

Abstract
Background
Microbial colonisation in infants is initially dependent on the mother and is affected by the mode of delivery. Understanding these impacts is crucial as the early-life gut microbiota plays a vital role in immune development, metabolism, and overall health. Early-life infant gut microbiota is diverse among populations and geographic origins. However, in this context, only a few studies have explored the impact of the mode of delivery on the intestinal microbiome in children in Guangzhou, China. Therefore, this study aimed to investigate the influence of birth mode on the intestinal microbiota of healthy infants in Guangzhou, China.
Methods
Faecal samples were collected once from 20 healthy full-term infants aged 1–6 months, delivered via either caesarean section (CS) or vaginal delivery (VD), post-enrolment. The intestinal microbiota were characterised using full-length 16S rRNA gene sequencing. Bacterial quantity and community composition were compared between the two groups.
Results
No significant differences in gut bacterial diversity and richness were observed between the CS and VD groups. The Pseudomonadota phylum (44.15 ± 33.05% vs 15.62 ± 15.60%, p = 0.028) and Enterobacteriaceae family (44.00 ± 33.11% vs 15.31 ± 15.47%, p = 0.028) were more abundant in the CS group than in the VD group. The VD group exhibited a higher abundance of the Bacillota phylum (40.51 ± 32.77% vs 75.57 ± 27.83%, p = 0.019),
Conclusions
The early stage of intestinal bacterial colonisation was altered in the CS group as compared with the VD group. Our findings provide evidence that CS has the potential to disrupt the maturation of intestinal microbial communities in infants by influencing the colonisation of specific microorganisms. Further comprehensive studies that consider geographical locations are necessary to elucidate the progression of microbiota in infants born via different delivery modes.
PLAIN LANGUAGE SUMMARY
Microbial colonisation in infants is affected by the mode of delivery. Early-life infant gut microbiota is diverse among populations and geographic origins. Faecal samples were collected once from 20 healthy full-term infants aged 1–6 months that were delivered via either caesarean section (CS) or vaginal delivery (VD), and intestinal microbiota were compared between the two groups. No significant differences in gut bacterial diversity and richness were observed between the two groups; however, we did note that certain types of bacteria were more abundant in the CS group, while others were more abundant in the VD group. This suggests that CS may disturb intestinal microbial maturation in infants by affecting the colonisation of specific microorganisms. Further research is needed to fully understand this relationship.
Introduction
The intestinal microbiota is a complex micro-ecosystem composed of microorganisms living in a symbiotic relationship with their host that can impact host phenotypes later in life. Recent research has provided insight into the diverse functions of intestinal microbiota in chronic diseases including autoimmune, metabolic, and inflammatory diseases (Tilg et al. Citation2020). According to the literature, intestinal microbiota composition during early life is linked to the onset of diseases in childhood and at later stages of life (Healy et al. Citation2022), indicating that early colonisation by specific microorganisms can influence long-term health trajectories and predict health outcomes.
In a recent study with strong evidence, researchers collected foetal meconium using rectal swabs during selective breech caesarean section (CS), without labour and before antibiotic administration, and did not detect any signs of microbial life, confirming that intestinal microbial colonisation in the foetus is more likely to occur during or after birth but not before (Kennedy et al. Citation2021). Various factors can disturb the colonisation of intestinal microbiota. Among these, the mode of delivery is thought to be a major determinant. In recent years, the incidence of CS has markedly increased worldwide. The proportion of births via CS (estimated at 29.7 million) in 2015 was almost double the proportion in 2000 (21.1% vs 12.1%) (Boerma et al. Citation2018). There is abundant established evidence on the short- and long-term effects of CS on mothers and infants (Sandall et al. Citation2018, Keag et al. Citation2018). However, the effects of CS on the early stages of gut microbiota colonisation in infants remain controversial. Chu et al. compared samples collected from different body sites, including the nostrils, stool, skin, vagina, and oral gingiva. Analysis with whole-genome shotgun sequencing revealed that the infant bacterial community and functional capacity within the first 6 weeks of life were affected by body sites and not by the mode of delivery (Chu et al. Citation2017). Shao et al. showed that CS reduced the transmission of maternal bacteria (Bacteroides strains) and led to increased colonisation by Enterococcus, Enterobacter, and Klebsiella species, which are associated with the hospital environment. This suggests that the mode of delivery is a major factor affecting the composition of infant gut microbiota from the neonatal period to infancy (Shao et al. Citation2019). Geography (origin and location) also has a significant influence on the dominant bacterial community in the gut; different regions with diverse living environments, cultures, and dietary habits present a diversity of complex mechanisms for colonisation and development of gut microbiota. Thus, conflicting results observed in several previous studies can be partly attributed to variations in geographical location (Kemppainen et al. Citation2015, Sun et al. Citation2020). The geographic origin of subjects should be explicitly considered in microbiome studies. Guangzhou is a typical southern city in China, with a unique lifestyle, culture, diet habits, and environment; however, few studies have explored the impact of the mode of delivery on the intestinal microbiome of children in this single geographical location. Therefore, we aimed to determine the impact of birth mode on the gut microbiota of 1–6-month-old infants in Guangzhou, China.
Methods
Study design and participants
The participants and their data for the current study were taken from a cross-sectional study that investigated the intestinal microbiota of infants with dermatitis and healthy controls (Yu et al. Citation2021). Healthy, full-term, breastfeeding infants aged 1–6 months who were delivered via CS or VD were recruited during their routine physical examination. The following infants were excluded from the study: those receiving formula feeding or mixed feeding; premature infants; those with heart, liver, or kidney disease; those with intestinal, congenital genetic, blood system, endocrine, and metabolic diseases; and those who were administered probiotics or antibiotics within 1 month of sampling. Patients were divided according to mode of delivery into the CS and VD groups.
Sample collection and analysis
A single stool sample was collected from each infant post-enrolment for bacterial DNA extraction and subsequent microbiome analysis. Total genome DNA from samples was extracted using the cetyltrimethylammonium bromide/sodium dodecyl sulphate method. DNA concentration and purity were monitored on 1% agarose gels. According to the concentration, DNA was diluted to 1 ng/μL using sterile water (Wu et al. Citation2022). Polymerase chain reaction (PCR) was performed to amplify DNA samples using specific primers (5′-AGAGTTTGATCCTGGCTCAG-3′ and 5′- GNTACCTTGTTACGACTT-3′) to obtain the full-length 16S rRNA gene (Yu et al. Citation2021). All PCR reactions were carried out using TransStart® FastPfu DNA Polymerase (TransGen Biotech, Beijing, China). PCR amplification products were combined with the same volume of 1X loading buffer (containing SYB green) and electrophoresed on a 2% agarose gel. PCR products were mixed in equal density ratios and purified with the QIAquick Gel Extraction Kit (QIAGEN). Sequencing libraries targeting the full-length 16S rRNA gene were generated using the SMRTbell™ Template Prep Kit (PacBio) and assessed using the Qubit® 2.0 Fluorometer (Thermo Scientific) and FEMTO Pulse system, followed by sequencing on the PacBio Sequel platform. Raw data from the sequencing libraries were processed through the PacBio SMRT portal in the following steps: (1) sequences were filtered for a minimum of three passes and a minimum predicted accuracy of 90% (minFullPass = 3, minPredictedAccuacy = 0.9); (2) sequences outside the expected amplicon size (minLength = 1,340 bp, maxLength = 1,640 bp) were removed; (3) barcode and primer sequences were cut off; and (4) reads were compared with the reference database to detect chimaera sequences which were removed (Haas et al. Citation2011). After obtaining clean reads, we assigned sequences with a similarity of ≥97% to the same operational taxonomic unit (OTUs) using the UPARSE software (Uparse 7.0.1001, http://drive5.com/uparse/) (Edgar Citation2013). Based on Mothur’s algorithm, a representative sequence for each OTU was screened for further annotation using the SSU rRNA datasets of the SILVA database (https://www.arb-silva.de/) (Quast et al. Citation2013). Taxonomic information was obtained, and samples at kingdom, phylum, class, order, family, genus, and species levels were identified to show community composition.
The ‘Metagenomic Biomarker Discovery and Explanation’ approach with linear discriminant analysis (LDA) effect size algorithm (LEfSe) was used to analyse biomarkers for species with significant statistical differences between the groups. By calculating the correlation index (Spearman’s correlation coefficient or Pearson correlation coefficient) for all samples, the species correlation coefficient table was obtained, the absolute value of the correlation coefficient was filtered with a cut-off value of 0.6, and the map was combined with species abundance data.
Statistical analysis
Descriptive data are presented as mean ± standard deviation for continuous variables and as percentages for categorical variables. Differences in infant characteristics between the CS and VD groups were tested using the chi-squared test or t-test. The t-test was used to determine differences in average alpha diversity and relative bacterial abundance at each classification level between the groups. The Wilcoxon test was used to determine differences in beta diversity between the groups. The Adonis analysis was conducted to examine whether mode of delivery, gestational age at birth, maternal age, infant age, infant gender, and infant birth weight/height/head circumference had significant effects on the microbiome community composition. Statistical significance was set at p < 0.05. All data were processed using QIIME (v1.9.1), and analyses were performed using the R software (v2.15.3, http://www.R-project.org). This observational study was conducted in accordance with the ethical principles of the Declaration of Helsinki. Ethical approval for this study was granted by the Medical Research Ethics Committee of Guangdong Women and Children Hospital (No. 201,701,068). Written informed consent was obtained from each participant’s parents prior to stool sample collection.
Results
Characteristics of study participants
We recruited 20 healthy full-term infants (10 delivered via VD and 10 via CS) and collected 20 stool samples (). All infants were exclusively breastfed on demand without formula supplementation at the time of sampling. There were no differences in maternal age, gestational age, infant birth weight, infant birth height, and head circumference between the CS and VD groups. The sex ratio and the age of the infants were also similar between the two groups (). A total of 1,82,029 optimised reads and 176 OTUs were obtained from the 20 samples using full-length 16S rRNA sequencing. Out of all OTUs detected, 44 were unique to the CS group, 85 were unique to the VD group, and 47 were shared by both groups. The rarefaction curve and rank-abundance curve of the gut microbiota in different groups are shown in Figure S1.
Figure 1. Flow chart of the study. CS: caesarean section; N: number; VD: vaginal delivery.
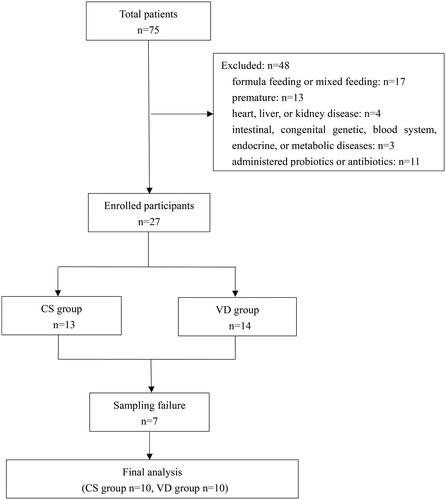
Table 1. Characteristics of infants included in the study.
Community composition of gut microbiome
Alpha diversity (19.41 ± 8.34 vs 28.27 ± 23.08) as measured by the ace estimator and Simpson’s diversity index (0.50 ± 0.26 vs 0.47 ± 0.30) were statistically insignificant (p = 0.796 and 0.971) (). Illustrations of principal coordinate analysis and weighted UniFrac distances between the CS and VD groups did not show far-apart clustering (). The clustering differences of samples by group were supported by analyses of molecular variance (p = 0.048). Based on the Adonis analysis using Bray-Curtis, the results indicate that none of the variables studied, including mode of delivery, gestational age at birth, maternal age, infant age, infant sex, and infant birth weight/height/head circumference, had a significant effect on beta diversity among the groups ().
Figure 2. Gut microbial alpha diversity and beta diversity in the CS (A) and VD (B) groups.Ace: abundance based coverage estimator; PC: principal component; PCoA: principal co-ordinates analysis. (a) Distribution of ace estimator, (b) Distribution of Simpson’s diversity index, (c) Principal coordinate analysis based on weight, (d) Weighted UniFrac metrics of faecal samples in the two groups.
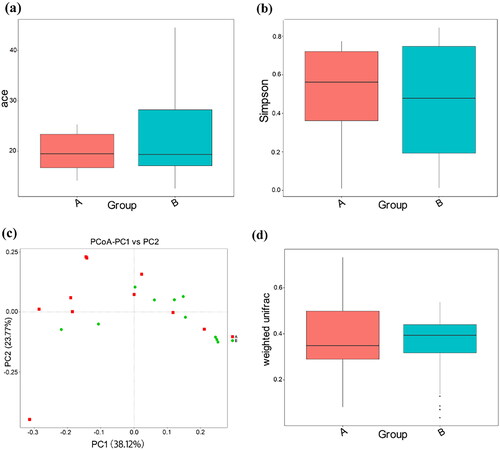
Table 2. Adonis analysis based on the Bray-Curtis.
The abundance of the relative taxa of predominant microbes at the phylum, family, genus, and species levels between the CS and VD groups is presented in and Figure S2. A total of nine phyla were found, and the three most dominant phyla in the CS and VD groups were Bacillota, Pseudomonadota, and Bacteroidota, collectively accounting for 99.64% and 95.36% of the entire community in the CS and VD groups, respectively. Additionally, rare bacterial phyla including Verrucomicrobiota, Actinomycetota, unidentified bacteria, Acidobacteriota, and Gemmatimonadota, were present in much lower proportions in both groups. A comparison of the taxonomic data revealed that infants in the CS group exhibited a relative underrepresentation of the Bacillota phylum and a relatively high abundance of the Pseudomonadota phylum. The differences included a 35.06% decrease in the relative abundance of Bacillota and a 28.53% increase in the relative abundance of Pseudomonadota in the CS group (p = 0.019 and 0.028, respectively) (). There were no significant differences in the abundance of other phyla including Bacteroidota between the CS and VD groups (p > 0.05).
Figure 3. Diagram of species differences between groups according to t-test at phylum (a) and family (b) levels. A: CS group, B: VD group.
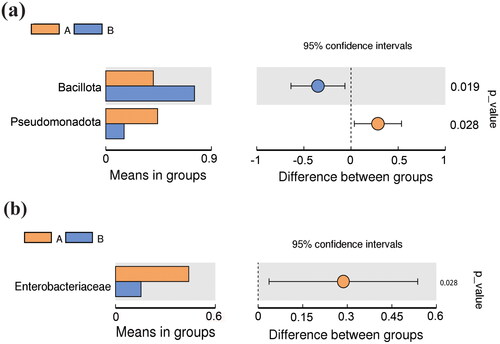
Table 3. Relative abundance of dominant bacterial taxa in the CS and VD groups.
At the family level, 41 bacterial families were identified, with 26 and 37 in the CS and VD groups, respectively. The CS and VD groups shared 23 bacterial families. The 10 predominant families were Enterobacteriaceae, Streptococcaceae, Bacteroidaceae, Veillonellaceae, Staphylococcaceae, Lachnospiraceae, unidentified Clostridiales, Akkermansiaceae, Micrococcaceae, and Tannerellaceae, collectively accounting for 91.76% and 78.36% of the total sequence in the CS and VD groups, respectively. At the family level, the CS group had higher levels of Enterobacteriaceae than the VD group (44.00 ± 33.11% vs 15.31 ± 15.47%, p = 0.028). With respect to the remaining bacterial families, relative taxa abundance was not significantly different between the CS and VD groups (p > 0.05) ().
Among the 63 genera identified, 33 and 57 were detected in the CS and VD groups, respectively. The two groups shared 28 genera. In contrast, 29 genera including Akkermansia and Lachnoclostridium were detected only in the VD group. The 10 predominant bacterial genera were unidentified Enterobacteriaceae, Streptococcus, Klebsiella, Bacteroides, Veillonella, Staphylococcus, unidentified Clostridiales, Akkermansia, Lachnoclostridium, and Rothia, collectively accounting for 88.95% and 74.20% of the total DNA sequence in the CS and VD groups, respectively. However, there were no differences in relative taxa abundance between the two groups at the genus level (p > 0.05).
At the species level, 65 species were identified. Of these, 41 and 47 were detected in the CS and VD groups, respectively. In contrast, 22 species were detected only in the VD group and 16 only in the CS group. Among the 10 predominant abundant species, excluding Clostridium butyricum, Bacteroides thetaiotaomicron, Bacteroides dorei, and Ruminococcus gnavus, each of the shared bacterial species accounted for >1% of the total DNA sequence in both groups. There were no significant differences in relative taxa abundance between the two groups at the species level (p > 0.05).
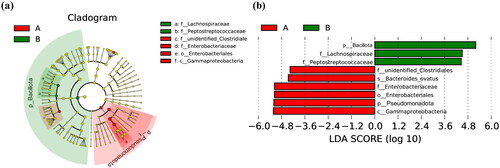
LEfSe was applied to determine the differences in relative bacterial abundance between the CS and VD groups and to assess the effect size of each differentially abundant taxon. The most differentially abundant taxa are illustrated in a cladogram () and by LDA coupled with effect size measurements (). A total of nine differentially enriched bacterial colonisers (six in the CS group and three in the VD group) were identified using LEfSe analysis. The analysis showed that the unidentified Clostridiales family, Bacteroides ovatus species, Enterobacteriaceae family, Enterobacteriales order, Pseudomonadota phylum, and Gammaproteobacteria class were enriched in the CS group. Sequences from the Bacillota phylum, Lachnospiraceae family, and Peptostreptococcaceae family were enriched in the VD group.
Figure 4. Cladogram plot (a) and linear discriminant analysis (LDA) score plot (b) from LDA effect size algorithm (LEfSe) analysis of the intestinal microbiota composition in the CS (A) and VD (B) groups. Concentric rings from inside to outside represent phylum, class, order, family, and genus, in order. The red circle represents biomarkers in group A, and the green circle represents biomarkers in group B. LEfSe identifies the most differentially abundant taxa between the two groups. Enriched taxa in group A are indicated with a positive LDA score (red), and enriched taxa in group B have a negative score (green). The taxa that meet the LDA significant threshold exceeding four are shown. The length of the histogram represents the impact of different species (LDA score). A: CS group, B: VD group. LDA: linear discriminant analysis.
Discussion
We found that the intestinal microbiota compositions in these infants were dominated by Bacillota, Pseudomonadota, and Bacteroidota at the phylum level, regardless of the mode of delivery. The microbiota in the CS group had a lower abundance of the Bacillota phylum and a higher abundance of the Pseudomonadota phylum. There was no significant difference in the abundance of Bacteroidota between the CS and VD groups. However, the findings of previous studies have shown different results. For instance, Murata C et al. reported that faecal samples collected from CS-delivered infants at 3–4 months after birth had a high abundance of Firmicutes and a low abundance of Bacteroidetes, resulting in a high Firmicutes/Bacteroidetes ratio in Mexico (Murata et al. Citation2020). A significantly low abundance of Bacteroides in CS-delivered infants during their first year of life has also been reported by Bokulich NA et al. in the USA (Bokulich et al. Citation2016). The gut microbiota during infancy is derived from various sources, is affected by various determinants, and is highly dynamic. This microbiota undergoes changes in diversity, stability, and maturation with temporal patterns that are unique to each infant (Thursby & Juge Citation2017). Galazzo G et al. longitudinally analysed gut microbial composition in 440 infants born via CS or VD at 5, 13, 21, and 31 weeks of life and in children aged 6–11 years using amplicon sequencing. This study showed that the variance in microbial composition was explained by the mode of delivery at 5 weeks of life, whereas feeding mode became a more significant factor affecting variance in the bacterial community at 13 weeks (Galazzo et al. Citation2020). The heterogeneity between our study and previous studies can be partly attributed to the diversity in age during sampling (Bokulich et al. Citation2016, Murata et al. Citation2020). In our study, we reported that the abundance of the Enterobacteriaceae family was higher in the CS group than in the VD group. This is consistent with the findings of Lundgren et al. who stated that CS-delivered infants had a higher abundance of the Enterobacteriaceae family than VD-delivered infants (Lundgren et al. Citation2018).
As nosocomial acquisition of hospital strains of Enterobacteriaceae in neonates is not uncommon in hospitals, it is a possible factor in our findings (Murono et al. Citation1993). A high abundance of facultative anaerobic Enterobacteriaceae indicates delayed maturation of the gastrointestinal microbiota in infants (Sim et al. Citation2015). A higher relative abundance of Enterobacteriaceae in the stool sample of CS-delivered infants suggests that CS disturbs the intestinal microbiota composition or abundance. However, some studies have shown that inter-individual variation of early development of children’s gut microbiota caused by delivery mode decreased significantly over time. A systematic review by Rutayisire et al. suggested that differences in gut microbiota diversity and colonisation patterns that are significantly associated with delivery mode are observed only in the first three months of life and disappear after 6 months of infant life (Rutayisire et al. Citation2016). A prospective cohort study found similar results, with marked changes in gut microbiota composition at 1 week and 1 month of age for caesarean delivery, but with only minor differences persisting by 1 year of age compared to vaginal delivery (Stokholm et al. Citation2020).
CS disturbs normal colonisation of the infant intestinal microbiota, whereas exposure to breast milk promotes a ‘healthy’ intestinal microbiota. Exclusive breastfeeding can partially restore intestinal microbiota in CS-delivered infants, as confirmed by Liu et al.(Liu et al. Citation2019). The duration of breastfeeding has a strong impact on the restoration of a health-promoting microbiota composition. In a recent study, the abundance of Lactobacillus and Bacteroides fragilis increased with the duration of breastfeeding in CS-delivered infants (Coker et al. Citation2021). The insignificant difference in the abundance of Bacteroidota between the two modes of delivery may be due to the long duration of breastfeeding in both groups. The abundance of Lactobacillus was not significantly different between the CS and VD groups in our study, although the most commonly distributed bacterial genus in the mother’s vagina (Komesu et al. Citation2020). This supports the opinion that infant microbiota colonises not only via vaginal microbiota seeding but also via maternal skin/stool seeding and/or environmental seeding (Reyman et al. Citation2019). Additionally, the introduction of solid foods and the composition of the infant’s diet may also impact the microbiota composition and diversity. The introduction of solid foods during infancy will cause profound shifts in the gut microbial composition of infants towards a more adult-like state (Homann et al. Citation2021).
Among the many factors that jointly explain the diversity of gut bacterial composition, geographical location is an important factor in the dominant gut bacterial community and has an even greater influence than ethnicity (Lin et al. Citation2020). A recent study found that when a subject travelled from a developed country to a developing country, there was an increase in the Bacteroidetes/Firmicutes ratio in the gut. This suggests that visits to other geographic locations, even for a short duration, can influence the gut microbial community structure (David et al. Citation2014). Recruiting both CS-delivered and VD-delivered infants within the same geographic location may have contributed to our results.
Our study has some limitations in showing the relationship between infant gut microbiota and mode of delivery. First, the small sample size reduced the power of the analysis. Second, stool samples were collected at only one time point, and there was a lack of longitudinal samples from every infant. Future studies should assess the infant gut microbiota serially to reveal its composition over time. Third, we did not sample the breast milk or maternal vaginal microbial communities for comparison with the infant gut microbiota, and therefore could not comment on how or if they were related. Fourth, the lack of clinical information on maternal factors, such as the use of antibiotics and the reason for delivery by CS, hampers the interpretation of the results.
Conclusions
Our study indicates that the early stage of infant intestinal bacterial colonisation is affected by the mode of delivery. CS may disturb the intestinal microbial maturation by affecting the colonisation of specific microorganisms, which varies by geographic location. To address the limitations of the present study, future investigations should be designed to thoroughly characterise and understand microbiota succession in infants with different modes of delivery.
Author contributions
Li Yu, Yong Guo, and Jie-Ling Wu contributed to the conception of the study. Li Yu and Yong Guo performed the experiment. Li Yu contributed significantly to the analysis and manuscript preparation. Li Yu and Yong Guo performed the data analyses and wrote the manuscript. Jie-Ling Wu provided valuable input during the analysis and discussions.
Supplemental Material
Download TIFF Image (14.3 MB)Supplemental Material
Download TIFF Image (352.2 KB)Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Disclosure statement
The authors have declared that no competing interest exists.
Data availability statement
The datasets that support the findings of the current study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Boerma, T., et al., 2018. Global epidemiology of use of and disparities in caesarean sections. Lancet, 392 (10155), 1341–1348.
- Bokulich, N. A., et al., 2016. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Science Translational Medicine, 8 (343), 343ra82.
- Chu, D. M., et al., 2017. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nature Medicine, 23 (3), 314–326.
- Coker, M. O., et al., 2021. Infant feeding alters the longitudinal impact of birth mode on the development of the gut microbiota in the first year of life. Frontiers in Microbiology, 12, 642197.
- David, L. A., et al., 2014. Host lifestyle affects human microbiota on daily timescales. Genome Biology, 15 (7), R89.
- Edgar, R. C., 2013. Uparse: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10 (10), 996–998.
- Galazzo, G., GI-MDH consortium., et al., 2020. Development of the microbiota and associations with birth mode, diet, and atopic disorders in a longitudinal analysis of stool samples, collected from infancy through early childhood. Gastroenterology, 158 (6), 1584–1596.
- Haas, B. J., Human Microbiome Consortium., et al., 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research, 21 (3), 494–504.
- Healy, D. B., et al., 2022. Clinical implications of preterm infant gut microbiome development. Nature Microbiology, 7 (1), 22–33.
- Homann, C.-M., et al., 2021. Infants’ first solid foods: Impact on gut microbiota development in two intercontinental cohorts. Nutrients, 13 (8), 2639.
- Keag, O. E., Norman, J. E. and Stock, S. J., 2018. Long-term risks and benefits associated with cesarean delivery for mother, baby, and subsequent pregnancies: Systematic review and meta-analysis. PLoS Medicine, 15 (1), e1002494.
- Kemppainen, K. M., TEDDY Study Group., et al., 2015. Early childhood gut microbiomes show strong geographic differences among subjects at high risk for type 1 diabetes. Diabetes Care, 38 (2), 329–332.,
- Kennedy, K. M., et al., 2021. Fetal meconium does not have a detectable microbiota before birth. Nature Microbiology, 6 (7), 865–873.
- Komesu, Y. M., Eunice Kennedy Shriver National Institute of Child Health and Human Development Pelvic Floor Disorders Network., et al., 2020. Defining the relationship between vaginal and urinary microbiomes. American Journal of Obstetrics and Gynecology, 222 (2), 154.e1–154.e10.,
- Lin, D., et al., 2020. The core and distinction of the gut microbiota in Chinese populations across geography and ethnicity. Microorganisms, 8 (10), 1579.
- Liu, Y., et al., 2019. The perturbation of infant gut microbiota caused by Cesarean delivery is partially restored by exclusive breastfeeding. Frontiers in Microbiology, 10, 598.
- Lundgren, S. N., et al., 2018. Maternal diet during pregnancy is related with the infant stool microbiome in a delivery mode-dependent manner. Microbiome, 6 (1), 109.
- Murata, C., et al., 2020. Delivery mode-associated gut microbiota in the first 3 months of life in a country with high obesity rates: A descriptive study. Medicine, 99 (40), e22442.
- Murono, K., et al., 1993. Acquisition of nonmaternal Enterobacteriaceae by infants delivered in hospitals. The Journal of Pediatrics, 122 (1), 120–125.
- Quast, C., et al., 2013. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research, 41 (Database issue), D590–D596.
- Reyman, M., et al., 2019. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nature Communications, 10 (1), 4997.
- Rutayisire, E., et al., 2016. The mode of delivery affects the diversity and colonization pattern of the gut microbiota during the first year of infants’ life: A systematic review. BMC Gastroenterology, 16 (1), 86.
- Sandall, J., et al., 2018. Short-term and long-term effects of caesarean section on the health of women and children. Lancet (London, England), 392 (10155), 1349–1357.
- Shao, Y., et al., 2019. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature, 574 (7776), 117–121.
- Sim, K., et al., 2015. Dysbiosis anticipating necrotizing enterocolitis in very premature infants. Clinical Infectious Diseases, 60 (3), 389–397.
- Stokholm, J., et al., 2020. Delivery mode and gut microbial changes correlate with an increased risk of childhood asthma. Science Translational Medicine, 12 (569), eaax9929.
- Sun, S., et al., 2020. Does geographical variation confound the relationship between host factors and the human gut microbiota: A population-based study in China. BMJ Open, 10 (11), e038163.
- Thursby, E. and Juge, N., 2017. Introduction to the human gut microbiota. The Biochemical Journal, 474 (11), 1823–1836.
- Tilg, H., et al., 2020. The intestinal microbiota fuelling metabolic inflammation. Nature Reviews. Immunology, 20 (1), 40–54.
- Wu, Z., et al., 2022. The microbiome of lower respiratory tract and tumor tissue in lung cancer manifested as radiological ground-glass opacity. Frontiers in Bioengineering and Biotechnology, 10, 892613.
- Yu, L., et al., 2021. Comparison of gut microbiota between infants with atopic dermatitis and healthy controls in Guangzhou, China. Journal of Asthma and Allergy, 14, 493–500.