
ABSTRACT
Traumatic, inherited, and age-related degenerative diseases of the retina, such as retinal detachment, retinitis pigmentosa, and age-related macular degeneration, are characterized by the irreversible loss of retinal neurons. While current treatments aim to prevent neuronal degeneration, there are no available treatments to restore neurons after loss. Cultured murine neuroretinal tissue explants model retinal injury and offer a high throughput approach to identify experimental interventions capable of regenerating neurons. Formalin-fixed paraffin-embedded (FFPE) preparations of murine neuroretinal explants can be used to identify cells throughout the retinal layers to provide information on proliferation and activity following exposure to therapeutics. However, retinal explants are friable, particularly after ex vivo culture, sample handling and FFPE processing steps can result in tissue loss and damage. Friability also prohibits bisecting samples post-culture to display more than one region of interest for analysis. We developed a sample handling and embedding technique for cultured murine neuroretinal explants using HistogelTM in combination with a post-processing trimming step that eliminates tissue loss, increases cross-sectional retinal representation, and captures proximal and central retina on one slide to facilitate analysis of explants subjected to neurotrophic compounds.
Introduction
Explant culture is a technique in which tissues or whole organs are removed from plants or animals and cultured ex vivo. Explant cultures bridge the gap between using dissociated cells in culture (in vitro) and using animal models (in vivo). Dissociated cell cultures can be used for high throughput screening and offer a high degree of experimental control. However, they typically only provide information on a single-cell type’s response to experimental manipulation and do not maintain the tissue architecture and cell interactions seen in vivo. Animal models show high fidelity, but can be time consuming, may take long periods of time to develop disease or injury, and can require large numbers of animals to validate findings. Additionally, there is a lack of precise control over experimental conditions, and investigators must consider pharmacodynamics in whole animals [Citation1,Citation2].
Ex vivo tissue explants supply a physiologic environment that maintains the tissue architecture present in vivo while allowing for a range of experimental manipulation in a controlled environment. The retina is a highly organized tissue consisting of layers of cells whose specialized interactions are vital to perform visual signal transduction. Retinal explants maintain this specialized structure ex vivo, and explants from various species and ages can be used to study retinal development, neurodegeneration, neuroprotection, neuroregeneration, and genetic modification [Citation1–3]. Many retinal diseases result from cellular loss or the disruption of cell–cell interactions, and explants may serve as a platform to screen novel therapeutics for the treatment of retinal diseases.
Rodent retinal explants are commonly harvested due to the lack of available human eye tissues, similarities between rodent and human retinas, and the ability to manipulate genes in rodent models [Citation4]. Explants are isolated by dissecting the retinal sheet from the underlying retinal pigment epithelium (RPE) and are cultured photoreceptor side down on a microporous membrane within a tissue culture well containing media [Citation5–7]. Incisions are made to allow the naturally cup-shaped retina to lay flat along the membrane, resulting in a flower-like structure. To assess experimental treatments for neuroregeneration, explants are harvested from mature mice and act as a natural injury model as mature cells show signs of degeneration within less than 4 days in culture. Explants from immature mice, used in retinal development studies, show better cytoarchitecture, display signs of normal cellular development, and are viable for longer periods of time in culture [Citation1,Citation8]. Variations in culture conditions and duration of culture can further affect explant quality.
Retinal explants can be analyzed by techniques including whole mount staining, electrophysiology, western blot, or histology [Citation3,Citation6,Citation9,Citation10]. For histologic analysis, retinal explants from mice can be fixed in 4% paraformaldehyde and immersed in graded sucrose solutions for cryosectioning [Citation11–14] or processed for formalin-fixed paraffin-embedded (FFPE) sectioning [Citation5]. Due to degeneration ex vivo as well as effects of culture conditions, retinal explants are friable, and sample handling and processing required in FFPE preparations can cause further damage and loss. The unwieldy flower-shaped explant must be embedded on edge for cross-sectional analysis, and, if the sample is too friable to bisect, it must be embedded along one petal edge, limiting on slide display to peripheral retina only.
In order to overcome the friable nature of the explants and provide cross sections of the entire retina, we introduced the use of HistogelTM (#HG-4000-012, Richard-Allan Scientific, USA) in a 2-step embedding method. Gel-based preparations such as agarose or HistogelTM are used to create cell blocks from fine needle aspirates, urine, and non-gynecologic specimens as well as cell pellets from needle biopsies or fragmented tissues in the clinical laboratory [Citation15]. HistogelTM is primarily composed of hydroxyethyl agarose and appears as a translucent pink liquid at 60°C and as a clear pink solid at room temperature (RT). HistogelTM encapsulates specimens or fluids while allowing processing chemicals to penetrate. It does not retain histology stains that could easily cause background discoloration on slides and sections. For small, friable veterinary tissues, such as routine biopsies of the nasal turbinates, gastrointestinal tract, and cerebellum, HistogelTM was shown to provide better tissue support and orientation compared to non HistogelTM-embedded FFPE samples. Additionally, HistogelTM-embedded tissue blocks were noted to be easy to section, and there was no difference in immunoreactivity compared to non HistogelTM-embedded FFPE tissues [Citation16].
In our laboratory, we routinely use HistogelTM to create cultured cell line pellets and decided to apply a similar approach to retinal explants. Here, we describe our previous retinal explant embedding and processing approaches and the new HistogelTM method. We recorded total sample loss and damage, scored stained slides for quality metrics, and used image analysis to quantify differences in total retinal area and length across three methods. We demonstrate that use of Histogel consistently eliminates tissue loss, greatly reduces tissue fragmentation, captures full retinal layer thickness, and captures more tissue area and retinal length per slide.
Materials and methods
Retinal explant harvest and culture
Mice used for explant experiments were housed in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). All animal procedures were conducted under a protocol approved by the Institutional Animal Care and Use Committee in accordance with the Guide for the Care and Use of Laboratory Animals and applicable laws and regulations.
Retinal explants were isolated and cultured as previously described [Citation8,Citation17,Citation18]. Eyes were enucleated from male and female 6-8 week-old, wild-type 129X1/SvJ or Rosa26.LSL.tdTomato.cki._RLBP1.Cre.ERT2.tg mice. A circumferential incision was made in the cornea using Vannas Spring Scissors (#15018-10, Fine Scientific Tools, USA), and the retina was ejected from the globe by applying pressure posteriorly to anteriorly to the back of the eye. The neural retina was isolated from any remaining ciliary body, iris, or lens and cut into a flower shape with four petals by making four 1–2 mm radial incisions from the periphery towards the central retina with Vannas Spring Scissors (). Each explant measured approximately 4 mm in diameter and 160–180-μm in thickness, and each petal was about 2 mm wide. A widened pipette tip (3 mm diameter; #30389212, Rainin, USA) was used to transfer the retina onto a tissue culture insert containing a microporous membrane (0.4 μM, 30 mm diameter; PICM0RG50, Millipore, USA) with the ganglion cell layer facing up. The insert with the sample was placed into a 6-well plate (#3516, Corning Inc., USA), and media was added under the insert membrane to establish an air-liquid interface. Each well contained 1 ml of culture media: 50% MEM/HEPES (#12360-038, Thermo Fisher Scientific, USA), 25% HBSS (#14025-092, Thermo Fisher Scientific), 25% heat-inactivated horse serum (#26050088, Life Technologies, USA), 200 μM L-glutamine (#25030-081, Thermo Fisher Scientific), and 5.75 mg/ml glucose (A24940-01, Thermo Fisher Scientific). Media changes were performed every other day, and explants were maintained at 34°C in a humidified atmosphere of 5% CO2 and 95% air. Most explants were cultured 4 days, treated with test compounds or a vehicle control, then fixed, processed, embedded, sectioned, and evaluated for morphology by routine hematoxylin and eosin (H&E) staining. A small subset of samples was harvested with no or minimal (4 h) culturing and processed for FFPE to use as a baseline for comparison to cultured samples.
Figure 1. Schematic representation of murine retinal explant culture and three different sample handling and embedding methods. (a) The neural retina was isolated from the enucleated mouse eye as a cup shape and four radial incisions made to flatten the retina into a flower shape. The retinal explant was cultured on a microporous membrane of a cell culture insert with the retinal ganglion cell (RGC) layer facing up. (b) The initial two approaches to sample processing involved three sample handling steps, including transfer to a 24-well plate for fixation, transfer to a hard plastic mesh cassette (Method #1) or biopsy cassette insert (Method #2) for processing, and positioning the whole explant along one petal edge for embedding. (c-i) The Histogel embedding method (Method #3) eliminated sample handling by fixing the explants on the culture inserts and transferring the entire insert onto a cold plate (d). Histogel was pipetted directly onto and encapsulated the explant (e). A weighing spatula was used to transfer the embedded explant onto a biopsy wrap, which was folded over and placed into a cassette (f). Following processing, the Histogel was clear and firm (g). A razor blade was used to bisect the explant so that each half contained central and peripheral retina (h). Each half was embedded on edge diagonally in an embedding mold (i).
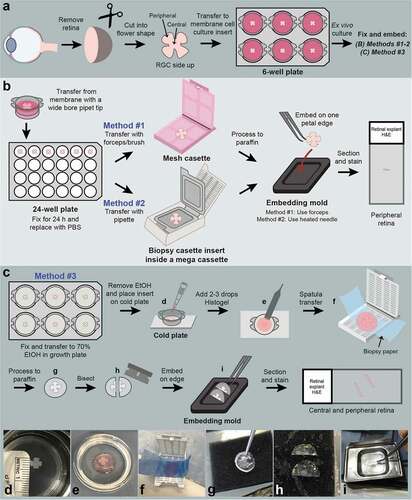
Traditional histology embedding methods
For initial retinal explant studies, samples were transferred from the microporous culture membrane with a widened pipette tip (3 mm diameter) into a 24-well plate (#353047, Corning Inc.) and fixed in 10% neutral buffered formalin (NBF) (#16004, VWR, USA) at RT for 24 h (). 10% NBF was replaced with Dulbecco’s Phosphate Buffered Saline (DPBS) (#55-031-PC, Corning Inc.) for 1 day at RT during transit to the Histology Core Laboratory in order to limit formaldehyde cross-linking that can affect epitopes in subsequent immunofluorescence assays. Samples were then processed using one of two methods:
Method #1: Samples were transferred to a hard plastic mesh cassette (#4306, Sakura Finetek, USA) using forceps (High Precision Straight Broad Strong Point Forceps, #12-000-128, Fisher Scientific, USA) or an artists’ brush (#1).
Method #2: The end of a disposable transfer pipette (#16001-180, VWR, USA) was cut with a razor blade to enlarge the diameter to ~3–4 mm. The explant was gently aspirated into the pipette containing 0.25 ml PBS and transferred to an open biopsy cassette insert (CellSafe+ Biopsy Capsule, #EBE-0201-02A, CellPath, UK), which has a hinged frame with extra fine nylon mesh to secure samples. The support of the mesh allowed the PBS to pool underneath the sample so tissue folds could be gently teased out with an #1 artist brush. The open biopsy insert was placed on top of a light-weight towel (WypAll L30, Kimberly Clark, USA) to absorb excess PBS and help the sample flatten, then closed and placed inside of a labeled mega-cassette (#4173, Sakura Finetek).
Samples from Methods #1 and #2 were processed through 40 min incubations on a Sakura Finetek Tissue-Tek VIP 5 automated tissue processor programmed for no agitation or vacuum to limit disruption of friable samples (see Supplementary Table 1 for processing protocol and reagent details). Flex reagents (Epredia, USA), which are patented formulations of methyl and isopropyl alcohol, were used in place of reagent alcohols as they can reduce excess tissue dryness and facilitate better sectioning.
Following processing, the mesh cassette or the biopsy insert within the mega-cassette was removed from the processor and opened. One ml of molten paraffin was dispensed into a 24 × 24 × 5 mm embedding mold (#4163, Sakura Finetek) on the 70°C hot surface of the HistoStar Embedding Center (A81010100, Thermo Scientific). The cassette or biopsy insert containing the sample was inverted and submerged into the mold to allow the sample to float into the paraffin. For the mesh cassettes (Method #1), more paraffin was dispensed on top to facilitate sample transfer into the mold, taking care not to let the samples float out of the mold. The biopsy inserts (Method #2) were gently tapped with the the straight, broad point forceps to allow the sample to float out. Once in the mold, samples were gently supported by forceps (Method #1) or a heated needle tip from a 1 ml tuberculin syringe (#309626, Becton Dickinson, USA) (Method #2) while the bottom third of the mold was placed on the 5°C cold spot at a 30° angle. As the paraffin hardened towards the sample, the forceps or needle was used to orient the explant along one edge of the petal (~2 mm). The cassette lid was placed on top of the mold, topped off with paraffin, and placed on the cold plate for 15 min or until it was easy to remove the block for sectioning.
HistogelTM embedding method
In our new embedding method using HistogelTM (Method #3), explants were fixed on the tissue culture inserts in the 6-well plate by adding 10% NBF below and above the insert. Samples were fixed at RT overnight. After fixation, the samples were rinsed with PBS, and the wells were filled with 70% ethanol (EtOH) (KOPTEC #V1401, Decon Labs, Inc., USA) just to the top level of the membrane (). Samples were left in 70% EtOH at 4°C for 1 day. On the day of embedding, Histogel was removed from refrigeration and heated in a 60°C oven for approximately 3 h until molten, then kept in an insulating block. The tissue culture plate was placed on the 5°C cold plate of the embedding center. A #1 artist brush was used to gently orient each sample into the middle of the membrane in preparation for Histogel embedding. A transfer pipette was used to aspirate 70% EtOH from the bottom of the well between the membrane and the well to not disturb the tissue. The insert was then lifted from the well with fine forceps (High Precision Straight Slender Fine Point Forceps, #12-000-127, Fisher Scientific) and blotted on an absorbent pad to remove excess 70% EtOH. The insert was placed directly onto the cold plate after first scraping any ice from this embedding center surface to better visualize the retinal explant against the darker blue cold plate surface. Two to three drops (0.1 to 0.15 ml) of molten HistogelTM were pipetted directly onto the top of the explant. Adding more HistogelTM can make the explant more difficult to see to bisect evenly. Since the explant was not adherent to the membrane, some HistogelTM flowed under the sample, encapsulating the retinal explant (). The HistogelTM was left to harden on the cold plate for 5 min until the HistogelTM was set. A weighing spatula was used to lift the embedded explant and transfer it onto a 2” × 3” biopsy wrap (Surgipath Bio-Wraps, #3801090, Leica Biosystems, USA), which was then folded over letter style and placed into a cassette (#8170, Sakura Finetek) (). Samples were placed in 70% EtOH and processed using the same protocol as described for the other methods (Supplementary Table 1).
Following processing, samples were removed from the biopsy wrap and prepared for paraffin embedding. The processed HistogelTM was still translucent but turned clear rather than pink, and the retinal sample remained encapsulated in the center of the HistogelTM (). The samples were firm enough to manipulate with forceps, but not hardened or friable, which can occur during dehydration steps in processing, particularly if Flex reagents are not used.
The straight broad point forceps or Curved Medium Point (#16-100-110, High Precision, Fisher Scientific) were used to pick up the edge surrounding the encapsulated sample and move it onto the 70°C hot spot of the embedding center. Prior to processing, the contrast provided by the pink HistogelTM made it easy to center and embed the retinal explant; however, after processing, it was somewhat difficult to visualize the sample in the clear HistogelTM. If the sample was left to cool, the HistogelTM became opaque, and the sample was not visible. The flower shape of the retinal explant became visible within the HistogelTM as it warmed, but the embedding center warming plate did not provide enough contrast, and a jeweler’s eye loupe was needed to ensure even bisecting. A 0.23 mm razor blade (Personna #94-0140, AccuTec Blades, Inc., USA) was used to bisect the explant flower such that each half contained central and peripheral retina. Each half was lifted and transferred onto a biopsy pad (Black Foam, #339014, Bioindustrial Products, USA) (). A new blade was used for each sample. The raised surface of the biopsy pad facilitated easy transfer of both explant halves, one at a time, to an embedding mold using forceps, while being careful not to crush the tissue inside the gel (). The first half was embedded on edge diagonally by dispensing a small amount of molten paraffin into the mold, placing the mold onto the embedding center cold spot and allowing the sample to anchor to the hardening paraffin. The mold was then briefly removed from this cold spot so the paraffin remained soft enough to embed the second half parallel to the first in the same plane for sectioning. The mold with a cassette lid was placed onto the cold plate as described previously.
Sectioning and staining
FFPE blocks were soaked on icy water for 5 min and sectioned at 4 μm on a Leica HistoCore AUTOCUT microtome (Leica Microsystems, USA) using low profile, disposable microtome blades (#4689, Sakura Finetek). Sections were placed on a 47°C water bath (TFB-S, Triangle Biomedical Sciences Inc, USA) for up to 1 min in order to flatten folds in the explant.
Sections were lifted onto SuperFrost PlusTM Slides (#6776214, Epredia), air dried in racks on top of a 60°C hot plate for 20 min, then placed in an Isotemp Incubator (Model 1602D, #11-690-650D, Fisher Scientific) at 60°C for 20 min. Slides were regressively stained with Gill’s Hematoxylin III (HXGHE, American Master Tech, USA), as suggested by the manufacturer, and counterstained with Eosin Y (STE0157, American Master Tech) on a Leica AutoStainer XL.
Sample cohort
The samples in this study consisted of 244 retinal explants submitted for processing, embedding, and sectioning over the course of 20 consecutive experiments during a 2-year period. Among minimally cultured or non-cultured samples, there were 6 samples embedded with Method #1 and 12 samples with Method #2 from one experiment each and 17 samples embedded with Method #3 from two independent experiments. Among 4-day cultured samples, there were n = 12 samples embedded with Method #1 across two independent experiments, 167 samples from Method #2 across eleven independent experiments, and 30 samples from Method #3 across three independent experiments. H&E sections from 94% (229/244) of samples were available for histologic evaluation. Fifteen samples embedded using Method #2 (2 minimally cultured samples and 13 cultured samples) were lost during processing, embedding, or sectioning.
Whole slide imaging and slide scoring
All slides were scanned at 20x magnification over a period of five months on a NanoZoomer S360 Digital slide scanner (#C13220-01, Hamamatsu, USA). The scanner’s automated focus point software assisted in focus point selection during scanning. After a quality control pass, revisions were performed on folds, instances of blur, and lightly stained tissue. To assess the quality of each embedding method, two independent readers (S.H. and E.S.) scored the digital H&E-stained slides of mouse retinal explants for the following features: (1) fragmentation of sample into multiple pieces, (2) proper tissue orientation (i.e. on edge), (3) presence of contiguous retinal layers without separation between individual layers, and (4) presence of a full thickness retinal cross section. To evaluate fragmentation, the following metric was used: fragmentation value = number of fragments observed/number of fragments submitted for processing and embedding (one in Methods #1 and #2, two in Method #3). This normalized value accounted for differences in the number of fragments embedded in Methods #1 and #2 (one fragment) versus Method #3 (two bisected halves). The fragmentation value was then used to assign a fragment score: ‘none/mild’ for 1–2, ‘moderate’ for 3–5, ‘severe’ for 6–10, and ‘extreme’ for >10. The remaining three metrics were scored based on the percent of tissue on the slide that exhibited that feature: ‘none’ for 0%, ‘few’ for >0–50%, ‘partial’ for >50–75%, ‘most’ for >75% but <100%, and ‘all’ for 100%. Evaluation of a full retinal cross section was based on the presence of the outer nuclear layer (ONL), the inner nuclear layer (INL), and the retinal ganglion cell (RGC) layer. The RGC layer was assessed as present even if minimal since this layer varied in cellularity due to the degenerative nature of the explant model. Supplementary Table 2 summarizes these metrics across 229 total slides evaluated across all three methods, including 33 samples that were immediately embedded after isolation (not cultured) or cultured only for 4 h, and 196 samples cultured ex vivo for 4 days.
Quantitative image analysis
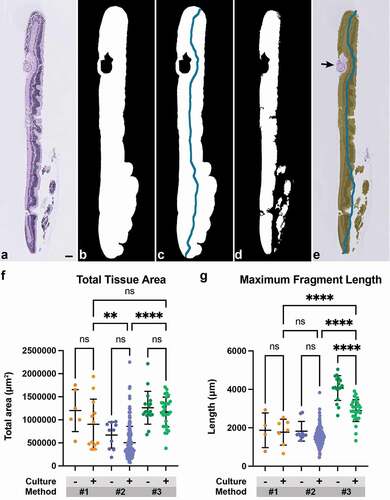
Total tissue area and maximum fragment length were captured for each H&E-stained sample using Python’s scikit-image package [Citation19] (Supplementary Table 3). The area was obtained by converting images to grayscale and Otsu thresholding. Through a series of morphological openings and closings, a binary mask of area was produced (). A topological thinning was performed on this mask, followed by pruning [Citation20] to obtain a filament representing tissue length for each fragment on the slide (). To remove noise, large whitespacing due to tissue fragmentation that interfered with and artificially inflated the area and length were removed and recorded throughout the stages of the stated process (). Additional artifacts on the slide, including fragments of lens capsule or non-cellular debris, were manually annotated and excluded from the captured area (). The code used to perform image analysis can be found on GitHub at https://github.com/kamcbk/retinal-explant-section-metrics. Total tissue area and the maximum fragment length were compared between cultured and non-cultured samples within an individual method and across embedding methods for cultured samples. One-way ANOVA with post-hoc Šidak’s test for multiple comparisons was used to test for significance between groups. For the length measurement, 7/18 samples from Method #1 (2 non-cultured, 5 cultured) and 4/164 samples from Method #2 (all cultured) were excluded from analysis as extensive specimen fragmentation precluded accurate evaluation.
Results
Gross observations
Gross differences in sample integrity were observed across the different sample handling and embedding approaches. Several types of damage were noted during sample transfer to the processing cassettes for Methods #1 and #2 that affected both non-cultured and cultured samples. Explants were fragmented into multiple pieces on receipt, stuck to the well plate, rolled up or folded over, or occasionally fell apart at transfer. Following processing, in Method #1, it was sometimes necessary to dispense more molten paraffin on top of the inverted cassette to help eject the sample, causing damage. Additional challenges at embedding included sample crumbling, difficulty in embedding on edge, and the need to embed multiple small fragments. Method #2 provided several improvements through the use of the flexible biopsy insert, which could be gently tapped to release the visible sample, and use of a heated needle rather than forceps to help orient the sample upright during embedding. Nevertheless, since the samples were not rigid, the top part of the retinal flower occasionally folded over at embedding, resulting in incomplete retinal sections or doubled over retinal layers.
Among samples embedded using Method #1, 50% (n = 3/6) of non-cultured samples and 50% (n = 6/12) of cultured samples were grossly fragmented into small pieces at the time of embedding. Among samples embedded using Method #2, 17% (n = 2/12) of minimally cultured explants and 8% (n = 13/167) of cultured explants were lost between initial sample handling and sectioning or were excluded from image analysis due to severe damage. An additional 50% (n = 6/12) of minimally cultured samples and 19% (n = 31/167) of cultured samples were damaged but had sufficient tissue for H&E evaluation, with the majority noted as being grossly fragmented into smaller pieces or folded.
In contrast, none of the explants processed using Method #3 showed gross fragmentation. Only 13% (n = 4/30) of cultured explants embedded using Method #3 showed some folding or tissue tears when they shifted during transit and became stuck between the membrane of the tissue culture insert and the well, necessitating some sample handling prior to stabilization in HistogelTM. HistogelTM also facilitated FFPE sectioning as its lighter color in the paraffin block enabled easy identification of tissue location, reducing the chance of sectioning past the small explant (). Overall, these gross differences demonstrate that use of HistogelTM significantly reduces sample damage and loss by eliminating direct sample handling steps and improving tissue visualization during sectioning.
Figure 2. Histogel improves visualization of tissue in formalin-fixed paraffin embedded (FFPE) blocks. (a) Representative FFPE blocks of retinal explants embedded with and without Histogel. Histogel appears whiter than paraffin, enabling easy identification of tissue in the block (red arrows). (b) Improved visualization reduces the chance of sectioning past explant tissue.

Histology
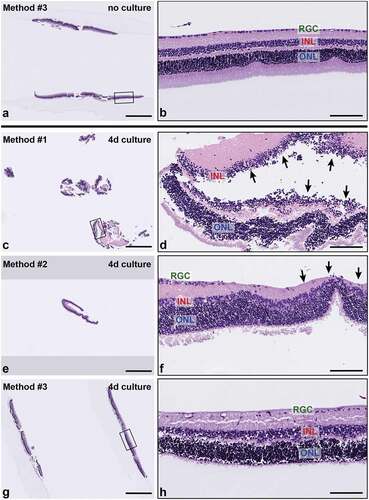
Samples from all methods consisted of normal retinas with variable representation of the main layers of the neural retina, including the ONL, INL, and RGC layers. No RPE was present in any samples, as expected. Samples that were not cultured or minimally cultured showed normal histomorphology on H&E sections, with normal cellularity and thickness of all retinal layers (). In contrast, cultured samples, regardless of embedding method () or experimental treatment group, exhibited variable degeneration of retinal ganglion cells as well as cell-cell dyshesion, occasionally resulting in artifactual displacement of photoreceptor nuclei from the ONL into the INL. The photoreceptor layer was often not fully visualized due to fragmentation and degeneration. Separation within () and between retinal layers, most often between the INL and ONL, was observed in both non-cultured and cultured explants but occurred more frequently in the ONL. Variability in H&E sections obtained from the different embedding methods primarily related to sample integrity and tissue visualization. Severe fragmentation and tangential sectioning present in samples from Method #1, in particular (), and from Method #2, to a lesser degree, limited morphologic assessment for those sections. Notably, samples from Methods #1 and #2 primarily consisted of fragments of peripheral retina () due to the retinas being embedded whole and the nature of the sectioning technique, which first samples the peripheral retina and does not capture the central retina without significant leveling (~2 mm) into the block. In contrast, samples embedded in HistogelTM consistently displayed complete cross sections containing both peripheral and central retina on the same slide ().
Figure 3. Representative H&E-stained sections of retinal explants from each sample handling and embedding approach. (a-b) Explant. no culture, embedded with Method #3. Non-cultured retinal explants embedded with Method #3 demonstrated all layers of the neural retina with normal morphology and cellularity. (c-h) Cultured explants showed variable degeneration of retinal ganglion cells and photoreceptor processes as well as cell-cell dyshesion. (c) Sample embedded using Method #1 shows severe fragmentation and tangential sectioning (d) and frequent separation of retinal layers (black arrows) (e-f). Sample embedded using Method #2 shows smaller sections of peripheral retina with less fragmentation, improved orientation and layer continuity. Black arrows in (f) indicate focal tangential sectioning. (g-h) Samples embedded using Method #3 show minimal fragmentation, proper orientation, and improved representation of full-thickness retinal cross sections. INL, inner nuclear layer, ONL; outer nuclear layer; and RGC, retinal ganglion cell layer. Scale bar = 1000 μm (a, c, e, g); 100 μm (b, d, f, h).
Slide scoring and image analysis
H&E sections of samples from all three embedding methods were next evaluated for tissue quality and sample integrity based on four metrics: lateral fragmentation, orientation, layer separation, and cell layer representation (summarized in and Supplementary Table 2). Both non-cultured and cultured samples showed similar trends across embedding methods. However, among samples embedded using the same method, those not cultured or minimally cultured showed a higher percentage of tissue was embedded on edge and demonstrated contiguous layers, without separation within or between layers. These findings suggest that degeneration associated with ex vivo culture contributes to layer separation. Among samples from Methods #1 and #2, the non-cultured or minimally cultured explants also more frequently displayed a full thickness retinal cross-section as compared to cultured explants.
Figure 4. Histogel embedding improves multiple metrics of sample integrity and tissue quality. (a-d) H&E-stained sections of retinal explants from each embedding method were scored based on four metrics: lateral fragmentation (a), tissue orientation on edge (b), continuity of retinal layers (c), and the percentage of tissue with a full-thickness cross section (d). Within each embedding method, samples were divided into those not cultured or minimally cultured (‘-’) and those cultured ex vivo for 4 days (‘+’). Histogel-embedded samples show superior performance in the extent of sample fragmentation, sample orientation, and full thickness layer representation.
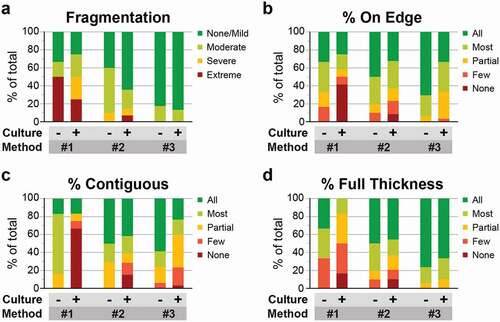
Among cultured samples, Method #1 showed the highest degree of lateral fragmentation, with 50% showing severe or extreme fragmentation (). In contrast, only 36% of cultured explants from Method #2 and 13% from Method #3 showed any fragmentation. The fragmentation in Method #3 samples, which was not observed during fixation, likely occurred at embedding. Sample orientation also progressively improved from Method #1 to #3. Among Method #1 samples, only 42% of sections showed 75% or more of the tissue embedded on edge (classified as ‘most’ or ‘all’), and another 42% showed no tissue properly embedded on edge (). Methods #2 and #3 showed a similar, higher distribution of tissues correctly embedded, with 63% and 67% of samples showing most or all of the tissue embedded on edge. Moreover, 97% of Histogel-embedded samples showed >50% of the tissue correctly embedded. The percentage of tissue with contiguous layers similarly improved from Method #1 to Methods #2 and #3 (). Among samples in Method #1, 67% completely lacked contiguous layers (classified as ‘none’). Among samples in Methods #2 (71%) and #3 (77%), demonstrated contiguous layers in >50% of the tissue. In this metric, Method #2 was superior, with 61% of samples classified as ‘most’ or ‘all’ compared to 40% in Method #3. This discrepancy may be associated with the larger size of sampled tissue in Method #3 or differences in layer separation from peripheral to central retina. Finally, the number of samples that displayed a full thickness retinal cross section progressively improved across methods and was highest with Method #3, with 90% of slides showing all retinal layers in 75% or more of the tissue, as compared to 17% with Method #1 and 63% with Method #2 (). Notably, Method #3 Histogel-embedded samples had a higher percent of full thickness retinal layers regardless of whether the samples were cultured. Taken together, the findings support that HistogelTM embedding decreases sample fragmentation and improves the yield of properly oriented, full thickness retinal cross sections.
Total cross-sectional tissue area and maximum fragment length per slide were next compared across embedding methods (, Supplementary Table 3). No significant differences in total tissue area were observed between non-cultured and cultured samples within each method. Among cultured samples, the mean total tissue area from Method #1 (903,226 μm2) and Method #3 (1,169,271 μm2) was similar and significantly greater (~2-fold) than the mean total tissue area from Method #2 (503,457 μm2) (p = 0.0017 for Methods #1 versus #2; p < 0.0001 for Methods #2 versus #3) (). The presence of approximately twice the amount of tissue area in samples from Method #3 as compared to Method #2 is consistent with the fact that two fragments of bisected retina were embedded in the HistogelTM approach compared to one edge of peripheral retina in the latter approach. Though Method #1 samples often showed a similar high tissue area, this was predominantly due to improper embedding, sample fragmentation, and tangential sectioning. In contrast to area, cultured samples from Method #3 showed a significantly higher mean maximum fragment length (2,895 μm) than samples from both Method #1 (1,773 μm) and Method #2 (1,538 μm) (p < 0.0001 for Methods #1 versus #3 and for Methods #2 versus #3) (). Similar to area, the fragment length was approximately twice as long in Method #3 as in Method #2. Interestingly, non-cultured HistogelTM-embedded explants showed a significantly higher mean maximum length (4,069 μm) as compared to cultured Histogel-embedded explants (p < 0.0001), suggesting possible sample contraction with ex vivo culture or increased susceptibility to shrinkage during processing. Overall, these data demonstrate that the use of HistogelTM significantly increases the amount of analyzable tissue per slide.
Figure 5. Use of Histogel increases the amount of analyzable tissue per slide. (a) Representative H&E-stained section of a retinal explant. (b) Tissue area was obtained by converting images to grayscale and Otsu thresholding to produce a binary mask. (c) Topological thinning and pruning were performed on this binary mask to obtain a filament representing tissue length for each fragment. (d) Whitespace was removed from both area and length measurements. (e) Additional artifacts, such as fragment of lens capsule (black arrow), were manually excluded from the captured area. (f) Total tissue area and (g) maximum fragment length were compared among minimally cultured or non-cultured (‘-’) and cultured (‘+’) samples across threeembedding methods. Method #3 Histogel-embedded cultured explants has significantly higher tissue area per slide than Method #2 samples and higher fragment length compared to Methods #1 and #2 (One-way ANOVA with post-hoc Šidak’s test for multiple comparisons; **, p < 0.01; ****, p < 0.0001). Scale bar = 100 μm.
Discussion
Loss and damage of cultured retinal explants during FFPE tissue handling can occur at sample transfer, embedding, and, more rarely, at sectioning, inhibiting proper histologic evaluation of experimental treatment groups. These barriers can necessitate repeat studies that involve significant time, labor and require more animals. The HistogelTM double embedding method offers a way to more reliably generate full thickness retinal explant sections so the advantages associated with FFPE tissue can be exploited for explant research. These advantages include better morphology as compared to frozen sections and the ability to generate thinner sections (4 μm) compared to whole mount, vibratome sections (50 μm) [Citation12], and frozen explant preparations (typically 10–14 μm) [Citation21–23], to allow better antibody penetration and shorter incubation times for immunohistochemical assays. The 4 μm cross sections obtained with FFPE can facilitate easier visualization and localization of a signal to a specific retinal layer. In contrast, imaging whole mount samples presents challenges in visualizing cellular layers as cross sections need to be reconstructed from the xz or yz plane, which has lower resolution. Furthermore, in whole mount preparations, the entire tissue is assayed for one set of markers, while FFPE tissue blocks can supply hundreds of sections for multiple immunostaining runs with multiple markers.
For FFPE preparations of retinal explants, use of HistogelTM can ameliorate a number of factors that can affect the quality of histologic sections. Retinal explants are flimsy, thin, and highly sensitive to manipulation. Tissue culture membrane inserts provide support of an explant during experimental manipulations, but this support is typically removed prior to FFPE tissue processing, which can damage the already fragile tissue [Citation1,Citation2,Citation12,Citation14]. In our laboratory’s experience, explants stick to biopsy paper and pads routinely used to keep samples flat during tissue processing. Compared to our initial Method #1 sample handling and embedding protoco adoption of the biopsy insert with a soft, protective mesh as opposed to a hard plastic mesh cassette, and use of a transfer pipette for sample manipulation improved section quality (Method #2). Even with these modifications, these samples were often still damaged. In contrast, applying HistogelTM (Method #3) to the sample directly on top of the tissue culture insert stabilized the tissue as the first step. Subsequent tissue processing did not affect sample orientation, and handling was limited to solidified HistogelTM rather than to the explant itself, eliminating damage caused by a brush, forceps, or pipette transfer.
Notably, the explant occasionally detached from solidified HistogelTM after bisection making it difficult to embed an unsupported sample on a level plane and resulted in folding artifacts. This challenge may have contributed to a subset of samples receiving scores of ‘moderate’ for percent on edge and ‘most’ rather than ‘all’ for percentage of tissue displaying full thickness retina. Causes of explant detachment from HistogelTM included addition of HistogelTM droplets onto the sample too slowly, resulting in formation of HistogelTM layers and uneven embedment of the explant. In addition, high volumes of 70% EtOH in the culture plate occasionally caused explants to float off of the tissue culture insert during sample transit and become displaced between the membrane of the insert and the well. To minimize sample handling, these samples were embedded in HistogelTM within the well but were more likely to later detach since the HistogelTM could not flow underneath and fully encase the sample.
Another challenge in working with these thin samples is obtaining a properly oriented cross section of the tissue on edge. Frozen blocks may be placed into an object holder mechanism (#63055-10, Electron Microscopy Sciences, USA), permitting x, y, and z adjustments to orient the specimen at any plane. Similarly, explants may be embedded in agarose and glued onto the specimen stage 90° to the vibratome blade for cross sections [Citation9,Citation12]. Although there is a universal block holder for microtomes, the holder does not permit x, y, and z adjustments for paraffin blocks; therefore, to obtain retinal cross sections, the thin, flower-shaped explant must be embedded on edge in paraffin. Importantly, the rigidity of HistogelTM-encased explants made it possible to bisect the sample as if it were a solid piece of tissue and embed two cross-sectional pieces that both include the central and peripheral retina, increasing the tissue area per slide. This sample rigidity could also facilitate alternate embedding and sectioning schemes. One possibility is to embed the sample on one of the peripheral edges in a 10 mm deep embedding mold (#4166, Sakura Finetek) for serial sections or levels through the entire 4 mm of tissue (or approximately 800 4 μm serial sections).
Finally, HistogelTM provided two key advantages for sample sectioning. First, since HistogelTM is a visible shade of color different from paraffin, it was easy to confirm the tissue location in a block, reducing the chance of sectioning past the explant. In contrast, for Methods #1 and #2, unstained sections needed to be examined microscopically to confirm that the sample was present and evaluate whether deeper levels were required. Second, the HistogelTM-embedded samples contained central and peripheral retina at the same level within the initial sections cut into the block. Capturing the central retina in non HistogelTM-embedded samples required sectioning an additional 2 mm into the block. Having central and peripheral retina on one slide for analysis allows investigators to study cell populations that differ in abundance across the retina as well as cell processes that occur along a central to peripheral gradient e.g., as for the timing of neurogenesis during retinal development.
Conclusions
Our results demonstrate the multiple benefits of HistogelTM embedding: less tissue loss/damage due to stabilizing friable samples in HistogelTM prior to processing; virtually no tissue fragmentation; representation of central and peripheral retina on one slide; longer fragment length for analysis; and reliable demonstration of the full thickness neural retina. There was no improvement in retinal layer separation compared to our previous embedding methods, but layer continuity in non-cultured samples suggests this finding is likely associated with the degenerative nature of mature retinal explant culture. Notably, a trend of better sample scores with later studies using Methods #2 and #3 also suggested technical improvement over time with additional experience. Overall, given the multiple advantages offered by HistogelTM, our data supports its use to ameliorate damage associated with FFPE processing for cultured retinal explants from mature mice. The HistogelTM embedding scheme presented here may allow investigators to expand the usage of FFPE samples for retinal studies. Similar embedding schemes could also be applied to other thin, friable tissues to allow for more reliable sample quality and bisection.
Acknowledgments
We thank Carmina Espiritu for her laboratory support and encouragement; Sarajane Saturnio-Nghiem for her contribution of careful laboratory processing notes for historical retinal explant studies; Xiurong Zhang for contributing to sectioning of samples; Charles Havnar for sharing his knowledge of Histogel use; Joanna Yung, Renee Raman, and William Lin for whole slide scanning of all slides used in this study; and Frank Peale and Josh Webster for helpful comments on this manuscript.
Disclosure statement
All authors are current employees of Genentech and may be shareholders of Roche.
References
- Rettinger CL, Wang H-C. Current advancements in the development and characterization of full-thickness adult neuroretina organotypic culture systems. Cells Tissues Organs. 2019;206(3):119–132.
- Li Y, Zhang Y, Qi S, et al. Retinal organotypic culture – a candidate for research on retinas. Tissue Cell. 2018;51:1–7.
- Schaeffer J, Delpech C, Albert F, et al. Adult mouse retina explants: from ex vivo to in vivo model of central nervous system injuries. Front Mol Neurosci. 2020;13:599948.
- Johnson TV, DeKorver NW, Levasseur VA, et al. Identification of retinal ganglion cell neuroprotection conferred by platelet-derived growth factor through analysis of the mesenchymal stem cell secretome. Brain. 2014;137(2):503–519.
- Caffé AR, Visser H, Jansen HG, et al. Histotypic differentiation of neonatal mouse retina in organ culture. Curr Eye Res. 2009;8(10):1083–1092.
- Sawamiphak S, Ritter M, Acker-Palmer A. Preparation of retinal explant cultures to study ex vivo tip endothelial cell responses. Nat Protoc. 2010;5(10):1659–1665.
- Müller B. Mouse cell culture, methods and protocols. Methods Mol Biol. 2019;1940:181–191.
- Hatakeyama J, Kageyama R. Retrovirus-mediated gene transfer to retinal explants. Methods. 2002;28(4):387–395.
- Belhadj S, Tolone A, Christensen G, et al. Long-term, serum-free cultivation of organotypic mouse retina explants with intact retinal pigment epithelium. J Vis Exp. 2020(165). doi:10.3791/61868.
- Alarautalahti V, Ragauskas S, Hakkarainen JJ, et al. Viability of mouse retinal explant cultures assessed by preservation of functionality and morphology. Investigative Opthalmol Vis Sci. 2019;60(6):1914.
- Akita J, Takahashi M, Hojo M, et al. Neuronal differentiation of adult rat hippocampus-derived neural stem cells transplanted into embryonic rat explanted retinas with retinoic acid pretreatment. Brain Res. 2002;954(2):286–293.
- Wang J, Kolomeyer AM, Zarbin MA, et al. Organotypic culture of full-thickness adult porcine retina. J Vis Exp. 2011(49). doi:10.3791/2655.
- Rettinger CL, Wang H-C. Quantitative assessment of retina explant viability in a porcine ex vivo neuroretina model. J Ocul Pharmacol Th. 2018;34:521–530.
- Johnson TV, Martin KR. Development and characterization of an adult retinal explant organotypic tissue culture system as an in vitro intraocular stem cell transplantation model. Investigative Opthalmol Vis Sci. 2008;49(8):3503.
- Shidham VB. CellBlockistry: chemistry and art of cell-block making – a detailed review of various historical options with recent advances. Cytojournal. 2019;16:12.
- Joiner KS, Spangler EA. Evaluation of HistoGelTM-embedded specimens for use in veterinary diagnostic pathology. J Vet Diagn Invest. 2012;24(4):710–715.
- Osakada F, Ooto S, Akagi T, et al. Wnt signaling promotes regeneration in the retina of adult mammals. J Neurosci. 2007;27(15):4210–4219.
- Ooto S, Akagi T, Kageyama R, et al. Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. Proc Natl Acad Sci U S A. 2004;101(37):13654–13659.
- van der Walt S, Schönberger JL, Nunez-Iglesias J, et al. scikit-image: image processing in Python. Peerj. 2014;2:e453.
- Koch EW, Rosolowsky EW. Filament identification through mathematical morphology. Arxiv. 2015
- Léger H, Santana E, Beltran WA, et al. Preparation of mouse retinal cryo-sections for immunohistochemistry. J Vis Exp. 2019(149). doi:10.3791/59683.
- Xin H, Yannazzo J-AS, Duncan RS, et al. A novel organotypic culture model of the postnatal mouse retina allows the study of glutamate-mediated excitotoxicity. J Neurosci Meth. 2007;159(1):35–42.
- Schaeffer J, Tardy C, Albert F, et al. Adult mouse retina explants: an ex vivo window to explore central nervous system diseases. Biorxiv. 2020; 2020 Feb 22. 960609.