
ABSTRACT
Microbially mediated reduction of Fe(III) in chlorite and biotite by Shewanella oneidensis MR-1 leads to a significant reduction in sorption of both Cs and Sr compared to the abiotic systems. As seen in previous studies, biotite is a more efficient sorbent than chlorite. Reduction of the mineral-associated Fe(III) causes increased dissolution of both minerals and reduces the sites available for Cs and Sr sorption. As this dissolution progresses it causes desorption of Cs and Sr from chlorite, but not biotite. Subsequent exposure to air increases sorption due to precipitation of secondary Fe(III) oxyhydroxide minerals derived from the Fe(II) released by bio-reduction. In contrast to successful bioremediation of redox active elements, this study suggests that microbial Fe(III) reduction could enhance the migration of Cs and Sr through phyllosilicate-dominated strata.
Introduction
The radioactive isotopes of caesium and strontium (caesium-137 and strontium-90) are high-yield fission products that accumulate in nuclear fuel rods. They are a major component of the radioactivity in spent fuel and in waste from spent fuel storage ponds and fuel reprocessing plants, such as at Sellafield, UK (Thorpe et al. Citation2012), and the Hanford Site, USA (McKinley et al. Citation2007; Zachara et al. Citation2002). Cs-137 and Sr-90 are released into the environment through planned discharges or accidental releases (Renshaw et al. Citation2011) and, with have half-lives of ∼30 years, they are hazardous in the environment for a few hundred years after release.
In addition to direct exposure to these radionuclides in water systems and near-surface sediments, Cs-137 can enter the food chain due to similarities in its biogeochemical behavior to the essential nutrient K (Hinton et al. Citation2006), and Sr-90 readily exchanges for Ca in living organisms, e.g., in hydroxyapatite (bone) (Handley-Sidhu et al. Citation2011; McKinley et al. Citation2007). Thus, radioactive Cs and Sr will be present in high level nuclear wastes (HLW) intended for interim storage and geological disposal and their behavior in both near surface and natural environments needs to be understood to safely manage HLW. In all these environments, nitrate (NO3−) will be present as either a co-contaminant from the wastes or as a pre-existing component (e.g., derived from agricultural use).
Radioactive caesium, and strontium can be mobile in the subsurface (Brookshaw et al. Citation2012). Remediation options for these radionuclides rely on their sorption to pre-existing minerals or incorporation into newly formed secondary minerals (Brookshaw et al. Citation2012), e.g., solid-phase capture of Sr in calcite (Fujita et al. Citation2004), hydroxyapatite (Handley-Sidhu et al. Citation2011) and Fe(III) oxide minerals (Ferris et al. Citation2000). However, such Sr incorporation may be applicable only under constrained geochemical conditions, such as high pH or elevated carbonate concentrations (Thorpe et al. Citation2012).
Sorption, however, may offer immobilization under a greater range of conditions, and is influenced by the reactive surfaces available and the concentration of competing ions, in addition to geochemical factors such as pH. A large body of research has been undertaken over the past forty years regarding adsorption of these radionuclides to different Fe(III) oxyhydroxides (Ferris et al. Citation2000; Langley et al. Citation2009) and clay minerals (Bostick et al. Citation2002; Cornell Citation1993; Kemner et al. Citation1997). Phyllosilicates are ubiquitous in near-surface sediments, and micas and chlorite are present in varying amounts in the lithological environments being considered for the siting of nuclear waste disposal facilities.
Their high surface area and negative surface charges mean the phyllosilicate components of rocks and sediments can contribute significantly to the removal of contaminants from solution; in granitic lithologies biotite will be the main sorbing phase and in many rock types, including mature mudstones and altered basic rocks, chlorite will be very important (Baik et al. Citation2003; Tsai et al. Citation2009; Zachara et al. Citation2002). Thus, the importance of the relationship between Cs and Sr and phyllosilicates has been well recognized, and many studies have investigated the sorption behavior of these radionuclides to clays such as montmorillonite (Bostick et al. Citation2002; Lu and Mason Citation2001), smectites in general (Galambos et al. Citation2012), illite and chlorite (Hinton et al. Citation2006) as well as micas such as muscovite and biotite (Cho and Komarneni Citation2009; McKinley et al. Citation2004; Meleshyn Citation2010; Stout et al. Citation2006; Taylor et al. Citation2000; West et al. Citation1991; Zachara et al. Citation2002).
High-specificity interactions between Cs and phyllosilicates can offer relatively long-term immobilization of this radionuclide (Cornell Citation1993). Cs may be sorbed to high affinity sites at the phyllosilicate mineral edges (Chang et al. Citation2011; Steefel et al. Citation2003), typically forming very stable inner sphere complexes (Bostick et al. Citation2002). Cs may also be taken up into the interlayer regions of swelling clays such as montmorillonite, and dehydration of interlayers can make this sorption highly resistant to desorption (Fuller et al. Citation2015). In contrast, Sr typically retains its hydration sphere and forms outer-sphere complexes at the solution-mineral interface (Sahai et al. Citation2000). These sorption mechanisms are sensitive to many environmental factors including perturbations in pH, changes in ionic strength, and the introduction of competing cations.
Bacteria are commonly present in the shallow subsurface. Microbial metabolism can involve the reduction of nitrate as well as a number of transition metals including manganese (as Mn(IV)) and iron (as Fe(III)) contained within existing minerals. Microbial activity can change the geochemical conditions such as pH (Thorpe et al. Citation2012) or lead to the formation of reducing (Fe(II)) or oxidizing (nitrite, NO2−) species. Microbial reduction of Fe(III) can also lead to the dissolution of Fe(III) oxides and the precipitation or recrystallization of secondary minerals (Fredrickson et al. Citation1998; O'Loughlin et al. Citation2007).
In the case of silicate minerals, there is evidence that microbial reduction affects the structural Fe and results in a limited amount of mineral dissolution (Brookshaw et al. Citation2013; Kukkadapu et al. Citation2006; Ribeiro et al. Citation2009). This will modify the surface charge and possibly the microscale surface structure of these minerals, altering the numbers and properties of surface sorption sites. However, the interplay between these biogeochemical processes and their effects on the sorption of Cs and Sr to biotite and chlorite are currently not well understood. Biotite is a 2:1 phyllosilicate with layered structure comprising a sheet of silica-oxygen tetrahedra (T) (with some Al) forming either side of a sheet of octahedral cations (Fe(II), Fe(III), Mg and Mn) to form a T-O-T layer; the T-O-T structures are joined by weakly bonded interlayer K atoms. Chlorite is a 2:1:1 phyllosilicate comprising T-O-T layers joined by “brucite-like” layers of Mg(+ Al)-OH.
The present study is aimed at developing an understanding of how the microbial reduction of Fe(III)-containing biotite and chlorite, and subsequent re-oxidation scenarios, affects mineral properties, and the impact that this has on the sorption behavior of Cs and Sr. To develop a comprehensive understanding of the fate of Cs and Sr in natural and anthropogenic systems, it is essential to examine the coupled processes in microbe-(bio)mineral system that prevail in potential HLW environments.
Methods
Minerals
The minerals were supplied by the Excalibur Mineral Company, New York and sourced from Silver Crater Mine, Cardiff, Ontario, Canada (biotite) and Michigamme, Michigan, USA (chlorite). Biotite flakes were ground using an agate ball mill and separated into different size fractions using a shaking sieve stack. Biotite powder in the size range 180–500 µm was used in these experiments. The powder may include particles of < 180 µm due to particle clumping because of electrostatic attraction between small flakes. Chlorite particles of < 100 µm were used in the experiments. The compositions of the minerals were determined by electron probe microanalysis (EPMA) using a CAMECA SX100 instrument, operating at an accelerating voltage of 20 kV and a 20-nA beam current and using silicate standards. The minerals were also characterized by powder X-ray diffraction (XRD), using a Burker X'Pert diffractometer, and the surface areas of the powder fractions used in the experiments were determined by Brunauer-Emmett-Teller (BET) analysis using N2 gas adsorption. Further details of the mineral preparation and characterization have been described previously (Brookshaw et al. Citation2013).
Solution chemistry
The chemistry of the solutions was the same for all experiments, unless detailed otherwise. These solutions were prepared using distilled deionized water (18 Ω) and analytical-grade chemicals. Solutions were buffered using 3 × 10−2 M final concentration 3-(N-morpholino) propanesulfonic acid (MOPS). The pH of the buffer was corrected by dropwise addition of 10 mol l−1 NaOH until it reached 7 ± 0.1. MOPS buffer was used in all experiments to prevent supersaturation conditions with respect to carbonate phases (including SrCO3) (Thorpe et al. Citation2012). Na-lactate was added to give a final concentration of 1 × 10−2 M and acted as electron donor for microbial reduction in experiments containing bacteria.
Abiotic sorption experiments
Abiotic interactions between biotite or chlorite and Cs or Sr were characterized in batch anaerobic experiments. The kinetics of sorption of Cs (5 × 10−4 mol l−1) and Sr (5 × 10−4 mol l−1) were studied in experiments with a 1:40 mineral to solution ratio by weight. Aliquots of slurry sample were extracted via a degassed syringe at 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h and 96 h after the contaminants were added. The samples were centrifuged at 16,160 g (Sigma Benchtop Microfuge) for 5 minutes, and the supernatant separated for further analysis of cation concentrations. The equilibrium sorption capacity of the minerals was studied in experiments spiked with a range of concentrations of Cs or Sr from 5 × 10−5 to 5 × 10−3 mol l−1. The experiments were sampled after 2 weeks (allowing sufficient time for the reaction to reach equilibrium) and the concentrations of Cs and Sr in the supernatant determined by ICP-MS.
Biotic sorption experiments
Batch anaerobic bottles were prepared with the same mineral-solution ratio as for the abiotic sorption experiments. These were equilibrated with Cs (5 × 10−4 mol l−1) or Sr (5 × 10−4 mol l−1) for at least 1 h before addition of bacterial cultures. A new set of parallel abiotic experiments (‘without bacteria’) were performed for direct comparison.
The Fe(III)-reducing bacterium S. oneidensis MR-1 was used to reduce bioavailable Fe(III) in biotite and chlorite. The bacterium was cultured aerobically and at late log phase of growth, aliquots were re-inoculated and cultured in defined minimal medium under anaerobic conditions with lactate as the electron donor and fumarate as the electron acceptor (von Canstein et al. Citation2008). Cells were harvested by centrifugation at 5000 g for 20 min and washed twice in a 3 × 10−2 mol l−1 MOPS buffer (pH 7). Washed cells were added to each experimental bottle to a final optical density at 600 nm (OD600) of ∼0.3. Suspensions were then incubated at 30°C in the dark.
After microbial Fe(III) reduction had peaked, and Fe(II) levels had stabilized, selected treatments were re-oxidized. Triplicates of each set of conditions were aerated by piercing the butyl rubber stoppers of the bottles using wide needles and injecting ∼20 ml of air via a syringe, daily. Triplicates of each set of conditions were also amended by the addition of nitrate (1 × 10−2 mol l−1 final concentration) to stimulate oxidation of Fe(II) coupled to denitrification.
The experiments were sampled at 0, 1, 4, 7, 14, 25 and 32 days. Approximately 0.7 ml of slurry sample was extracted via a degassed syringe at each sampling point, and analyzed for Fe(II), pH, Eh and cation concentrations.
Solution chemistry
Ferrozine analysis
At each time point an aliquot of the slurry was added to 0.5 mol l−1 HCl. After 1 h, an aliquot was added to ferrozine solution (Stookey Citation1970) buffered to pH 7 and the OD562 measured to quantify the amount of ‘bioavailable’ Fe(II) (Lovley and Phillips Citation1987). An excess of the reducing agent hydroxylamine hydrochloride (final concentration of 0.25 mol l−1) was then added to the sample and allowed to react for another hour. A further aliquot was reacted with the ferrozine solution before measuring the OD562, to give the total acid-extractable Fe.
pH and Eh
At each time point the pH and Eh of the slurry samples was measured. Aliquots of slurry samples were shaken before the measurements were made. The Eh measurements were carried out within 1 h of the sample being obtained to prevent significant changes due to aeration of the samples.
ICP-MS analysis
Aliquots of sample supernatants were added to 2% nitric acid and analyzed by inductively coupled plasma mass spectrometry (ICP-MS) on a Agilent 7500CX instrument for concentrations of Cs or Sr, as well as concentrations of the major cations present in the minerals: Si, Al, Mg, K, Ca, Fe and Mn.
Calculations
The distribution coefficient for each concentration of Cs or Sr was calculated according to the equation:(1) where Kd is the distribution coefficient (l kg−1), [Cs] is the amount of the contaminant sorbed to the solid (mmol kg−1) and [Caq] is the concentration of the contaminant in solution (mmol l−1) at equilibrium. The amount of Cs or Sr taken up by the mineral was determined as the difference between the input concentration and the concentration in solution in the sample at equilibrium.
The sorption behavior was modelled with a Langmuir sorption isotherm:(2) where α relates to the affinity of sites for the contaminant (l mmol−1), and β is the maximum number of sorption sites (mmol kg−1). The data were plotted to give a straight line and a regression line was fitted. The parameters were estimated from the regression line, with the slope giving 1/β and intercept of the line with the Y axis, 1/αβ (Fetter Citation1999). The Langmuir distribution coefficient, Kdl, was calculated using:
(3)
Results and discussion
Mineral characterization
The powdered minerals were confirmed to be monomineralic Fe-rich biotite (phologopite) and chlorite (clinochlore) by XRD (Brookshaw et al. Citation2013). The compositions of the two minerals (as weight percent, wt%, major cations) are shown in . The mineral fractions used in these experiments had relatively similar surface areas (9.02 m2/g for biotite and 6.43 m2/g for chlorite) allowing comparison between experiments with the two minerals.
Table 1. Biotite and chlorite compositions (wt% oxide), determined by EPMA.
Sorption analysis
The kinetics of the sorption of Cs and Sr by biotite and chlorite were determined in batch abiotic experiments, and compared to previous work. In these experiments, pH remained stable between 7 and 8, and the ionic strength of the solutions was 0.055 mol l−1 and 0.057 mol l−1 for Cs and Sr experiments, respectively. In all four treatments, there was an immediate initial uptake of Cs and Sr (, ), with >85% and >70% of the maximum sorption to biotite and chlorite, respectively, occurring within 4 h after spiking. This was followed by continued slow sorption over the remainder of the experiment. Biotite was found to be the more efficient sorbent compared to chlorite, with the partitioning of 65% of the added Cs and 49% of the Sr to the solid phase over a period of 72 h, compared to the sorption by chlorite of 19% of the starting Cs and 19% of the starting Sr concentration over the same period.
Figure 1. Concentrations of Cs and Sr in solution in experiments with (A) biotite and (B) chlorite in abiotic experiments. In adsorption isotherms (C) each point represents the average of triplicate measurements with error bars giving the standard error for the measurements. Calculated Kd values (D) in relation to the starting concentration [C0] (the X-axis is shown as log10 of [C0] for ease of viewing of the data).
![Figure 1. Concentrations of Cs and Sr in solution in experiments with (A) biotite and (B) chlorite in abiotic experiments. In adsorption isotherms (C) each point represents the average of triplicate measurements with error bars giving the standard error for the measurements. Calculated Kd values (D) in relation to the starting concentration [C0] (the X-axis is shown as log10 of [C0] for ease of viewing of the data).](/cms/asset/ddc5aea6-37bf-4b00-8582-d3f17c144c8d/ugmb_a_1076543_f0001_b.gif)
In sorption isotherm experiments, the pH remained stable and did not exceed pH 8. The ionic strength of the solutions ranged from ∼0.055 mol l−1 to 0.06 mol l−1 for Cs and from 0.057 mol l−1 to 0.075 mol l−1 for Sr. Isotherms for the equilibrium concentrations of Cs and Sr in relation to the calculated concentrations on the solid show nonlinear (L-shaped) sorption of the contaminants to the minerals (). In all treatments, the greatest percentage sorption occurred at lower concentrations of the contaminants, suggesting saturation of the high affinity sorption sites of the mineral and continued sorption to lower affinity sites. The decrease in sorption rate may also be due to a diffusion process, rather than an electrostatic sorption process (Cornell Citation1993; McKinley et al. Citation2004). The distribution coefficients calculated at each concentration of the contaminant are plotted in . There is a decrease in the distribution coefficient with increasing starting concentration, consistent with previously observed trends (Tsai et al. Citation2009).
The Langmuir sorption isotherm provided a good fit to the experimental data (with R2 values above 0.95, ). The fast sorption in the first 4 h followed by continued slow sorption over the remainder of the experiment reinforces the likely presence of at least two sorption sites on both minerals, a high affinity and a low affinity site. The values for α (calculated using Equation Equation2(2) detailed in Methods) for biotite were similar for Cs and Sr (1.19 l mmol−1 and 0.97 l mmol−1 respectively) indicating that the affinity of the sorption sites in this mineral taken as a whole (without differentiating between different sites), was similar for both the monovalent and divalent contaminants studied.
Table 2. Langmuir sorption isotherm parameters.
The greater sorption of Cs to biotite compared to Sr is explained by a greater sorption density of Cs (67 mmol kg−1) than Sr (31.6 mmol kg−1). In contrast, sorption sites in chlorite have a greater affinity for Sr (1.18 mmol−1) than Cs (0.47 mmol−1), but sorption was much lower than in biotite due to a significantly lower site density (7.3 mmol kg−1). The values of these parameters were used to estimate the distribution coefficients (Kdl) for Cs and Sr partitioning to the two minerals (). These calculated Kdl values were similar to the Kd values for the experiments at contaminant concentration of 5×10−4 mol l−1, indicating that the sorption processes at this concentration are representative of the sorption behavior over the range of concentrations studied.
Sorption during microbial reduction
Sorption of Cs and Sr to biotite and chlorite was studied in experiments where S. oneidensis MR-1 was added to mediate the reduction of mineral-associated Fe(III). Increase in the Fe(II) percentage of the extractable iron, accompanied by a significant decrease in Eh (from 60 ± 27 mV to −150 ± 34 mV (biotite) and from 100 ± 2 mV to −185 ± 7 mV (chlorite)), shows that significant bioreduction occurred in treatments where the bacteria were added (). To understand the impact of the bacteria on contaminant behavior, contaminant sorption is examined in the context of the consequential microbial Fe(III) reduction and resulting solution chemistry change; experiments were conducted with and without bacteria.
Figure 2. Cs and Sr behavior in microbial reduction experiments: (A). Fe(II) concentration as a percentage of the total acid-extractable iron; (B). Cs remaining in solution in experiments with biotite and chlorite in the presence and absence of bacteria; (C). Sr remaining in solution in experiments with biotite and chlorite in the presence and absence of bacteria; (D). pH evolution during the experiments. Each point represents the average of triplicate measurements and the error bars – the standard error. The phases of microbial reduction, as discussed in the text, are highlighted: dark grey– early Fe(III) reduction; pale grey– sustained Fe(III) reduction; medium grey– no further reduction.
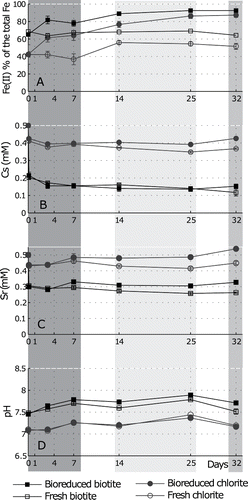
After an initial instantaneous sorption of Cs to biotite and chlorite, slow sorption of Cs continued until day 7. In that time, there was no difference in the concentrations of Cs in treatments with and without bacteria despite significant changes in solution chemistry due to microbial reduction (significant decrease in Eh and notable increase in pH, also accompanied by a rapid increase in acid-extractable Fe(II)). Sustained Fe(III)-reducing conditions were established in treatments with added cells of S. oneidensis between 14 and 25 days, and the maximum Fe(II) percentages were recorded as 92 ± 2% in biotite and 86 ± 3% in chlorite. During this stage of reduction, Eh levels were typical for Fe(III)-reducing conditions (−70 mV to −225 mV) and pH remained relatively stable, with little difference between the pH in experiments with and without added bacteria. During this period there was slow continued removal of Cs in experiments with biotite with no difference between treatments with and without bacteria (0.14 mmol l−1 Cs remained in solution in both on day 25). There was no further Cs sorption to chlorite in treatments containing bacteria (Cs concentration remained 0.40 mmol l−1 throughout this phase) while, interestingly, some further slow removal of Cs was observed (from 0.40 mmol l−1 on day 7 to 0.35 mmol l−1 on day 25) was observed in the uninoculated treatments with this mineral. No significant increase in Fe(II) was observed after day 25, suggesting the end of microbial Fe(III) reduction had occurred by day 25.
Initial instant Sr sorption by both minerals was followed, on day 7, by detectable desorption of Sr in experiments with bacteria, although this latter process was not a feature of the experiments without bacteria. During the 14–25-day sustained Fe(III) reduction phase, no further sorption of Sr was observed in bioreduction treatments, but there was continued slow removal of Sr by the minerals in the experiments without bacteria.
After 32 days, sorption of Cs to biotite in the presence of the added bacteria did not differ significantly to that in systems without the microbial inoculum (with Kd32 of 90.7 l kg−1in experiments with bacteria and 130.9 l kg−1 in uninoculated controls). In contrast, sorption of Cs to chlorite was actually lower in experiments with added bacteria (Kd32 = 6.9 l kg−1) compared to the equivalent uninoculated treatments (Kd32 = 14.5 l kg−1). No further sorption of Sr to biotite had occurred in experiments without bacteria (Kd32 = 36 l kg−1), yet in all other experiments there was desorption (inoculated biotite: Kd32 = 21 l kg−1; uninoculated chlorite: Kd32 = 4.54 l kg−1). Indeed, greatest desorption of Sr occurred in experiments with chlorite where bacteria were present, and all of the initial sorbed Sr was desorbed by the end of the experiment (day 32). In the experiments containing Cs and Sr during bioreduction of Fe(III), in all cases the apparent distribution coefficients at day 32 were lower than those for the equivalent abiotic experiments without bacteria.
Whereas there were no significant differences between the amount of Cs and Sr adsorbed to chlorite, biotite showed greater affinity for Cs than Sr. This points to differences in the Cs sorption sites available on the two minerals studied. The nonlinear distribution of Kd values further supports the likely change of the dominance of sorption sites as low-number, high-affinity sites became occupied at greater contaminant concentrations (Barnett et al. Citation2000), but sorption to high number, low-affinity sites continued. The typically observed sorption of Cs to high affinity edge sites on biotite surfaces (Tsai et al. Citation2009) may have prevented significant desorption due to the presence and activity of bacteria. The lack of bacterial-stimulated desorption further suggests that stable inner sphere complexes were formed at these sites. The marked difference in behavior of Sr compared to Cs is consistent with a less “secure” sorption process occurring in these systems, likely adsorption by the formation of outer sphere complexes (Sahai et al. Citation2000).
Our results suggest that, surprisingly, Cs and Sr sorption to biotite and chlorite was not affected by the changes in the chemistry of the solution due to microbial Fe(III) reduction (such as an increase in pH (Thorpe et al. Citation2012). Bacteria may provide biosorption sites on the added biomass, but this effect was not observed in additional control experiments containing just biomass and no mineral substrate. Both the physical presence of the bacteria and biogeochemical processes such as microbially mediated Fe(III) reduction appear to lead to the observed differences between experiments with and without bacteria. The unexpected result was the neutral or deleterious effects on Cs and Sr sorption.
Organic matter associated with respiring bacteria has been shown to modify the sorption properties of biotite and chlorite (Bellenger and Staunton Citation2008). However, these authors observed enhanced sorption of Sr to phyllosilicates, unlike the decrease in sorption observed in the present study. Conversely, decreased sorption of Cs during microbial sulphate reduction has been attributed to bacteria physically blocking sorption sites on minerals (West et al. Citation1991), but a more recent study suggested that microbial activity, rather than the physical presence of bacterial cells, was responsible for the lower sorption seen in experiments with respiring bacteria (Russell et al. Citation2004). Microbial Fe(III) reduction potentially directly modifies the mineral structure and disrupt layer charges which could alter the site density and affinity for the contaminants, causing, for example, desorption of outer-sphere complexes from low affinity sites.
Evolution of solution chemistry
In are shown the concentrations of the cations that may be involved in mineral equilibration, microbial mineral reduction and cation exchange processes, (Si, Al, Ca, K, Mg, Fe, and Mn), associated with the different treatments at 0, 4, 14 and 25 days. Their release in solution is considered in the context of cation location within the mineral structure to understand the effect of microbial Fe(III) reduction on the stability of the different mineral layers and infer the potential effects on mineral charge and distribution of sorption sites.
Figure 3. Comparisons of cation concentrations for different treatments at time points 0, 3, 14 and 25 days. Error bars represent the standard error of triplicate measurements.
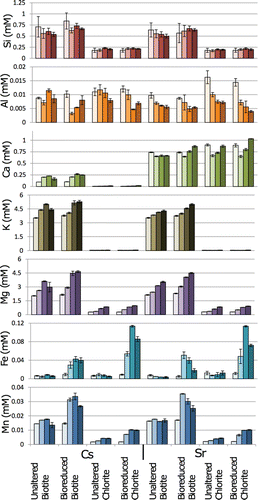
The mineral present and its treatment affected the concentration in the aqueous phase of cations representative of the tetrahedral layer (Si) and the octahedral layer (Mg). Greater release of Si (significant at the 5% confidence level based on triplicate sample measurements, p < 0.05, 3), Mg and Mn (p < 0.001, 3) occurred in the experiments with biotite compared to those with chlorite, the former being more labile in the experimental solutions (Brookshaw et al. Citation2013). The concentrations in solution of the octahedral cation Mg (both minerals) and in biotite, the interlayer cation K, increased as the experiment progressed due to mineral equilibration with the solution (seen in experiments both with and without bacteria). Of great importance is the observation that microbial reduction of the Fe(III) in both minerals caused a significant increase in the aqueous concentrations of both Mg and K compared to the equivalent uninoculated controls (in all cases, p < 0.05, 3); this can be directly related to the destabilization of the O layer and cation exchange in the interlayers due to microbial reduction of Fe(III) in this layer.
In experiments without bacteria, there was no significant change in the concentration over time of Fe and Mn in solution. Mn in biotite was approximately twice as labile as that in chlorite (1.3% of the biotite-associated Mn dissolved compared to 0.3–0.6% of the chlorite-associated Mn). The concentrations of Fe in solution were similar for the two minerals when related to the amount of Fe available in each mineral, but very low, suggesting most of the reduced Fe remained associated with the solid phase (sorbed to the minerals or as structural Fe). In experiments with bacteria, microbially mediated Mn(IV) reduction and Fe(III) reduction led to a significant increase in the solution concentration of Fe and Mn, with maxima detected typically on day 14 in both minerals. Microbially mediated metal reduction enhanced the release of Mn two-fold compared to the abiotic controls. In experiments with both minerals where bacteria were added, soluble Fe was an order of magnitude higher than in the equivalent abiotic experiments on days 4 and 14, but represented less than 0.1% of the Fe available in the bulk mineral (less than 0.12 mmol l−1, and typical concentrations recorded by Ferrozine extraction were 100 mmol l−1), indicating that most of the reduced Fe remained sorbed to the mineral or locked within the mineral structure.
The Ca concentrations in different experiments can be correlated with the type of contaminant present, but is not affected by the type of mineral present or the presence of bacteria. The solution concentrations of Ca in experiments with biotite and Cs (0.097–0.277 mmol l−1) exceeded the amount of Ca available in the biotite based on EPMA results (∼0.033 mmol l−1; see ), suggesting the dissolution of secondary Ca-containing mineral such as calcite (that may have been present as a microscopic, minor impurity in the experiments). Significantly higher concentrations of Ca were detected in all experiments with Sr (0.602–0.867 mmol l−1 and 0.650–1.031 mmol l−1 in experiments with biotite and chlorite, respectively). These Ca concentrations are higher than the maximum Sr concentrations used in the experiments; however, the majority of the added Sr remained in solution. This suggests that, whereas Sr may enhance the dissolution of Ca-containing minerals, removal of Sr from solution is controlled by the stable mineral surfaces present (biotite and chlorite) and not an exchange/incorporation interaction with another Ca-containing mineral.
Re-oxidation experiments
After maximum levels of microbial Fe(III) reduction were revealed on day 25 ( and , ), one set of the biominerals were oxidized by addition of air, and another set by the addition of nitrate, as shown by the grey box on . Oxidation of Fe(II) in the acid-extractable fraction occurred in both biotite and chlorite. The Fe(II) concentrations at the end of the experiment (56.2 ± 56.2% and 49.7 ± 1.8%, respectively) are not significantly different from the Fe(II) concentrations in the acid-extractable iron in the respective minerals in the uninoculated controls (p > 0.05, open symbols on ). These concentrations are also very similar to the starting Fe(II) of the acid-extractable iron in the minerals, suggesting that only the bioreduced Fe(II) was re-oxidized by the addition of air.
Figure 4. Cs and Sr behavior during reoxidation. (A and B) Fe(II) concentration as % of total extractable Fe during oxidation of biotite and chlorite respectively. (C and D) Cs concentrations in solution normalized to the concentrations in abiotic controls (where a value of 1 shows that both concentrations are equal, < 1 shows that there is less Cs in solution in the experiment compared to the abiotic control, and > shows that there is more Cs in solution than in the abiotic control). (E&F) Sr concentrations remaining in solution, normalized to concentrations in the abiotic controls. The bars represent the average value and the error bars the standard error of triplicate measurements.
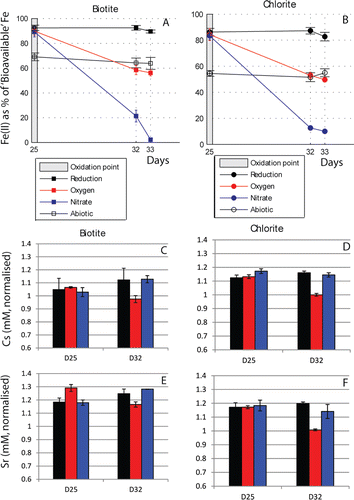
The oxidation of the minerals by aeration led to increased removal of Cs and Sr in all experiments, as can be seen by the decrease in the concentration of these contaminants from day 25 to day 32 (). This indicates that O2-driven oxidation increased the sorption capacity of these systems (e.g., by precipitation of secondary insoluble oxides) compared to both the uninoculated experiments and those where reducing conditions were maintained; for example, by the formation of secondary Fe(III)- and Mn(III/IV)-oxyhydroxides. Indeed, the addition of air was accompanied by a significant decrease in the solution concentration of both Fe and Mn from day 25 to day 32 () (pmax < 0.05, 3 for Fe in all treatments, and p < 0.001, 3 for Mn).
Table 3. Mn and Fe concentrations at day 25 and day 32 (ICP-MS).
No enhanced removal of Cs or Sr was observed when re-oxidation was caused by nitrite released from the microbial reduction of nitrate; the concentrations of Cs and Sr remained comparable to those in experiments where Fe(III) reducing conditions were maintained. In experiments with biotite, there was no significant difference (at the 5% level) in the Fe and Mn aqueous concentrations before and after oxidation via addition of nitrite. Interestingly, in chlorite, nitrate oxidation was associated with a decrease in the aqueous concentrations of Fe and Mn, significant at the 1% level (p < 0.01, 3). As no significant increase in the sorption capacity of this system was observed, it is unlikely that this decrease resulted from the formation of secondary, potentially sorbing, phases. Instead, the addition of nitrate may have caused cation exchange leading to sorption of Fe and Mn from the aqueous phase. However, since the solution concentrations of Fe(II) and Mn (II) were so low compared to the concentrations of the competing cations (), such an exchange process did not lead to a significant change in the concentration of Cs or Sr in these experiments.
Conclusions
The sorption of Cs and Sr onto biotite and chlorite reveals fast sorption to high affinity, high density sites and slower sorption to lower affinity, lower density sites. Microbially mediated reduction of Fe(III) associated with biotite and chlorite is extensive but leads to either no change or less sorption of Cs and Sr than abiotic systems. For Sr, the initial sorption is followed by desorption if bacteria are present. The trends seen do not correlate with significant changes in pH, and are unlikely to result from any physical effect due to the presence of bacteria, such as the blocking of sorption sites. Instead, microbially mediated Fe(III) reduction appears to cause significant mineral dissolution (including greater loss of Fe, Mg, Mn and K from the minerals than in bacteria-free conditions), which alters and removes the types of sorption sites available for Cs and Sr contaminants.
For example, a simple change in the mineral charge may have caused desorption of outer-sphere complexes of Cs and Sr from the basal planes of chlorite or Sr from biotite. The re-oxidation of the samples (by air) increases the sorption capacity of the system, probably through the formation of secondary Fe(III) and Mn(III/IV) oxyhydroxides, whereas complete oxidation of acid-extractable Fe by the addition of nitrate does not a detectable change in the solution concentrations of Cs and Sr. Microbial metal reduction is an important remediation strategy, imm-obilizing redox-active contaminants. However in the microbe-mineral systems examined here, Cs and Sr contamination is not retarded by microbial reduction activity. This reinforces the need for examination of coupled systems relevant to radionuclide behavior in subsurface environments. In particular, a further understanding of the mechanisms by which microbial Fe(III) reduction in key minerals such as biotite and chlorite affects their sorption properties is needed to make the large body of information on sorption processes relevant to environmental studies, and useful predictive models for complex wastes.
Acknowledgments
We thank Paul Lythgoe for ICP-MS analysis and John Waters for XRD and BET analysis.
Funding
We thank EPSRC for funding this research through Grant (EP/G063699/1) as part of the BANDD consortium (Biogeochemical Applications in Nuclear Decommissioning and Waste Disposal).
References
- Baik MH, Hyun SP, Cho WJ, Hahn PS. 2003. Contribution of minerals to the sorption of U(VI) on granite. Migration Conference; Sept 21–26, 2003; Gyeongju, South Korea: R Oldenbourg Verlag, p663–669.
- Barnett MO, Jardine PM, Brooks SC, Selim HM. 2000. Adsorption and transport of uranium(VI) in subsurface media. Soil Sci Soc Amer J 64:908–917.
- Bellenger JP, Staunton S. 2008. Adsorption and desorption of Sr-85 and (CS)-C-137 on reference minerals, with and without inorganic and organic surface coatings. J Environ Radioact 99:831–840.
- Bostick BC, Vairavamurthy MA, Karthikeyan KG, Chorover J. 2002. Cesium adsorption on clay minerals: An EXAFS spectroscopic investigation. Environ Sci Technol 36:2670–2676.
- Brookshaw DR, Lloyd JR, Vaughan DJ, Pattrick RAD. 2013. Bioreduction of biotite and chlorite by a Shewanella species. Amer Mineral 99, 1746–1754.
- Brookshaw DR, Pattrick RAD, Lloyd JR, Vaughan DJ. 2012. Microbial effects on mineral–radionuclide interactions and radionuclide solid-phase capture processes. Mineral Mag 76:777–806.
- Chang HS, Um W, Rod K, Serne RJ, Thompson A, Perdrial N, Steefel CI, Chorover J. 2011. Strontium and cesium release mechanisms during unsaturated flow through waste-weathered hanford sediments. Environ Sci Technol 45:8313–8320.
- Cho Y, Komarneni S. 2009. Cation exchange equilibria of cesium and strontium with K-depleted biotite and muscovite. App Clay Sci 44:15–20.
- Cornell RM. 1993. Adsorption of cesium on minerals—A review. J Radioanal Nucl Chem 171:483–500.
- Ferris FG, Hallberg RO, Lyven B, Pedersen K. 2000. Retention of strontium, cesium, lead and uranium by bacterial iron oxides from a subterranean environment. Appl Geochem 15:1035–1042.
- Fetter CW. 1999. Contaminant Hydrogeology. Englewood Cliffs, New Jersey: Prentice Hall.
- Fredrickson JK, Zachara JM, Kennedy DW, Dong HL, Onstott TC, Hinman NW, Li SM. 1998. Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium. Geochim Cosmochim Acta 62:3239–3257.
- Fujita Y, Redden GD, Ingram JC, Cortez MM, Ferris FG, Smith RW. 2004. Strontium incorporation into calcite generated by bacterial ureolysis. Geochim Cosmochim Acta 68:3261–3270.
- Fuller AJ, Shaw S, Ward MB, Haigh SJ, Mosselmans JFW, Peacock CL, Stackhouse S, Dent AJ, Trivedi D, Burke IT. 2015. Caesium incorporation and retention in illite interlayers. Appl Clay Sci 108:128–134.
- Galambos M, Osacky M, Rosskopfova O, Krajnak A, Rajec P. 2012. Comparative study of strontium adsorption on dioctahedral and trioctahedral smectites. Journal of Radioanal Nucl Chem 293:889–897.
- Handley-Sidhu S, Renshaw JC, Yong P, Kerley R, Macaskie LE. 2011. Nano-crystalline hydroxyapatite bio-mineral for the treatment of strontium from aqueous solutions. Biotechnol Lett 33:79–87.
- Hinton TG, Kaplan DI, Knox AS, Coughlin DP, Nascimento RV, Watson SI, Fletcher DE, Koo BJ. 2006.Use of illite clay for in situ remediation of Cs-137-contaminated water bodies: Field demonstration of reduced biological uptake. Environ Sci Technol 40:4500–4505.
- Kemner KM, Hunter DB, Bertsch PM, Kirkland JP, Elam WT. 1997. Determination of site specific binding environments of surface sorbed cesium on clay minerals by Cs-EXAFS. J De Phys Iv 7(C2):777–779.
- Kukkadapu RK, Zachara JM, Fredrickson JK, McKinley JP, Kennedy DW, Smith SC, Dong HL. 2006. Reductive biotransformation of Fe in shale-limestone saprolite containing Fe(III) oxides and Fe(II)/Fe(III) phyllosilicates. Geochim Cosmochim Acta 70:3662–3676.
- Langley S, Gault AG, Ibrahim A, Takahashi Y, Renaud R, Fortin D, Clark ID, Ferris FG. 2009. Sorption of strontium onto bacteriogenic iron oxides. Environ Sci Technol 43:1008–1014.
- Lovley DR, Phillips EJP. 1987. Rapid assay for microbially reducible ferric iron in aquatic sediments. Applied and Environmental Microbiology 53:1536–1540.
- Lu NP, Mason CFV. 2001. Sorption-desorption behavior of strontium-85 onto montmorillonite and silica colloids. Appl Geochem 16:1653–1662.
- McKinley JP, Zachara JM, Heald SM, Dohnalkova A, Newville MG, Sutton SR. 2004. Microscale distribution of cesium sorbed to biotite and muscovite. Environ Sci Technol 38:1017–1023.
- McKinley JP, Zachara JM, Smith SC, Liu C. 2007. Cation exchange reactions controlling desorption of Sr-90(2+) from coarse-grained contaminated sediments at the Hanford site, Washington. Geochim Cosmochim Acta 71:305–325.
- Meleshyn A. 2010. Adsorption of Sr2+ and Ba2+ at the cleaved mica-water interface: Free energy profiles and interfacial structure. Geochim Cosmochim Acta 74:1485–1497.
- O'Loughlin EJ, Larese-Casanova P, Scherer M, Cook R. 2007. Green rust formation from the bioreduction of gamma-FeOOH (lepidocrocite): Comparison of several Shewanella species. Geomicrobiol J 24:211–230.
- Renshaw JC, Handley-Sidhu S, Brookshaw DR. 2011. Chapter 7. Pathways of Radioactive substances in the environment. Nuclear Power and the Environment. London, UK: The Royal Society of Chemistry, p152–176.
- Ribeiro FR, Fabris JD, Kostka JE, Komadel P, Stucki JW. 2009. Comparisons of structural iron reduction in smectites by bacteria and dithionite: II. A variable-temperature Mössbauer spectroscopic study of Garfield nontronite. Pure Appl Chem 81:1499–1509.
- Russell RA, Holden PJ, Payne TE, McOrist GD. 2004. The effect of sulfate-reducing bacteria on adsorption of Cs-137 by soils from arid and tropical regions. J Environ Radioact 74:151–158.
- Sahai N, Carroll SA, Roberts S, O'Day PA. 2000. X-ray absorption spectroscopy of strontium(II) coordination - II. Sorption and precipitation at kaolinite, amorphous silica, and goethite surfaces. J Coll Interf Sci 222:198–212.
- Steefel CI, Carroll S, Zhao PH, Roberts S. 2003. Cesium migration in Hanford sediment: a multisite cation exchange model based on laboratory transport experiments. J Contam Hydrol 67:219–246.
- Stookey LL. 1970. Ferrozine—A new spectrophotometric reagent for iron. Anal Chem 42:779–781.
- Stout SA, Cho YC, Komarneni S. 2006. Uptake of cesium and strontium cations by potassium-depleted phlogopite. Appl Clay Sci 31:306–313.
- Taylor AS, Blum JD, Lasaga AC, MacInnis IN. 2000. Kinetics of dissolution and Sr release during biotite and phlogopite weathering. Geochim Cosmochim Acta 64:1191–1208.
- Thorpe CL, Lloyd JR, Law GTW, Burke IT, Shaw S, Bryan ND, Morris K. 2012. Strontium sorption and precipitation behavior during bioreduction in nitrate impacted sediments. Chem Geol 306–307:114–122.
- Tsai SC, Wang TH, Li MH, Wei YY, Teng SP. 2009. Cesium adsorption and distribution onto crushed granite under different physicochemical conditions. J Hazard Mater 161:854–861.
- von Canstein H, Ogawa J, Shimizu S, Lloyd JR. 2008. Secretion of flavins by Shewanella species and their role in extracellular electron transfer. Appl Environ Microbiol 74:615–623.
- West JM, Haigh DG, Hooker PJ, Rowe EJ. 1991. Microbial influence on the sorption of Cs-137 onto materials relevant to the geological disposal of radioactive waste. Experientia 47:549–552.
- Zachara JM, Smith SC, Liu CX, McKinley JP, Serne RJ, Gassman PL. 2002. Sorption of Cs+ to micaceous subsurface sediments from the Hanford site, USA. Geochim Cosmochim Acta 66:193–211.