1643, 1563 cm−1 for Ebeit; 1641, 1564, 1526 cm−1 for Ebpit; and 1681, 1566 cm−1 for Mbeit strongly suggests a variable degree of delocalization of the π-electron density within the heterocyclic thione [Citation49, Citation53]. The thioamide V bands which are due to ʋs(C = S)+ʋas(C = S) appeared at 670, 514 cm−1 for Ebeit; 707, 675, 665, 532 cm−1 for Ebpit and 715, 515 cm−1 for Mbeit. These are characteristically perturbed when the metal coordinates through the thione sulfur [Citation52–54]. The ESI-HRMS spectra of both 2a and 2b (i.e. Ebeit and Ebpit) gave mainly pseudo molecular [M + H]+ ions at m/z values very close to the calculated values.
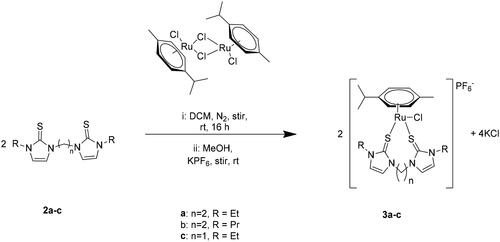
2.2. Synthesis of half-sandwich ruthenium complexes
The reaction of two equivalents of each neutral thione ligand Ebeit, Ebpit, and Mbeit with [Ru(p-cymene)Cl2]2 at ambient temperature in dichloromethane (DCM), and subsequent precipitation of the corresponding complexes with excess aqueous KPF6, afforded brown crystals of the half-sandwich ruthenium complexes formulated as [η6-arene-Ru(L)Cl]PF6 (3a: L = 2a), η6-arene-Ru(L)Cl]PF6 (3b: L = 2b) and η6-arene-Ru(L)Cl]PF6 (3c: L = 2c) (Scheme 2). All three arene ruthenium complexes were air and moisture-stable in the solid state. They also showed excellent solubility in polar aprotic organic solvents like MeCN, DMF, and acetone. The complexes are characterized by a combination of NMR spectroscopy, mass spectrometry, and X-ray crystallography.
Scheme 2. Synthesis of the chelating (SS) half-sandwich η6-arene-Ru(II) complexes with organochalcogen ligands 3a–c.
The 1H NMR spectra of these complexes exhibited resonances at δ 2.82–2.90 and 5.32–6.02 ppm due to the isopropyl protons CH(CH3)3 and aromatic protons η6-C6H4, respectively, on the p-cymene moiety in CD3CN solution. The characteristic resonance signals assigned to the backbone olefinic protons in the imidazolyl moiety of the thione ligands were at δ 7.03–7.43 ppm in the 1H NMR spectra of the complexes. The 1H NMR spectra also showed the CH2–CH2 proton signals of the ethylene bridged complexes 3a and 3b as multiplets. This was attributed to their appearance as either syn or anti to the chloride ligand [Citation29]. The two protons in the bridging methylene backbone of 3c appeared to be diastereotopic [Citation29, Citation56, Citation57]. In contrast, the coupling of these germinal protons is observed in the 1H NMR spectrum at approximately 264.3 Hz, with the protons resonating in the range of 5.83 to 6.49 ppm. The formation of the S-Ru coordination in the complexes was confirmed by the significant upfield shift of the 13C NMR resonance corresponding to the C = S group to the range δ 151.15 to 158.33 ppm in spectra of all the complexes.
An important feature in the infrared spectra of the complexes is the significant perturbation of ʋ(C = C)+ʋ(C = N) and ʋs(C = S)+ʋas(C = S) bands that indicate the thione sulfur coordination to the metal [Citation51, Citation54, Citation55]. The spectra of free ligands consisted of ʋ(C = C) + ʋ(C = N) bands in the 1641 − 1681 cm−1 and 1526 − 1566 cm−1 regions. The peaks in the former region collapsed, while the corresponding peaks in the latter underwent a slight blue shift in the Fourier-transform infrared spectroscopy (FTIR) spectra of the three arene-Ru complexes. This suggested a decrease in the delocalization of π-electrons within the thione heterocyclic, consequently weakening the ʋ(C = N) contribution upon coordination. The complexes ʋs(C = S)+ʋas(C = S) band in the region 554 − 743 cm−1 is the most affected as it has the greatest C = S contribution [Citation52, Citation53]. The appearance of a sharp peak between 824 and 826 cm−1, which is assignable to υ(P-F) [Citation58, Citation59], confirms the presence of the PF6 counter ion in the structure of the complexes. Furthermore, the ESI-MS analyses of all the complexes contained molecular ion peaks [M – PF6]+, confirming their identity, with the observed m/z values within acceptable ranges to calculated values.
2.3. Molecular structures
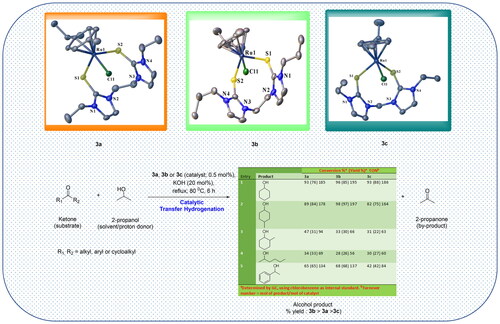
Single crystals of 3a, 3b, and 3c suitable for solid-state X-ray diffraction analysis were grown by slow diffusion of diethyl ether into concentrated acetonitrile solutions of the respective complexes. The crystallographic data for 3a-3c are summarized in , and selected bond lengths and angles are given in . depict the molecular structures for 3a, 3b, and 3c, respectively. All the structures showed eight or nine-membered chelate rings of the ligand framework around the Ru center via coordination through two S atoms. Thus, the soft donor atoms in the three half-sandwich complexes make pseudo-octahedral piano-stool-type configurations around the Ru centers. The arrangement around the metal center in the complexes entails the p-cymene ring occupying three octahedral sites. Further, two sites are occupied by the two S donors of the ligand, and the chloride (chlorido ligand) completes the coordination sphere (). Each compound then has one PF6- counter ion to balance the charge on each complex salt. To our knowledge, these half-sandwich Ru(II) complexes are the first reported of their kind bearing ligands containing alkane-bridged dithione donor groups. The three complexes, 3a, 3b, and 3c, crystallized in the monoclinic system (). Specifically, 3a and 3b form nine-membered macrocyclic rings ( and ), while 3c forms an eight-membered macrocyclic ring.
Figure 2. ORTEP plot showing the chelating SS^Ru(II) complex 3a with ellipsoids drawn at the 50% probability level. Hydrogen atoms and PF6- ions are omitted for clarity.
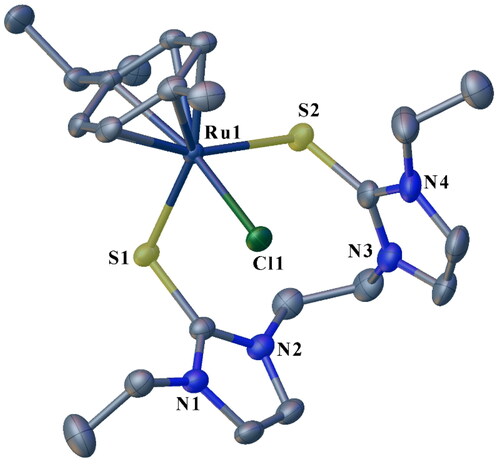
Figure 3. ORTEP plot of 3b showing the chelating SS ligand with ellipsoids drawn at the 50% probability level. Hydrogen atoms and PF6- ions are omitted for clarity.
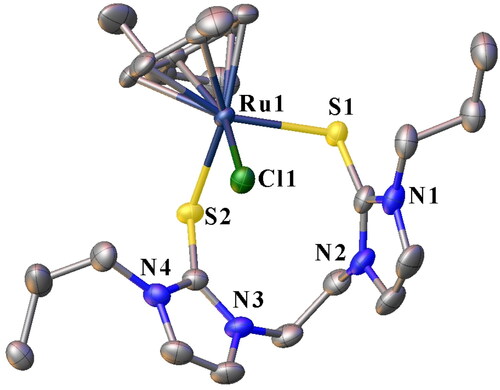
Figure 4. ORTEP plot of 3c showing the chelating SS ligand with ellipsoids drawn at the 50% probability level. Hydrogen atoms and PF6− ions are omitted for clarity.
Table 1. Crystallographic data and structure refinement parameters for 3a–c.
Table 2. Selected bond distances (Å) and angles (°) for 3a, 3b, and 3c.
The S(1)–Ru(1)–S(2) angle of 92.83(2)° recorded for 3a is slightly larger than the corresponding 91.02(2)° observed in 3b (). Comparatively, the two angles are larger than the measured 89.31(3)° in 3c. This is expected and is attributed to the larger macrocycle repulsion with an increase in ring size [Citation29]. The S(1)–Ru(1)–S(2) angles in the three complexes were lower than the 85.56(3)° reported for an analogous half-sandwich ruthenium complex bearing a tridentate pyridine-bridged thione ligand, [Ru(p-cymene)(Bmtp)]PF6 [Citation38]. The measured Ru − Cl bond lengths are similar for 3a, 3b and 3c i.e. 2.407(4) Å, 2.403(6) Å and 2.401(7) Å, respectively. The range for the bond lengths is shorter than those previously reported for similar complexes [Citation40]. The two sets of C-S bond lengths in each of the complexes, i.e. 1.720(2)–1.719(2) Å for 3a, 1.720(2)–1.719(2) Å for 3b and 1.718(3)–1.716(3) Å () for 3c are longer than the ranges of 1.692(4)–1.697(3) Å and 1.702(10)–1.713(6) Å reported for [Ru(p-cymene)(Bmtp)]PF6 [Citation38], and arene ruthenium(II) complexes with unsymmetrical bidentate thioether and thione ligands [Citation40], respectively. Comparatively, these C-S bond lengths are longer than a typical C = S bond [Citation16]. The lengthening of the C-S bonds upon Ru coordination is consistent with metal binding. It is attributed to the back donation from the metal and the increase in the contribution from the zwitterionic resonance structure of the thione ligand [Citation16].
The Ru-S bond lengths in the complexes range from 2.401(5) to 2.473(5) Å (), which is not significantly different from the literature values [Citation32, Citation38, Citation40, Citation60, Citation61]. The average Ru-S bond length of 2.456 Å for 3b (with propyl wingtips) is longer than the average bond length of 2.437 Å recorded for 3a and that of 2.440 Å in 3c, both of which have ethyl wingtips.
2.4. Catalytic transfer hydrogenation of ketone
2.4.1. Optimization reactions
Since ruthenium complexes are among those commonly used as catalysts for the transfer hydrogenation reaction [Citation21, Citation40, Citation62], it is natural to test the half-sandwich ruthenium complexes 3a–c for transfer hydrogenation (TH) of cyclohexanone to cyclohexanol. Complex 3c was used for optimization reactions; the results are presented in . The use of a base was observed to impact the catalytic performance significantly. Strong inorganic bases like NaOH and KOH significantly increased both the conversion and yield compared to the organic base Et3N (; entries 3, 5, and 6). Because KOH gave the highest yield over other inorganic bases (; entry 3), it was then used as the base in the subsequent reactions. A set of consecutive trials was performed to optimize the amount of the KOH. A base loading of 5 mol% resulted in lower conversion and yield (, entry 2). In contrast, a base loading of 20 mole% did not significantly improve the conversion of cyclohexanone to cyclohexanol (, entry 4). Therefore, a base loading of 10 mole% was chosen for subsequent reactions ( entries 2–4).
Efforts to lower the initial catalyst concentration to 0.25 mol% for a higher turnover number (TON) were successful but gave lower conversion (; entry 7). Subsequent efforts to achieve greater conversion by increasing the catalyst concentration to twice the initial 0.5 mol% of 2c resulted in no significant increase in the conversion and yield of the reduced product (; entries 3 and 8). Hence, the optimum catalyst concentration was set at 0.5 mol%. Complex 3c gave the highest cyclohexanol yield in 6 h (, entry 3), as shorter reaction times gave lower conversion and yield (, entry 10). Similarly, longer times showed no significant increase in conversion and yield (, entry 11). Lower yields were obtained when the reactions were performed without a base (, entry 1), or without the ruthenium catalyst (, entry 9), or at room temperature (, entry 12). This is expected as the presence of the base is necessary for supporting active reaction intermediates in the catalytic transfer hydrogenation reactions. 2-Propanol was chosen as the hydrogen donor because it is readily available, inexpensive, and simple to handle [Citation62]. It has also advantage of maintaining the optimum acidic pH in the base-promoted hydroxide ion transfer reduction [Citation63]. Thus, the optimal reaction conditions for the transfer hydrogenation of cyclohexanone are 0.5 mol% of the catalyst, alongside 10 mol% KOH as a base (auxiliary catalyst) in refluxing 2-propanol for 6 h.
Table 3. Optimization of reaction conditions for the transfer hydrogenation of cyclohexanone catalyzed by 3c.
With the derived optimized conditions, the transfer hydrogenation reactions of a range of ketone substrates are carried out using 3a–c as catalysts to understand the scope and generality of this catalytic system. The ketones were chosen to investigate the effects of electronic and steric variations on the substrate backbone, and the results are presented in . The findings showed that the length of the alkyl spacer and N-substituents on the ligand framework significantly affect the catalytic activity of 3a-c towards the transfer hydrogenation of ketones. Complexes 3a and 3b, bearing the ethylene spacer, were more active than 3c, which bears a methylene spacer. This is attributed to the ligand frameworks increased flexibility with the increased spacer length, which consequently changes to a more favorable bite angle during the catalytic cycle [Citation64]. Furthermore, changing the N-substituent from ethyl in 3a to propyl in 3b made it the most active, even though both have the same ethylene spacer in the ligand framework. N-propyl-substituted ligands have a stronger steric and electronic effect on the thione bond and thus influence the loss of the chloride from the coordination sphere to form the catalytic intermediate. This could be because of the electron-donating nature of the propyl group, which affects the Lewis acidity of the ruthenium center, making the formation of the hydride intermediate easier [Citation62]. It was further observed in a related study that the activity and selectivity of complexes increased with the steric demands of the wingtips [Citation65].
Table 4. Transfer hydrogenation of some ketones catalyzed by 3a-c.
In general, cyclic aliphatic ketones gave higher conversion than the acyclic example (). This can be attributed to steric hindrance in accessing the C = O bond [Citation66]. Changing the location of the substituent from the 4 to the 2 position of cyclohexanone resulted in a significant decrease in catalytic activity. This behavior pattern is common and has been reported in similar catalyst systems [Citation62, Citation66]. The ketonic substrates with substituent groups in the 2-position showed the least reactivity among the cyclic compounds. This is likely due to the steric interference of the substituents with the reactive C = O bond and poor electronic contribution from the methyl substituent (; entry 3).
3. Conclusion
Three ethylene and methylene-bridged bis-imidazole-2-thione ligands, 2a, 2b, and 2c, were synthesized and characterized. The first studies into their coordination chemistry have been carried out on half-sandwich ruthenium complexes. A combination of spectroscopic studies and X-ray crystallography confirmed the structures of the corresponding ruthenium complexes 3a, 3b, and 3c as half-sandwich ruthenium chloro complexes with the thione ligands coordinated in a bidentate mode. The synthesized complexes showed significant activity as catalysts in the transfer hydrogenation of various aliphatic and aromatic ketones. The catalytic activities of the complexes are influenced by bridge (alkyl spacer) length and steric bulk of the N-substituents.
4. Experimental
4.1. General
All manipulations were carried out under a dinitrogen gas atmosphere using standard Schlenk and vacuum-line techniques. All solvents are purified and degassed before use by standard procedures [Citation67]. The starting materials, [(η6-arene-Ru(μ-Cl)Cl]2 [Citation68, Citation69], 1,1′-diethyl-3,3-ethylenediimidazolium dibromide; 1a, 1,1′-dipropyl-3,3-ethylenediimidazolium dibromide; 1b and 1,1′-diethyl-3,3-methylenediimidazolium dibromide; 1c [Citation13, Citation42, Citation70, Citation71], were prepared according to the procedures described in the literature (note that arene = p-cymene). Other chemicals are of analytical grade and used as received. The NMR spectral data are obtained using a Bruker Top Spin 400 MHz spectrometer with sample dissolved in CDCl3 for ligands and in CD3CN for the complexes. The two deuterated solvents are obtained from Sigma-Aldrich. The NMR peaks are reported in ppm (δ) with tetramethylsilane (TMS at 0.00 ppm) as the internal standard. Infrared spectral data in the solid state are recorded between 4000 and 400 cm−1 using an ATR Perkin Elmer Spectrum 100 spectrophotometer. High-resolution mass spectra are recorded using a Waters Synapt G2 TOF-MS analyzer by direct ES in the positive mode. The melting points for the synthesized compounds are determined utilizing a Stuart Scientific melting point apparatus, SMP3. The transfer hydrogenation reaction was monitored by gas chromatography (GC) using a YL6500 instrument fitted with a DB wax polyethylene column (0.32 mm in diameter, 30 m in length, and 0.50 μm film thickness), a flame ionization detector, and hydrogen gas at a flow rate of 30 mL/min.
4.2. General procedure for the synthesis of alkane-bridged organochalcogen ligands
In a 100 mL round-bottomed flask fitted with a reflux condenser are placed 1,1′-diethyl-3,3-ethylenediimidazolium dibromide/1,1′-dipropyl-3,3-ethylenediimidazolium dibromide or 1,1′-diethyl-3,3-methylenediimidazolium dibromide (10 mmol of 1a-c, respectively), sulfur powder (20 mmol), K2CO3 (2.0 g) and dry methanol (50 mL) as solvent. The mixture is then refluxed for 24 h and concentrated with a rotary evaporator. All organic compounds are extracted by dissolving the residue in CH2Cl2 (2 × 30 mL), which was then filtered through a bed of Celite. Removal of all volatiles from the filtrate using a rotary evaporator afforded the pale yellow, orange, or colorless crystalline solids of the imidazole-2-thione ligands, 2a–c, in good yields (Scheme 1).
4.2.1. 1,1’-Ethylenebis(3-ethyl-imidazole-2-thione (Ebeit) (2a)
Yield: (1.84 g, 65%. m.p.: 131–133 °C).1H NMR (400 MHz, CDCl3): δ (ppm) 1.34 (t, J = 7.3 Hz, 2NCH2-CH3, 6H), 4.06 (m, 2NCH2CH3, 4H), 4,48 (s, 2NCH2-linker, 4H), 6.58 (d, J = 14.0 Hz, 4CH-imid, 4H).13C NMR (100 MHz, CDCl3): δ (ppm) 14.3 (CH2CH3), 42.9 (CH2CH3), 45.4 (NCH2-linker), 115.9 (NCH-imid), 118.1 (NCH-imid), 161.6 (C = S). FTIR (solid state): ʋ(=C-H) 3154, 3117, 3084 cm−1, ʋ(CH3) 2981, 2940 cm−1, ʋ(C = C)+ʋ(C = N) 1643, 1563 cm−1, ʋ(C-N) +ʋ(C = S) 1177 cm−1, ʋs(C = S)+ʋas(C = S) 670, 514 cm−1. ESI-HRMS (CH3CN): m/z found for [M + H]+: 283.1049; calculated: 283.1051.
4.2.2. 1,1’-Ethylene bis (3-propyl-imidazole-2-thione (Ebpit) (2b)
Yield: (1.86 g, 60%. m.p.: 91–92 °C). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.92 (t J = 7.4 Hz, 2NCH2CH2CH3, 6H), 1.77 (m, 2NCH2CH2CH3, 4H), 3.96 (t, J = 7.3 Hz, 2NCH2CH2CH3, 4H), 4.49 (s, 2NCH2-linker, 4H), 6.54 (d, J = 11.24 Hz, 4NCH-imid, 4H). 13C NMR (100 MHz, CDCl3): δ (ppm) 11.1 (CH2CH3), 22.3 (CH2CH3), 45.3 (NCH2), 49.5 (NCH2-linker) 116.7 (NCH-imid), 117.9 (NCH-imid), 161.8 (C = S). FTIR (solid state): ʋ(=C-H) 3163, 3127, 3097 cm−1, ʋ(CH3) 2959, 2933 cm−1, ʋ(C = C)+ʋ(C = N) 1641, 1564, 1526 cm−1, ʋ(C-N) +ʋ(C = S) 1179 cm−1, ʋs(C = S)+ʋas(C = S) 665, 532 cm−1. ESI-HRMS (CH3CN): m/z found for [M + H]+: 311.1364; calculated: 311.1364.
4.2.3. 1,1’-Methylene bis (3-ethyl-imidazole-2-thione (mbeit) (2c)
Yield: (1.88 g 70%. m.p.: 175–177 °C). 1H NMR (400 MHz, CDCl3): δ (ppm) 1.35 (t, J = 7.3 Hz, 2NCH2CH3, 6H), 4.05 (m, 2NCH2CH3, 4H), 6.33 (s, NCH2-linker, 2H), 6.62 (d, J = 2.3 Hz, 2NCH-imid, 2H), 7.64 (d, J = 2.3 Hz, 2NCH-imid, 2H). 13C NMR (100 MHz, CDCl3): δ (ppm) 14.1 (CH2CH3), 42.9 (CH2CH3), 55.8 (NCH2-linker), 116.0 (NCH-imid), 116.9 (NCH-imid), 162.9 (C = S). FTIR (solid state): ʋ(=C-H) 3109, 3080 cm−1, ʋ(CH3) 2989, 2945 cm−1, ʋ(C = C)+ʋ(C = N) 1681, 1566 cm−1, ʋ(C-N) +ʋ(C = S) 1070 cm−1, ʋs(C = S)+ʋas(C = S) 715, 515 cm−1. ESI-HRMS (CH3CN): m/z found for [M + H]+: 269.0904; calculated: 269.0850.
4.3. Synthesis of half-sandwich η6-arene-ruthenium complexes (arene = p-cymene) bearing chelating (SS) organochalcogen ligands (3a-c)
The Ru precursor, [Ru(p-cymene)(μ-Cl)Cl]2 (0.05 mmol), and the respective alkylene bridged organochalcogen ligand (0.1 mmol) were dissolved in DCM (6 mL) and stirred at room temperature for 16 h. The DCM is removed, and dry CH3OH (2 mL) is added to dissolve the solids. The subsequent addition of a saturated KPF6 aqueous solution to the methanolic solution precipitated the respective brown half-sandwich ruthenium complexes. These were then filtered off using vacuum filtration, washed successively with water and diethyl ether (3 × 2 mL), and finally dried in vacuo to afford the air and moisture-stable half-sandwich complexes 3a-c in good yields (Scheme 2).
4.3.1. η6-arene-Ru(L1)Cl]PF6 (3a)
Yield: (46 mg, 66%. m.p.: 137–139 °C decomp.). 1H NMR (400 MHz, CD3CN): δ (ppm) 1.33 (m, 2NCH2CH3, CH(CH3)2-p-cymene, 12H), 2.16 (s, C6H4CH3, 3H), 2.88 (m, CH(CH3)2-p-cymene, 1H), 3.87 − 4.38 (m, 2NCH2CH3, 2NCH2-linker, 8H), 5.32 (d, J = 5.7 Hz, η6-C6H4, 1H), 5.49 (d, J = 5.7 Hz, η6-C6H4, 1H), 5.69 (d, J = 6.0, η6-C6H4, 1H), 5.82 (d, J = 6.0 Hz, η6-C6H4, 1H), 7.05 (d, J = 21.6 Hz, 2NCH-imid, 2H), 7.25 (d, J = 9.4 Hz, 2NCH-imid, 2H). 13C NMR (100 MHz, CD3CN): δ (ppm) 14.8 (NCH2CH3), 18.9 (C6H4CH3), 22.9 (CH(CH3)2, 31.6 (CH(CH3)2), 45.2 (NCH2CH3), 47.7 (NCH2-linker), 84.5 (η6-C6H4), 85.3 (η6-C6H4), 101.2 (η6-C6H4), 104.04 (η6-C6H4), 120.7 (NCH-imid), 122.3 (NCH-imid), 155.2 (C = S). FTIR (solid state): ʋ(=C-H) 3172, 3133, 3100 cm−1, ʋ(CH3) 2970, 2882 cm−1, ʋ(C = C)+ʋ(C = N) 1564 cm−1, ʋ(C-N) +ʋ(C = S) 1198 cm−1, ʋ(P-F) 824 cm−1 ʋs(C = S)+ʋas(C = S) 737, 555 cm−1. ESI-HRMS (CH3CN): m/z found for [M-PF6]+: 553.0801; calculated: 553.0800.
4.3.2. η6-arene-Ru(L2)Cl]PF6 (3b)
Yield: (51 mg, 70%. m.p.: 125–127 °C decomp.). 1H NMR (400 MHz, CD3CN): δ (ppm) 0.91 (t, J = 7.4 Hz, 2NCH2CH2CH3), 6H), 1.32 (d, J = 6.9 Hz, CH(CH3)2, 6H), 1.76 (m, 2NCH2CH2CH3, 4H), 2.30 (s, C6H4CH3, 3H), 2.82 (m, CH(CH3)2, 1H), 3.78 (m, NCH2CH2CH3, 2H), 4.05 (m, NCH2CH2CH3, 2H), 4.47 (m, NCH2-linker, 2H), 5.32 (m, NCH2-linker, 2H), 5.86 (d, J = 5.8 Hz, η6-C6H4, 2H), 6.01 (d, J = 5.8 Hz, η6-C6H4, 2H), 7.23 (d, J = 2.1 Hz, 2NCH-imid, 2H), 7.33 (d, J = 2.1 Hz, 2NCH-imid, 2H). 13C NMR (100 MHz, CD3CN): δ (ppm) 11.4 (CH2CH3), 18.8 (C6H4CH3), 23.1 (CH2CH3), 24.0 (CH(CH3)2), 31.6 (CH(CH3)2), 48.9 (NCH2), 51.5 (NCH2-linker), 79.3 (η6-C6H4), 81.5 (η6-C6H4), 97.8 (η6-C6H4), 100.4 (η6-C6H4), 122.8 (NCH-imid), 124.6 (NCH-imid), 152.3 (C = S). FTIR (solid state): ʋ(=C-H) 3174 cm−1, ʋ(CH3) 2968, 2940 cm−1 cm−1, ʋ(C = C)+ʋ(C = N) 1565 cm−1, ʋ(C-N) +ʋ(C = S) 1185 cm−1, ʋ(P-F) 824 cm−1, ʋs(C = S)+ʋas(C = S) 685, 555 cm−1. ESI-HRMS (CH3CN): m/z found for [M-PF6]+: 581.1107; calculated: 581.1113.
4.3.3. η6-arene-Ru(L3)Cl]PF6 (3c)
Yield: (35 mg, 51%. m.p.: 161–163 °C) 1H NMR (400 MHz, CD3CN): δ (ppm) 1.29 (d, J = 6.9 Hz, CH(CH3)2, 6H), 1.39 (t, J = 7.8 Hz, 2NCH2CH3, 6H), 2.13 (s, C6H4CH3, 3H), 2.90 (m, CH(CH3)2, 1H), 4.08 (m, 2NCH2CH3, 4H), 5.33 (d, J = 5.8 Hz, η6-C6H4, 2H), 5.51 (d, J = 5.8 Hz, η6-C6H4, 2H), 6.16 (dd, J = 13.6, 13.8 Hz, NCH2-linker, 2H), 7.16 (d, J = 2.0 Hz, 2NCH-imid, 2H), 7.43 (d, J = 2.0 Hz, 2NCH-imid 2H). 13C NMR (100 MHz, CD3CN): δ (ppm) 15.1 (CH2CH3), 19.0 (C6H4CH3), 22.9 (CH(CH3)2), 31.2 (CHCH3), 45.53 (CH2CH3), 58.4 (CH2-linker), 84.7 (η6-C6H4), 85.0 (η6-C6H4), 101.3 (η6-C6H4), 104.2 (η6-C6H4), 120.9 (NCH-imid), 121.7 (NCH-imid), 158.3 (C = S). FTIR (solid state): ʋ(=C-H) 3174, 3150 cm−1, ʋ(CH3) 2965 cm−1, ʋ(C = C)+ʋ(C = N) 1570 cm−1, ʋ(C-N) +ʋ(C = S) 1080 cm–1, ʋ(P-F) 826 cm−1, ʋs(C = S)+ʋas(C = S) 743, 554 cm−1. ESI-HRMS (CH3CN): m/z for [M-PF6]+: 539.0646; calculated: 539.0644.
4.4. Single crystal X-ray diffraction analysis
X-ray diffraction data on suitable single crystals of 3a, 3b, and 3c were collected on a Bruker APEX-II CCD diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The crystals were kept at 149.99 K during the experiment, and all the data were collected using Olex2 [Citation72]. The structure was solved with the SHELXT [Citation73] structure solution program using Intrinsic Phasing and refined with the SHELXL [Citation74] refinement package using Least Squares minimization. Crystallographic data for the structures in this article have been deposited with the Cambridge Crystallographic Data Centre with deposition numbers 2302988–2302990 for 3a–c, respectively. The data can be obtained free of charge at http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223/336-033; E-mail: [email protected]).
4.5. General procedure for transfer hydrogenation of ketones
The reactions were all performed under the same conditions to allow for comparison of results. In a typical experiment, ketone (1 mmol) and one of the complexes, 3a, 3b, or 3c (0.5 mol%), are dissolved in 2-propanol (5 mL). The mixture was then stirred at 82 °C for 10 min, after which it was allowed to cool to room temperature. To the cooled flask contents, 0.5 mL of 0.2 M KOH (0.1 mmol) solution in 2-propanol was added, and the mixture was continuously stirred at 82 °C for 6 h. The progress of the reactions was monitored by GC.
Supplemental Material
Download MS Word (1.6 MB)Acknowledgment
We thank the NRF and UKZN for financial support and Mr. S. Zamisa for collecting X-ray data.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- A. Beheshti, E.S. Mousavifard, B. Zargar, P. Mayer, S.E. Rezatofighi. New J. Chem., 44, 8520 (2020).
- B.S. Stadelman, J.L. Brumaghim. ACS Symp. Ser. Am. Chem. Soc., 1152, 33 (2013).
- L. Maria, C. Moura, A. Paulo, I.C. Santos, I. Santos. J. Organomet. Chem., 691, 4773 (2006).
- H.M. Alvarez, T.B. Tran, M.A. Richter, D.M. Alyounes, D. Rabinovich, J.M. Tanski, M. Krawiec. Inorg Chem., 42, 2149 (2003).
- M. Garner, M.-A. Lehmann, J. Reglinski, M.D. Spicer. Organometallics, 20, 5233 (2001).
- C. Kimblin, T. Hascall, G. Parkin. Inorg. Chem., 36, 5680 (1997).
- I.R. Crossley, A.F. Hill, A.C. Willis. Organometallics, 25, 289 (2006).
- S.L. Kuan, W.K. Leong, L.Y. Goh, R.D. Webster. Organometallics, 24, 4639 (2005).
- I.R. Crossley, A.F. Hill, E.R. Humphrey, A.C. Willis. Organometallics, 24, 4083 (2005).
- M.M. Morlok, K.E. Janak, G. Zhu, D.A. Quarless, G. Parkin. J. Am Chem Soc., 127, 14039 (2005).
- I. Kuzu, I. Krummenacher, J. Meyer, F. Armbruster, F. Breher. Dalton Trans., 43, 5836 (2008).
- M.D. Spicer, J. Reglinski. Eur J. Inorg Chem., 2009, 1553 (2009).
- R.M. Silva, M.D. Smith, J.R. Gardinier. J. Org Chem., 70, 8755 (2005).
- W.G. Jia, Y.B. Huang, G.X. Jin. J. Organomet. Chem., 694, 3376 (2009).
- M. Slivarichova, R.C. Costa, J. Nunn, R. Ahmad, M.F. Haddow, H.A. Sparkes, T. Gray, G.R. Owen. J. Organomet. Chem., 847, 224 (2017).
- W.G. Jia, Y.B. Huang, Y.J. Lin, G.L. Wang, G.X. Jin. Eur J. Inorg Chem., 2008, 4063 (2008).
- C. Johnson, M. Albrecht. Coord. Chem. Rev., 352, 1 (2017).
- V. Cadierno, M.P. Gamasa, J. Gimeno. Coord. Chem. Rev., 248, 1627 (2004).
- D. Carmona, M.P. Lamata, L.A. Oro. Eur. J. Inorg. Chem., 2002, 2239 (2002).
- J. Popp, S. Hanf, E.H. Hawkins. ACS Omega, 4, 22540 (2019).
- J.M. Gichumbi, H.B. Friedrich. J. Organomet. Chem., 866, 123 (2018).
- T. Sarıdağ, K. Buldurun. Catal Lett., 154, 107 (2024).
- G. Gasser, I. Ott, N.M. Nolte. J. Med Chem., 54, 3 (2011).
- A.A. Nazarov, C.G. Hartinger, P.J. Dyson. J. Organomet. Chem., 751, 251 (2014).
- S.K. Singh, D.S. Pandey. RSC Adv., 4, 1819 (2014).
- M. Gozzi, B. Schwarze, E. Hey-Hawkins. Pure Appl. Chem., 91, 563 (2019).
- K. Buldurun, N. Turan, G. Mahmoudi, E. Bursal. J. Mol. Struct., 1262, 133075 (2022).
- P. Kumar, R.K. Gupta, D.S. Pandey. Chem. Soc. Rev., 43, 707 (2014).
- W.G. Jia, Y.B. Huang, Y.J. Lin, G.X. Jin. Dalton Trans., 5612 (2008).
- P.D. Akrivos. Coord. Chem. Rev., 213, 181 (2001).
- Y. Qin, Q. Ma, A.Q. Jia, Q. Chen, Q.F. Zhang. J. Coord. Chem., 66, 405 (2013).
- H. Zhu, Q. Ma, A.Q. Jia, Q. Chen, W.H. Leung, Q.F. Zhang. Inorg. Chim. Acta, 405, 427 (2013).
- P. Oswal, A. Arora, S. Singh, D. Nautiyal, S. Kumar, G.K. Rao, A. Kumar. Dalton. Trans., 49, 12503 (2020).
- L.-M. Zhang, H.-Y. Li, H.-X. Li, D.J. Young, Y. Wang, J.-P. Lang. Inorg. Chem., 56, 11230 (2017).
- P. Zhang, C.K.C. Chiu, H. Huang, Y.P.Y. Lam, A. Habtemariam, T. Malcomson, M.J. Paterson, G.J. Clarkson, P.B. O'Connor, H. Chao, P.J. Sadler. Angew. Chem. Int. Ed., 56, 14898 (2017).
- A.A.A. Seliman, M. Altaf, A.T. Onawole, S. Ahmad, M.Y. Ahmed, A. Hamad, S. Altuwaijiri, G. Bhatia, J. Singh, A.A. Isab. J. Organomet. Chem., 848, 75 (2017).
- M.Y. Jomaa, M. Altaf, S. Ahmad, A. Alhoshani, N. Baig, A.-N. Kawde, G. Bhatia, J. Singh, A.A. Isab. Polyhedron, 141, 360 (2018).
- W.G. Jia, T.T. Du, L.L. Gao, J. Du. Appl. Organomet. Chem., 34, e5651 (2020).
- W.G. Jia, L.L. Gao, X.T. Zhi, X.D. Li, Z.B. Wang, Y. Sun. Polyhedron, 195, 114978 (2021).
- A.K. Sharma, H. Joshi, K.N. Sharma, P.L. Gupta, A.K. Singh. Organometallics, 33, 3629 (2014).
- M. Poyatos, E. Mas-Marzá, M. Sanaú, E. Peris. Inorg Chem., 43, 1793 (2004).
- D.J. Williams, R.L. Jones, D.S. Menaldino. Inorg. Chim. Acta, 165, 173 (1989).
- X.-L. Tao, M. Lei, Y.-G. Wang. Synth Commun., 37, 399 (2007).
- Q. Liu, D. Shi, K. Yu, J. Xu. Acta Crystallogr. Sect. E. Struct. Rep. Online, 59, o356 (2003).
- G. Roy, P.N. Jayaram, G. Mugesh. Chem. Asian J., 8, 1910 (2013).
- Y.B. Huang, W.G. Jia, G.X. Jin. J. Organomet. Chem., 694, 86 (2009).
- A. Caballero, E. Diez-Barra, F.A. Jalon, S. Merino, J. Tejeda. J. Organomet. Chem., 395, 617 (2001). 618
- C.N.R. Rao, R. Venkataraghavan. Spectrochim. Acta A, 45, 299 (1989).
- E.S. Raper. Coord. Chem. Rev., 61, 115 (1985).
- C.N.R. Rao, R. Venkataraghavan, T.R. Kasturi. Can. J. Chem., 42, 36 (1964).
- E.S. Raper, I.W. Nowell. Inorg. Chim. Acta, 43, 165 (1980).
- B. Singh, K.P. Thakur, J. Inorg. Nucl. Chem., 36, 1735 (1974).
- E.S. Raper, J.L. Brook. J. Inorg. Nucl. Chem., 39, 2163 (1977).
- J.R. Creighton, D.J. Gardiner, A.C. Gorvin, C. Gutteridge, A.R.W. Jackson, E.S. Raper, P.M.A. Sherwood. Inorg. Chim. Acta, 3, 195 (1985).
- E.R. Atkinson, D.J. Gardiner, A.R.W. Jackson, E.S. Raper. Inorg. Chim. Acta, 98, 35 (1985).
- K.K.H. Tong, M. Hanif, J.H. Lovett, K. Hummitzsch, H.H. Harris, T. Sohnel, S.M.F. Jamieson, C.G. Hartiger. Molecules, 25, 3661 (2020).
- M. Albrecht, J.R. Miecznikowski, A. Samuel, J.W. Faller, R.H. Crabtree. Organometallics, 21, 3596 (2002).
- P. Singh, A.K. Singh. Organometallics, 29, 6433 (2010).
- J.M. Gichumbi, H.B. Friedrich, B. Omondi, M. Singh, K. Naicker, H.Y. Chenia. J. Coord. Chem., 69, 3531 (2016).
- M.R.S.J. Foreman, A.F. Hill, A.J.P. White, D.J. Williams. Organometallics, 23, 913 (2004).
- Y. Qin, Q. Ma, A.Q. Jia, Q. Chen, Q.F. Zhang. J. Coord. Chem., 66, 1405 (2013).
- J.M. Gichumbi, H.B. Friedrich, B. Omondi. J. Mol. Catal. A Chem., 416, 29 (2016).
- H. Ibrahim, M.D. Bala, H.B. Friedrich. Coord. Chem. Rev., 469, 214652 (2022).
- T.L. Brown, K.J. Lee. Coord. Chem. Rev., 128, 89 (1993).
- S.K.U. Riederer, P. Gigler, M.P. Högerl, E. Herdtweck, B. Bechlars, W.A. Herrmann, F.E. Kühn. Organometallics, 29, 5681 (2010).
- M.D. Bala, M.I. Ikhile. J. Mol. Catal. A Chem., 385, 98 (2014).
- W.L.F. Armarego, C.L.L. Chai. Purification of Laboratory Chemicals, 6th Edn, p 88, Butterworth-Heinemann, Oxford (2009).
- M.A. Bennet, T.-N. Huang, T.W. Matheson, A.K. Smith, S. Itel, W. Nickerson. Inorg. Synth., 17, 74 (2007).
- S.B. Jensen, S.J. Rodgers, M.D. Spicer. J. Organomet. Chem., 556, 151 (1998).
- C. Cao, Y. Zhuang, J. Zhao, H. Liu, P. Geng, G. Pang, Y. Shi. Synth. Commun., 42, 380 (2012).
- K.-M. Lee, J.C.C. Chen, I.J.B. Lin. J. Organomet. Chem., 617, 364 (2001).
- O.V. Dolomanov, L.J. Bourhis, R.J. Gildea, J.A.K. Howard, H. Puschmann. J. Appl Crystallogr., 42, 339 (2009).
- G.M. Sheldrick. Acta Crystallogr. A, 71, 3 (2015).
- G.M. Sheldrick. Acta Crystallogr. C: Struct. Chem., 71, 3 (2015).