

Abstract
Research shows an association between vaginal microbiota and the development of cervical cancer, but the role of altered microbiota in cancer development remains controversial. In this study, we attempted to reveal the vaginal microecological changes in cervical lesions by 16S rRNA gene sequencing. Vaginal secretions were collected from Hakka women in Meizhou City, Guangdong Province, China. The diversity, composition and the correlations among species of the vaginal microbiota were determined by sequencing the bacterial 16S rRNA gene. The microbial functional abundance was detected via KEGG and COG (Clusters of Orthologous Groups). The results showed that the Cancer group was characterised by evident changes in the composition of the vaginal microbiota, increased alpha diversity, and altered community structure distribution and microbial interaction network. Linear discriminant analysis (LDA) effect size showed that 21 bacterial species were abundant in the Cancer group. In addition, the loss of Lactobacillus stimulated other flora proliferation, resulting in a microecological disturbance. KEGG and COG analysis indicated the cancer group is mainly concentrated in energy metabolism. In short, the vaginal microecology of Hakka women in Meizhou City presents with different degrees of cervical lesions, and the flora imbalance is an important factor in the development of cervical cancer.
What is already known on this subject? Cervical cancer is one of the most common gynecological malignancies worldwide and has become a prominent public health problem.
What the results of this study add? Our study showed that the type of vaginal community status of Hakka women in Meizhou area was characterised by L. Iners predominates, and the gradual loss of Lactobacillus dominance in vaginal bacteria is key to microecological imbalance.
What the implications are of these findings for clinical practice and/or further research? Disturbances in vaginal microecology can stimulate energy metabolism and lipid metabolism to induce cervical cancer development.
IMPACT STATEMENT
Introduction
Cervical cancer, one of the most prevalent gynecological malignancies worldwide, has become a prominent public health problem (Colombo et al. Citation2012). It has been reported that the incidence of cervical cancer ranks second among gynecological malignancies, with the highest mortality rate among female genital tract malignancies. More terribly, cervical cancer was the second leading cause of cancer-related death in women aged 20 to 39 years in 2019, seriously threatening women’s health (Sung et al. Citation2021). Persistent cervical infection induced by high-risk human papillomavirus (HPV) is the leading cause of cervical cancers and direct precursor lesions. Fortunately, the advent of HPV vaccination and early screening programs have effectively managed and prevented cervical cancers. However, high incidence rates are still observed in low- and middle-income countries (Arbyn et al. Citation2020). Generally, surgery, chemotherapy and radiotherapy are the most common therapeutic regimens for cervical cancer. Nonetheless, the efficacy of chemotherapeutic agents is limited by the resistance of cervical cancer cells. Consequently, the 5-year survival rate in patients with cervical cancer is still unsatisfactory (Derks et al. Citation2018, Meijer and Snijders Citation2014). Therefore, further exploring the underlying mechanisms of cervical cancer pathogenesis is essential for the development of new cervical cancer biomarkers and therapeutic targets.
In addition to HPV infection, many other factors are responsible for cervical cancer development. Some researchers proposed the possibility of identifying early cervical cancers and providing a better prognosis for patients through specific biomarkers (Bizzarri et al. Citation2021; Dellino et al. Citation2022; Valenti et al. Citation2017). The vaginal microbiota has been found to be closely associated with gynecological diseases and reproductive health (Smith et al. Citation2003). Studies have reported that alterations in the microbiota are linked to the progression of many human diseases. For example, alterations in the microbiota disrupt the vaginal flora balance, thereby resulting in gynecological diseases such as cervical intraepithelial neoplasia (CIN) and cervical cancer (Brotman et al. Citation2014). The cervicovaginal microbiome has been classified as a community state type, usually a specific Lactobacillus species (Lactobacillus crispatus, Lactobacillus iners, Lactobacillus gasseri, or Lactobacillus jensenii), or a polymicrobial state (Ravel et al. Citation2011). The shift in the type of community state dominated by lactic acid bacteria was proved to produce deleterious health outcomes such as an increased risk of sexually transmitted infections (Bik et al. Citation2019) and premature delivery. Lee et al. reported that there was a correlation of increased vaginal microbial diversity with the prevalence of high-risk HPV infection and/or cervical abnormalities (relative to HPV negativity); in particular, Fusobacteria, including Sneathia spp., is strongly associated with HPV infection (Lee et al. Citation2013). In addition, both composition and diversity of the vaginal microbiota were influenced by host genetics, physiology, and other factors, such as menopause and oestrogen level; and in the meanwhile, the potential microbiological predictors reported could provide some insights into the pathogenesis of HPV and might be used as potential microbiological targets for novel diagnostic methodologies to improve women’s health status (Lee et al. Citation2013). L. curlatus is highly abundant and has been confirmed to be associated with lower HPV prevalence. The long-term application of vaginal probiotics containing Lactobacillus species can enhance HPV clearance compared with short-term ones (Palma et al. Citation2018). In short, the ecological structure of the vaginal microbiota is closely related to the occurrence and development of cervical cancer. Previous studies on vaginal microecology have verified an association between vaginal microbial diversity and cervical neoplasia. Furthermore, there are differences in the composition of vaginal microbial bacteria in adult women in different growth environments from different regions (Seo et al. Citation2016).
Currently, there is still little known about the complex interaction between cervical dysbiosis and cancer pathophysiology. To further clear the contributory roles of the microbiome in different degrees of cervical lesions, vaginal secretions collected from Hakka women in the Meizhou area were examined using a liquid-based cell detection system (LCT) and HPV. Through observing vaginal microecological changes of cervical lesions, we expected to identify new biomarkers and therapeutic targets for the treatment of cervical cancer.
Materials and methods
Sample collection
This study was approved by the Ethics Committee of Meizhou People’s Hospital (2019-C-83), and all patients involved in this study signed the informed consent form. Sixty women (age: 21–70 years) admitted to our gynaecology outpatient clinic from September 2019 to January 2020 were included in this study. Specifically, the inclusion criteria were shown as follows: patients (1) had no sexual intercourse in the last 24 h; (2) were mentally and cognitively normal, and could complete basic communication and exchange; (3) possessed complete clinical data. The exclusion criteria were displayed as follows: patients (1) had a history of vaginal irrigation and drug release; (2) were in menstruation; (3) were combined with acute gynecological inflammation or acute phase of immune diseases; (4) were in pregnancy or lactation.
Vaginal secretions were collected from 60 women using a sterile cotton swab for LCT and HPV examination. Patients with negative LCT and HPV test outcomes were regarded as the Control group and the positive ones as the experimental group. The latter was further divided into a low-grade CIN group (CIN I, Low group), a high-grade CIN group (CIN II/III, High group) and a cervical cancer group (Cancer group) in accordance with the pathological results. By the way, CIN I is an atypical hyperplasia; CIN II/III is a precancerous lesion of cervical cancer.
DNA Extraction and Amplification
DNA extraction was performed using the TIANamp Bacteria DNA Extraction Kit (Tiangen Biotech, Beijing, China), and microbial DNA was extracted from vaginal samples following the manufacturer’s protocol. The bacterial 16S rRNA gene in the V3 – V4 region was amplified by PCR (3 min at 95 °C, followed by 20 s at 98 °C, 15 s at 58 °C, 20 s at 72 °C, for a total of 30 cycles, and extending at 72 °C for 5 min finally). The reaction was carried out in 30 μL mixtures containing 15 μL 2 × KAPA Library Amplification ReadyMix, 343 F and 798 R primers each in 1 μL (10 μM) and template DNA and ddH2O 50 ng. The sequences of the primers used were shown as follows: 343 F 5′-TACGGRAGGCAGCAG-3′ and 798 R 5′-AGGGTATCTAATCCT−3′.
MiSeq Sequencing
Amplicons were gel-purified from 2% agarose gels using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, USA) according to the manufacturer’s instructions. The concentration of the samples’ DNA was quantified with the Qubit ® 2.0 (Invitrogen, USA). The prepared libraries were sequenced according to the tags on the MiSeq platform (Illumina Inc., USA), and after that, the paired-end reads (250 bp) were acquired. Notably, the MiSeq platform generated approximately 10 million reads. These DNAs overlapped at their 3’ends to constitute original long tags. DNA extraction, library construction, and sequencing were conducted at Oe Biotech Co., Ltd.
Process of data Sequencing
Labels, barcodes, length of primers and average base quality were checked and trimmed. The length of the 16S tag was restricted from 220 to 500 bp, and the average Phred score was not lower than 20 (Q20) and higher than 3 fuzzy N. The copy number of tags was enumerated, and the duplicates were removed. Only tags with frequencies >1 were considered reliable and could be clustered into operational taxonomic units (OTUs). Each data in the OTUs had a representative label. OTUs were clustered at 97% similarity via UPARSE (http://drive5.com/uparse/), and chimeric sequences were identified and removed using USEARCH (version 7.0). Besides, an RDP classifier (http://rdp.cme.msu.edu/) was employed for checking and classification of a single OTU representative sequence, with a confidence threshold of 0.8.
Statistical analysis
The SPSS 23.0 software was used for data analysis. Measurement data were presented as mean ± standard deviation (SD). Analysis of variance or Kruskal-Wallis test and independent sample t-test was adopted for comparisons among groups, and differences between two groups were further analysed via the least-squares difference test. The Mothur software was utilised to calculate α diversity index, and β diversity analysis was performed based on Unweighted unifrac distance. Microbial multivariate variable statistical analysis and prediction of microbial functional abundance in samples were conducted by analysis of variance (ANOVA), KEGG (Kyoto Encyclopaedia of Genes and Genomes) and COG (Clusters of Orthologous Groups) databases, respectively. Furthermore, a phylogenetic tree was constructed based on OTU representative sequences, followed by statistical analysis of changes in microbial communities in different groups of samples and multi-level species difference discriminant analysis based on linear discriminant analysis effect size (LEfSe). In addition, correlations between interspecies or environmental factors and species were analysed using Spearman correlation, and the R version 4.1.3 (https://www.r-project.org/) with Hmisc package was utilised for drawings. Finally, P < 0.05 indicated a significant difference.
Results
Composition and relative abundance of vaginal microbiota
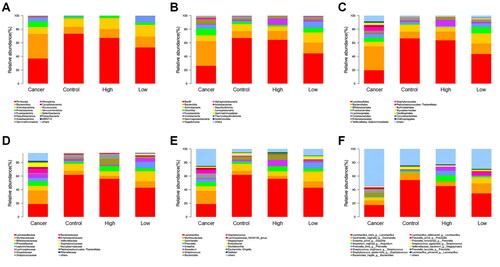
In this study, microbiological genomics analysis (16S rRNA gene sequencing) was performed based on clinical vaginal secretions from four groups (15 samples per group). The analysis results showed no significant difference in the types of vaginal microbiota among the groups but a significant difference in the relative abundance of microbial bacteria of each group based on different degrees of cervical lesions. At the phylum level, Firmicutes, Bacteroidota and Actinobacteriota were the dominant vaginal microbiota in the four group patients, while the relative abundance of Firmicutes and Actinobacteriota in the Cancer group was lower than that in the Control, Low and High groups (). At the class level, the core vaginal microbiota of patients included Bacilli, Bacteroidia, Actinobacteria, and Clostridia. Among the four groups, the Cancer group exhibited the lowest relative abundance of Bacilli and Actinobacteria and the highest level of Bacteroidia and Clostridia (). At the order level, Lactobacillales and Bifidobacteriales were the dominant microbiota in the vaginal microorganisms of the four groups; besides, a lower relative abundance of Lactobacillales and a higher abundance of Bifidobacteriales was observed in the Cancer group (). At the family level, the core microbiota in the four groups included Lactobacillaceae, Muri, and Bifdobacteriaceae (). Similarly, Lactobacillus, Muri, and Gardnerella were the dominant bacteria in the vaginal microorganisms of patients at the genus level (), and the trend of Lactobacillus and Gardnerella continued until the species level with their levels lowest in the Cancer group (). The above results suggested that the lesions of cervical cancer could be related to the abundance ratio of the dominant microbiota.
Figure 1. Composition classification and abundance comparison of vaginal microbiota.
Comparison of vaginal microbiota at the levels of phylum (A), class (B), order (C), family (D), genus (E) and species (F) in the cervical Cancer group, Control group, high-grade cervical intraepithelial neoplasia (CIN) group (High group) and low-grade CIN (Low group).
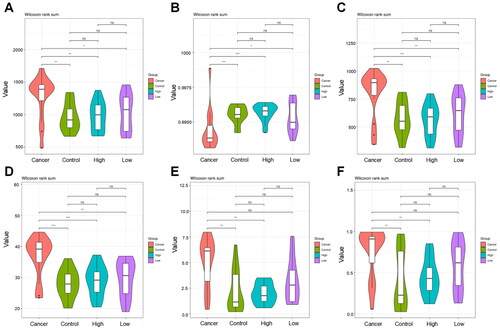
Diversity analysis of α
Subsequently, α diversity analysis was employed for the flora of samples in each group. The chao1 index revealed that the total number of vaginal microbiota in the Cancer group was significantly larger than that in the Control group (p = 0.0023), Low group (p = 0.0186) and High group (p = 0.0032), while among the other three groups, the difference was not significant (). The goods_coverage index indicated a significantly lower probability of sequence detection in the Cancer group relative to the Control group (p = 0.0007), Low group (p = 0.0128) and High group (p = 0.0002) (). In addition, the Cancer group had a remarkably higher abundance of vaginal microbiota than the Control group (p = 0.0012), Low group (p = 0.0037) and High group (p = 0.0010), as demonstrated by the observed_species results (). Furthermore, the Cancer group also displayed a higher index of PD_whole_tree (p = 0.0010), Shannon (p = 0.0023) and Simpson (p = 0.0043) than the Control group (). Collectively, compared with the Cancer group, the Control group not only had more species of the vaginal microbial bacterial community but also exhibited higher diversity, abundance and species uniformity.
Figure 2. Violin plot of operational taxonomic unit diversity index for inter-group comparison.
A: The chao1 estimator was used to evaluate the index of the number of operational taxonomic units (OTUs) contained in each group of samples, i.e., the total number of species; B: The detection probability of microbial sequences in each group of samples in this sequencing was shown by goods_coverage index; C: The number of microbiota contained in each group of samples was indicated by observed_species index, and the higher numerical value indicated the richer microbial abundance of samples; D: The phylogenetic relationship of species within the microbial community was signalled by PD_whole_tree index; E: The Shannon index was applied to estimate the microbial diversity and evenness in each group of samples. A higher index indicated a better evenness; F: The Simpson index was adopted to evaluate the microbial diversity in each group of samples, and the higher index indicated richer species diversity of samples and vice versa. The X-axis was the grouping; Cancer, cervical cancer; Control group, patients with negative LCT and HPV tests; High group, high-grade cervical intraepithelial neoplasia (CIN). Low group, low-grade CIN. The Y-axis is the diversity index value.
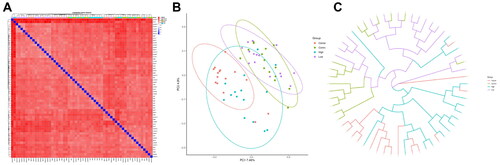
β Diversity analysis
Next, Unweighted unifrac distance was adopted for β diversity analysis of sample data, and it could be observed that the Cancer group was the furthest from the Control group but the closest to the High group (). Additionally, PCoA analysis was employed for the samples of the four groups based on a variety of distance algorithms. As shown in , the gut microbiota of the different groups was separated from each other, as the microbial community of the Cancer, High, Low and Control groups clustered in the red, blue, purple and green circles, respectively. Among them, the microbial community of the Low group was difficult to distinguish from the Control group; moreover, an intersection could be observed between the Low or Control group and the High group, except for the Cancer group. It was worth noting that there were many microbial communities in the High groups also appearing in the aggregation circle of the Cancer group, suggesting a close correlation between the two groups. As demonstrated more clearly by sample hierarchical cluster analysis, the Low and Control groups clustered with each other without significant differences. Interestingly, the High and Cancer group samples clustered separately first and finally clustered together ().
Figure 3. β diversity analysis of biomes of the cervical Cancer group, Control group, high-grade cervical intraepithelial neoplasia (CIN) group (High group) and low-grade CIN (Low group).
A: Sample distance heat map. Deeper blue colour indicated closer distance between samples, while higher similarity and deeper red colour indicated further distance; B: PCA analysis scatter diagram, abscissa (PC1) and ordinate (PC2) were the two main coordinates with the largest explanation of the difference between samples. The same colour represented the same group, a point indicated a sample, and similar samples would gather together; C: UPGMA hierarchical clustering tree between samples, closer branch distance suggested more similar samples.
Differences in flora of phylum, family, genus and species among the four groups
The flora differences of 4 groups was counted through ANOVA difference statistics. Briefly speaking, the relative abundance of the overall microbiota between the Cancer and the other three groups was different at phylum, family, genus and species levels. It was found that the sequences of Lactobacillaceae and Muribaculaceae at the phylum level were the most abundant in each group. Compared with the other three groups, the abundance of Lactobacillaceae in the Cancer group was significantly lower, but that of Murstreibaculaceae, Peptoptoculaceae, Chitinophagceae, Sphingomonadaceae, Moraxellaceae, Ruminocaceae, Clostridia and Erysipelotrichaceae was notably higher (Figure S1(A)). Interestingly, there was no evident difference in the the abundance of above species between the High and Control groups. At the family level, the Cancer group had a lower abundance of Lactobacillaceae and higher Muribaculaceae, Sphingomonas, Lachnoclostridium, Acinetobacter, Sediminibacterium and Muribaculum, relative to other groups; and as for the abundance of microbiota in the other three groups, the difference was not significant except Peptostreptococcus (Figure S1(B)). At the genus level, the abundance of Firmicutes was significantly reduced in the Cancer group, and that of Bacteroidota, Proteobacteria, Acidobacteriota, Myxococcota and MBNT15 increased with similarity observed among the other three groups (Figure S1(C)). At the species level, the Cancer group exhibited higher taxonomic ranks, including Bacteroides acidifaciens, Sediminibacterium sp, Lactobacillus jensenii, Acinetobacter bereziniae, Flexibacter Chitinophaga Lactobacillus murinus, Acetobacter pasteurianus, Bcteroides dorei, Burkholderiales, and Bacteroides thetaiota Omicron (Figure S1(D)).
Comparison of vaginal microbial community differences by LEfSe analysis
To analyse the differential bacteria in vaginal bacteria during cervical lesions, an LDA effect size (LEfSe) analysis was adopted to reveal the composition of differential species in the biomes of each group, with each layer of nodes indicating phylum/class/order/family/genus from inside to outside, respectively. The analysis identified the strains with abundance differences, including most strains with high taxonomic ranks in the Cancer group (21 strains). Specifically, the strains included P. Bacteroidota and P. proteobacteria at the phylum level, Corynebacterium and Bacteroides at the genus level, Corynebacteriaceae and Muribaculaceae at the family level, and Corynebacteriales and Clostridia at the class level. Additionally, the highest abundance of Escherichia Shigella appeared in the High group and the highest abundance of Sneathia and Leptptrichiaceae in the Low group (Figure S2).
Correlation analysis among species of vaginal bacterial community
To further explore the influencing factors of vaginal bacterial community change, we analysed the correlation among vaginal bacterial community species. The genus-level strains in the top 30 of abundance in the Cancer group were selected, then the correlation among species was analysed by the Spearman coefficient. The obtained numerical matrix was displayed in the heatmap map, and a negative correlation could be observed among most the species. However, the majority of those statistically significant correlations were positive. Of them, the strains whose abundance was positively correlated consisted of Lachnospiraceae UCG-001, Lachnoclostridium, Alistipes, Sphingomonas, Prevotellaceae, Enterobacter, Lachnospiraceae N4A136, Muribaculaceae, Ralstonia, Acinetobacter, and Sediminibacterium. However, the abundance of the above strains was negatively associated with that of Lactobacillus (Figure S(3)). In a nutshell, the mutual influence between these microbiotas might be one of the important reasons for their alteration.
Gene function prediction of bacterial communities based on 16S
Also, we predicted bacterial community function based on 16S analysis. First, KEGG analysis was performed according to the Kruskal-Wallis algorithm and the analysis result indicated 42 different prediction functions. To be specific, the Cancer group showed higher functional abundances of predicted energy metabolism, nervous system, metabolism of terpenoids, polyketides, lipid metabolism, transport and catabolism, and amino acid metabolism but lower abundances of immune system diseases, replication, and repair than the other three groups (Figure S4(A)). Besides, the abundances of the immune system, poorly characterised, genetic information, transcription and other functions were significantly higher in the High group than in the other three groups (Figure S4(A)). Based on the prediction of COG function, the abundances of COG0463, COG4974, COG0642, COG1309 and COG1028 were markedly higher, while those of COG0765, COG0474, COG0531 and COG0675 were notably lower in the Cancer group (Figure S4(B)), with similar abundances in the High and Control group.
Discussion
Over time, humans coexist with complex bacterial communities unique to specific ecological locations. The vagina is colonised by various microorganisms that form the vaginal microbiota (Martin Citation2012). To date, various microbial communities are believed markers of health. Nonetheless, infection-induced microbial colonisation of abnormal bacteria or overgrowth of normal vaginal bacteria is considered pathogens of diseases (Human Microbiome Project Citation2012). Recent studies have pointed out that abnormal vaginal microbiota is key to developing gynecological cancers such as cervical cancer (Masson et al. Citation2019). Furthermore, a better understanding of the factors affecting the composition and dynamics of vaginal microbial communities might facilitate the development of new strategies for diagnosing and personalised treatment of cervical cancer.
Of the collected samples in this paper, the composition of the bacterial community in the Cancer group was significantly changed. Specifically, the Cancer group exhibited a notable decrease in the number of Firmicutes at the phylum level, Bacillus at the class level, and Lactobacillus at the order level, relative to the other three groups; and such gradually decreased trend also presented in the number of Lactobacillus in the Low and High groups. Consequently, the proportion of Lactobacillus bacteria may be one of the factors inducing cervical cancer. Similarly, Kovachev also revealed a notable reduction in the proportion of Lactobacillus composition in vaginal secretions collected from patients with cervical cancer (Kovachev Citation2020). Besides, Li et al. discovered a significant increase in the number of Lactobacillus bacterial species in HPV16-infected cervical cancer patients after clinical treatment (Li et al. Citation2022). Notably, biological agents of Lactobacillus have been developed for the treatment of cervical cancer. In a recent study, HPV-infected cervical cancer patients who received oral LactobacillusM247 had a higher clearance of abnormal PAP smears than controls (Dellino et al. Citation2022). These findings demonstrate the important value of Lactobacillus in the treatment and screening of cervical cancer.
According to α diversity analysis, the number of species in the bacterial community of the Cancer group was higher than that in the Control group; also, the Cancer group showed the highest diversity, abundance, and species evenness compared with the other groups. Additionally, the β diversity analysis disclosed that the bacterial composition of the Cancer group was significantly different from that of the Control and Low groups, but was close to that of the High group. It could be speculated that during cancer development, the bacterial composition gradually showed colony structure development in the Cancer group. Several studies have reported a significant dysregulation in the vaginal flora of patients with cervical cancer. For example, Sodhani et al. reported a similar link between precancerous lesions and vaginal dysbacteriosis dominated by anaerobic bacteria (Sodhani et al. Citation2017). Audirac-Chalifour et al. investigated cervical microbial bacteria in women with histopathologically confirmed cervical precancerous lesions and cancer and then determined significant differences in microbial diversity between the two groups (Audirac-Chalifour et al. Citation2016). They found a significant difference in microbiota diversity in non-cervical lesions (NCL)-HPV-negative women versus those with squamous intraepithelial lesions (SIL) and cervical cancer (p = 0.006 and p = 0.036). Upon evaluating the β diversity, they observed that the cervical cancer samples showed the highest variation within groups and the largest distance compared to NCL-HPV-negative ones and concluded that the cervical microbiota might be implicated in cervical cancer pathology. All in all, the homeostasis of vaginal microecology is considered to be closely related to the health of women.
To further investigate the differences in microecology among the four groups, we counted the abundance differences of colonies in the Cancer, Control, High, and Low groups at the phylum, family, class and species levels by ANOVA differences. The outcomes indicated that the abundance of Lactobacillus was gradually reduced at all four levels in cervical cancer from low-grade to high-grade lesions and to cancer. Studies have pointed out that Lactobacillus is the first line of defense against pathogens (He et al. Citation2012). Actually, Lactobacillus can produce many protective peptides and metabolites, such as lactic acid and other acidic compounds, to inhibit pathogen adhesion and growth. The metabolites produced by Lactobacillus can not only maintain Lactobacillus colonisation growth but also resist the adhesion and growth of other bacteria (Horn et al. Citation2019; Li et al. Citation2020). The above indicates that Lactobacillus plays a protective role in maintaining vaginal health. However, as demonstrated by LEfSe analysis, lactobacilli lost their dominant position in Cancer, High and Low groups, while levels of Corynebacterium, Bacteroides and other genera increased significantly. Nevertheless, a reduced number of Lactobacillus might lead to anaerobic overgrowth (Bacteroides, Clostridia) and enhance the development of cervical cancer lesions (Xu et al. Citation2021).
We also analysed the species correlation by Spearman correlation and discovered that when the advantages of Lactobacillus were lost, other genera would grow through mutual stimulation, causing adverse effects on health. These findings indicate that dysbacteriosis might be a cofactor in the development of cervical cancer lesions. In addition, we observed that the status of L. Iners-predominated cervicovaginal microbiome of Hakka women in Meizhou City had a low protective effect against cervicovaginal infections (Mitra et al. Citation2016). Therefore, women in this area should pay attention to the prevention and treatment of cervical cancer.
Lastly, we found a close correlation between the vaginal bacteria and metabolic disorders during cancer development based on KEGG analysis and COG function prediction, including energy metabolism, lipid metabolism, transport and catabolism and amino acid metabolism. Of them, energy metabolism presented a higher abundance in the Cancer group. High levels of energy metabolism can remarkably stimulate the proliferation and development of tumour cells, and the methods to inhibit energy metabolism are regarded as an effective treatment for cervical cancer (Yang et al. Citation2018). Moreover, lipid metabolism can enhance the proliferation and metastasis of cervical cancer cells (Yang et al. Citation2021). Given that the metabolism of cancer cells differs from that of normal cells and several metabolic disorder phenotypes observed in T2DM patients, we hypothesise that the vaginal microbiota affects the host through metabolites. Although the identification of certain species such as L. Iners predominates and functional profiles during CIN progression to cervical cancer might contribute to improving early diagnostics for patients with the precancerous disease, further studies should be performed to elucidate the specific roles of bacteria, pathways and orthologs for enhanced understanding of the role of the cervical microbiome in cervical carcinogenesis. Moreover, further exploration may benefit the development of construction plans for the prevention and treatment of cervical cancer from the perspective of microbial diversity.
Conclusion
Collectively, the type of vaginal community status of Hakka women in the Meizhou area is dominated by L. Iners, and the gradual loss of Lactobacillus dominance in vaginal bacteria is the key to microecological imbalance. Besides, disturbances in vaginal microecology will stimulate energy metabolism and lipid metabolism and then to induce cervical cancer. However, the samples in our study were relatively limited and came from a single centre, so some inherent biases might not be avoided. To fully elucidate the role of vaginal microbiota in the development of cervical cancer, large-scale longitudinal and interventional studies are needed. Therefore, future studies can provide more scientific basis for the clinical application of Lactobacilluss by exploring in depth whether Lactobacillus exerts its therapeutic effects through the energy metabolism and lipid metabolism pathways based on in vitro and vivo experiments.
Ethical approval
The study was approved by the Ethics Committee of Meizhou people’s Hospital(2019-C-83), and all patients involved in this study signed the informed consent form.
Authors’ contributions
Conceptualisation: Weihong Zeng, Lishan Huang and Boming Wu; data collection: Ye Liang, Jingqing Zhou and Yaoxiang Zhong; formal analysis and original draft preparation: Haihong Lin, Haochang Liu and Haikun Yang; review and editing: Danfeng He, Jizhong Wen and Nanxiang Lei. All authors have read and agreed to the published version of the manuscript.
Supplemental Material
Download Zip (1.1 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request.
Additional information
Funding
References
- Arbyn, M., et al., 2020. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health, 8 (2), e191–e203.
- Audirac-Chalifour, A., et al., 2016. Cervical microbiome and cytokine profile at various stages of cervical cancer: a pilot study. PLoS One, 11 (4), e0153274.
- Bik, E.M., et al., 2019. A novel sequencing-based vaginal health assay combining self-sampling, HPV detection and genotyping, STI detection, and vaginal microbiome analysis. PLoS One, 14 (5), e0215945.
- Bizzarri, N., et al., 2021. Peritoneal HPV-DNA test in cervical cancer (PIONEER study): a proof of concept. International Journal of Cancer, 148 (5), 1197–1207.
- Brotman, R.M., et al., 2014. Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. Journal of Infectious Diseases, 210 (11), 1723–1733.
- Colombo, N., et al., 2012. Cervical cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol, 23 (Suppl 7), vii27–vii32.
- Dellino, M., et al., 2022. Lactobacillus crispatus M247 oral administration: Is it really an effective strategy in the management of papillomavirus-infected women? Infect Agent Cancer, 17, 53.
- Derks, M., et al., 2018. Surgical treatment of early-stage cervical cancer: a multi-institution experience in 2124 cases in The Netherlands over a 30-year Period. International Journal of Gynecologic Cancer, 28 (4), 757–763.
- He, R.Y., et al., 2012. The research of health management for prevention and treatment of cervical persistent high-risk HPV Infection. Clinical Medicine & Engineering, 8, 89–92.
- Horn, L.C., et al., 2019. Prognostic relevance of low-grade versus high-grade FIGO IB1 squamous cell uterine cervical carcinomas. Journal of Cancer Research and Clinical Oncology, 145 (2), 457–462.
- Human Microbiome Project C. 2012. Structure, function and diversity of the healthy human microbiome. Nature, 486, 207–214.
- Kovachev, S.M., 2020. Cervical cancer and vaginal microbiota changes. Archives of Microbiology, 202 (2), 323–327.
- Lee, J.E., et al., 2013. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS One, 8 (5), e63514.
- Li, C., et al., 2022. Changes in the cervicovaginal microbiota composition of HPV16-infected patients after clinical treatment. Cancer Medicine, 11 (24), 5037–5049.
- Li, D., et al., 2020. Vaginal microbiome analysis of healthy women during different periods of gestation. Bioscience Reports., 40, BSR20201766.
- Martin, D.H., 2012. The microbiota of the vagina and its influence on women’s health and disease. American Journal of the Medicine Science, 343 (1), 2–9.
- Masson, L., et al., 2019. Inflammatory cytokine biomarkers of asymptomatic sexually transmitted infections and vaginal dysbiosis: a multicentre validation study. Sexually Transmitted Infections, 95 (1), 5–12.
- Meijer, C.J. and Snijders, P.J., 2014. Cervical cancer in 2013: screening comes of age and treatment progress continues. Nature Reviews Clinical Oncology, 11 (2), 77–78.
- Mitra, A., et al., 2016. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: what do we know and where are we going next? Microbiome, 4 (1), 58.
- Palma, E., et al., 2018. Long-term Lactobacillus rhamnosus BMX 54 application to restore a balanced vaginal ecosystem: a promising solution against HPV-infection. BMC Infectious Diseases, 18 (1), 13.
- Ravel, J., et al., 2011. Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences, 108 (supplement_1), 4680–4687.
- Seo, S.S., et al., 2016. Combined effect of diet and cervical microbiome on the risk of cervical intraepithelial neoplasia. Clinical Nutrition, 35 (6), 1434–1441.
- Smith, J.S., et al., 2003. Cervical cancer and use of hormonal contraceptives: a systematic review. Lancet, 361 (9364), 1159–1167.
- Sodhani, P., et al., 2017. Bacterial vaginosis and cervical intraepithelial neoplasia: is there an association or is co-existence incidental? Asian Pacific Journal of Cancer Prevention, 18 (5), 1289–1292.
- Sung, H., et al., 2021. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. A Cancer Journal for Clinicians, 71 (3), 209–249.
- Valenti, G., et al., 2017. Tumor markers of uterine cervical cancer: a new scenario to guide surgical practice? Updates in Surgery, 69 (4), 441–449.
- Xu, B., et al., 2021. Bacterial diterpene synthases prenylate small molecules. ACS Catalysis, 11 (10), 5906–5915.
- Yang, L., et al., 2018. Hypoxia-induced miR-214 expression promotes tumour cell proliferation and migration by enhancing the Warburg effect in gastric carcinoma cells. Cancer Letters, 414, 44–56.
- Yang, Y., et al., 2021. Lipid metabolism regulator human hydroxysteroid dehydrogenase-like 2 (HSDL2) modulates cervical cancer cell proliferation and metastasis. Journal of Cellular and Molecular Medicine, 25 (10), 4846–4859.