
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In this work, we investigate the long-term (>30 days) competitive interaction of viable Gram-negative bacterium Shewanella putrefaciens and dissolved fulvic acids for aqueous cations of Mn(II) whose compounds play crucial role in geochemistry and mineralogy. It has been proved that the previously discovered bio-precipitates (Mn phosphate and Mn carbonate) were also formed in the presence of environmentally relevant concentrations of fulvic acids by the lower density bacterial cells during long-term sorption of dissolved Mn(II) under the same experimental conditions. However, fulvic acids altered the ratio between the above two compounds and caused the formation of an additional mineral, Mn hydrous oxides. The later finding put in order the entire mineralogical information about Mn-containing minerals dominated by Mn hydrous oxides; as a matter of fact, natural waters/soils always contain humic substances which affect biomineralization processes. The experiments with very high concentrations of fulvic acids showed that at equal binding powers of bacterial cells and aqueous fulvic acids, the latter completely depressed Mn(II) sorption by S. putrefaciens. This basic science study, however, for the first time demonstrated that the beginning of bio-precipitation processes is associated with an intermediate metabolic bacterial state in which the unicells are still viable but nonculturable anymore.
Introduction
Microorganisms and natural organic matter are widely present in every ‘nook and cranny’ of natural environment and consequently greatly influence the cycling of chemical elements on molecular-level and global scales via a variety of physico-chemical processes. It is not surprising that over the last decade, researchers focus on the interfacial reactions which involve either microbes or humic substances, such as sorption, redox transformation, mineralization, dissolution, biodegradation. As for the cycling of metal cations, investigators usually study the metal removal processes caused by either microbes or natural organic matter (NMO) (mainly humic substances as a major part of NMO) but they rarely include both, bacteria and organic matter, into the same experimental systems. In typical (simplified) works, the tasks have been to study one of the processes: adsorption of metal cations by microbial cells (solid-liquid interfaces) (Bahafid et al. Citation2013; Kulczycki et al. Citation2002; Merroun et al. Citation2001; Takenaka et al. Citation2007), chelating (coordination) ability of humic substances as interactions in dissolved state (Biswas et al. Citation2019; Kouhail et al. Citation2016; Town et al. Citation2012), adsorption of aqueous metal cations by undissolved (solid) humic acid/organic matter (Barnie et al. Citation2018; Nachtegaal and Sparks Citation2003) or adsorption of fulvic acids by bacterium without metal cations (Maurice et al. Citation2004).
Moreover, soil microorganisms produce humic compounds which in return influence the viability of their ‘creators.’ Simultaneous presence of both microorganisms and humic substances will always modify (one way or another) the interfacial processes compared with the simplified (1 + 1) systems. Due to the viability of microorganisms and variety of their metabolic pathways, we cannot theoretically predict the long-term fate of chemical elements in the environment affected by microbial interactions which requires experimental evidences to be gained by additional laboratory-based studies.
Recently, Gross and Harrison (Citation2019) highlighted the role of microbial processing in the protection of organic carbon substances, largely via soil aggregates and mineral sorption of DOC compounds, as well as the need to dig deeper into the mechanism of cycling of soil organic C associated with several processes among which are microbial activity, humic substances and minerals.
Since microorganisms and dissolved humic substances are always simultaneously present in natural waters and soils, there is much to be learnt about how they compete for aqueous metal cations during long-term interaction at the interface: bacterial cells – {aqueous metal cations + dissolved humic substances}, and how such complex interplay influence various biomineralization processes. However, Chowdhury et al. (Citation2012) revealed the combined impact of humic acids and bacteria on aggregation and deposition of nano-TiO2. Deonarine et al. (Citation2011) proved the role of humic substances in the formation/stabilization of ZnS nanoparticles. It was demonstrated that humic acids enhanced the formation of illite and talc (Liu et al. Citation2016), changed the removal organic phosphate esters by TiO2 (Tang et al. Citation2018) and altered the phosphorus uptake via struvite precipitation (Zhou et al. Citation2015). Webb and coauthors (2005) determined the fundamental role of simpler background electrolyte cations (Na+ and Ca2+) in the stabilization of the phyllomanganate biooxide structure produced by Bacillus sp., Strain SG-1. Influence of such background electrolyte as humic substances shall be stronger due to their organic chemical nature. In addition, they also contain Na+ (see the XPS data further).
Two transition metals, manganese and iron, play a principal role in geochemical cycling due to abundance of their minerals in Earth’s crust (Calvert and Pedersen Citation1996). Fe- and Mn-based compounds, mainly oxides, serve as adsorbents of ions and molecules which alter speciation and oxidation state of many chemical elements and change their bioavailability. Although participation of microorganisms in the formation of manganese oxides was evidenced in (Mayanna et al. Citation2015), the molecular-level mechanisms of Mn-containing mineral generation was poorly understood.
The previous works (Chubar et al. Citation2013, Citation2015) report a discovery of new biomineralization processes of Mn3(PO4)2 and MnCO3 occurred at the interface of viable Gram-negative bacterium Shewanella putrefaciens (known as Fe/Mn reducer) and aqueous Mn2+. The above novelty resulted from optimization of the experimental conditions, first of all, considerable extension of the contact time (from 2 to 48 h, adopted by most researchers, to 30 days), an increase of batch volume (and respectively, of a number of microorganisms) and of bacterial density. As a matter of fact, biomineralization did not start during a widely accepted contact time of 48 h (which was confirmed by FTIR spectra). First, we conducted the studies in smaller batches of 100 mL which resulted in one precipitate only, Mn(II) phosphate, formed at >4 days (Chubar et al. Citation2013). Initially, we did not expect any difference in larger batches, however, in kinetics studies, we had to increase the batch volumes which produced more precipitates. To our great surprise, we found out that in larger experiments of 1,000 mL, two compounds were bio-mineralized, Mn(II) phosphate and Mn carbonate (however, MnCO3 was never formed in 100 mL batches within 30 days). So, 10 times increase of batch size brought on a discovery of the second biomineralization process, MnCO3, which, during 30 days, was generated at 10, 22 and 30 °C but not at 5 °C, see Chubar et al. (Citation2015). It was demonstrated that the rate of biomineralization and the ratio between these two compounds was a function of the contact time, temperature, batch volume (number of cells) and Mn2+ loading. The experiments were conducted in background electrolyte 0.1 M NaCl which supported cation exchange processes but neither changed Mn(II) oxidation state nor competed with S. putrefaciens for aqueous Mn(II). We did not add any organic substances (for proper nutrition of microbes) to suspensions of viable cells to prevent competitive reactions in dissolved state (phase) which could stop/alter either adsorption of aqueous Mn2+ or bio-precipitation. The above works encouraged us to increase complexity of the experimental systems by adding dissolved humic substances (which play as important role in environmental cycling as microorganisms) to verify whether the same biomineralization processes will also occur. Naturally, large batches of 1000 mL were applied taking into account previous finding. Although humic substances is not a typical food for the nutrition of microorganisms, it is an organic matter which also can serve as a food for heterotrophs. On the other hand, proper (standard) food, given to microbes in laboratories, does not exist in natural environment. So, we added fulvic acids (also organic compounds) which is as widely spread in natural environment as microorganisms. We anticipated that living bacterial cells will definitely use the humic substances for their nutrition in absence of alternative organic food. Any organic matter such as humic substances is much preferred food for bacteria then NaCl (however, the latter is better support of microbial viability than deionized water). In addition, humic substances is strong complexing agent which will definitely compete with microbes for aqueous Mn(II) (with unpredictable theoretically outcome). So, fulvic acids added to the system {aqueous Mn2+ + living cells in NaCl} will play multiple role in the interfacial processes: background electrolyte (such as NaCl but with stronger potential to affect biomineralization), food (organic unlike NaCl) and strong competitor (which can even prevent sorption of Mn2+ by S. putrefaciens).
To summarize, in this study we extend the previous research tasks to investigate the influence of dissolved fulvic acids on biosorption and biomineralization of aqueous Mn(II) at the interface with viable S. putrefaciens. Several experiments were conducted to answer the following questions:
Whether the presence of the environmentally relevant concentrations of dissolved fulvic acids affects the long-term sorption of Mn2+ by viable S. putrefaciens;
If the formation of Mn(II) bio-precipitates (the previously discovered manganese phosphate and manganese carbonate) can be altered by dissolved fulvic acids in any way under the same experimental conditions (large batch volume of 1000 mL and contact time of 30 days);
How very high concentrations of fulvic acids (which do not exist in environment), whose cation exchange capacity is nearly equal to that of bacterial cells, can change the processes at the interface: S. putrefaciens – Mn2+ aqueous (a theoretical task).
Whether the same Mn-containing precipitates will be formed by much smaller number of bacterial cells suspended in the same volume of 1000 mL; bacterial density was an equivalent of an adsorbent dose of 1.0 and 0.5 grams of dry weight per liter (gdw/L) compared with the previously applied 2 gdw/L which resulted in discoveries.
This work is the first report which demonstrates that humic substances (widely present in every environmental system) promote the formation of Mn oxides additionally to (or maybe over) Mn phosphate and Mn carbonates at the end of 30-day experiment, making the former predominant Mn compounds on global scale, otherwise Mn phosphates and Mn carbonates might have dominated the Mn-based minerals in the Earth crust (Chubar et al. Citation2013, Citation2015) since the environmentally relevant contact time is months, years, decades and centuries (not 1–2 h). It is also the first study which points out on the principal role of an intermediate metabolic state of bacteria called viable but nonculturable (VBNC) (or stationary phase) in the formation of the Earth minerals. The results presented in this paper are useful for both areas, biogeochemistry/geomicrobiology and biomineralization/environmental engineering.
Materials and methods
Fulvic acids
In aquatic ecosystems, fulvic acids constitute >80% of humic substances (they dominate over humic acids), unlike in soils. Fulvic acids are characterized by lower molecular mass and greater abundance in carboxylic functional groups compared with humic acids which makes the former stronger chelating agent. Since the contents of humic substances in natural waters vary from 1 to 126 mg/L (Linnik et al. Citation2013), we chose an environmentally relevant concentration of fulvic acids of 45 mg/L; as higher than >100 mg/L rarely occurs in natural ecosystems. We also conducted the experiments with very high loading of fulvic acids of 500 mg/L to answer a fundamental research question (and to satisfy our scientific curiosity). The fulvic acids were purchased from the International Humic Substances Society (IHSS), IHSS Nordic Aquatic Fulvic Acid (1R105F).
Preparation of viable cells of S. putrefaciens for batch adsorption studies
Surface carboxylic groups are also the main functional groups in bacterial cells (like in fulvic acids) which makes them excellent cation exchangers and sorbents of aqueous metal cations. Viable cells of S. putrefaciens strain 200 R were prepared for the experiments using the widely accepted classical microbiological method as also described in the previous works (Chubar et al. Citation2013, Citation2015). Briefly, first, the bacterial cells were grown on agar plates of Luria–Bertani (LB) media after which their single colonies were inoculated into large volumes of liquid LB media and cultivated for 48 h to achieve the maximal concentration of functional groups on the cell surface Haas (Citation2004). The cells were harvested, pelletized by centrifugation at high speed for 20 min in 50 mL tubes, the pellets were washed five times with 0.1 M NaCl solutions, removed from the tubes and dispersed in fresh 0.1 M NaCl for the experiments.
To ensure a particular adsorbent dose, an interdependence between the cell’s optical density (measured at 660 nm) and the adsorbent dry weight (gdw) was preliminary calculated in separate experiments. To establish such correlation between optical density of microbial cells and their dry weight, a large volume of bacterial suspensions of 300–500 mL with the preliminary measured optical density was filtered, dried in oven at 110 °C for 24 h and weighted. This approach of sample preparation guarantees that the constant adsorbent dose in dry weight (gdw/L) will be preserved during the long term (>30 days) experiments in spite of inevitable change of cells’ viability. Unlike in most popular short-term studies (2–72 h contact time), in 1 month experiments the viability of bacterial cells will be amending naturally from fully viable to viable but nonculturable, and then to inactivated/dead cells. However, due to applying 110 °C to define an adsorbent dose in 1 litter, this value will be constant and independent of microbial viability. Whatever the ratio between the three states of microbial matter with different moisture each (simultaneously present in the same experimental batches), an adsorbent dose (measured in dry weight per liter) will be constant. The presence of three bacterial metabolic states in these experiments represents their existence in real natural environment in which viable, nonculturable and dead microorganisms simultaneously co-occur.
It is known that specific surface area plays an important role in adsorption, especially if physisorption mechanism dominates over chemisorption. In the above described experimental systems we could not measure a change of specific surface area of biosorbent S. putrefaciens during the entire experiments of 30 days which would be also useful. However, this issue is minor because of two particularities of the described interfacial processes: tiny surface area of microbes and chemisorption mechanism of the interactions: metal cation – microbial cells. Moreover, as reported in this work, biomineralization in such batches is a function of primarily bacterial viability.
Batch adsorption experiments
The experimental conditions applied in this work were set at 1000 mL of bacterial suspension with an adsorbent dose of 1.0 and 0.5 gdw/L, Mn2+ initial concentration of 200 mg/L, contact time of >30 days and background electrolyte 0.1 M NaCl. Pre-set amounts (volume of a particular concentration) of fulvic acids and stock solution of Mn2+ were simultaneously added to bacterial suspension to ensure the final concentration of fulvic acids of 0.5 g/L or 45 mg/L, and 200 mg/L of Mn(II). An exact concentration of bacterial cells was measured before and after adding the stock aqueous solution with Mn2+. To protect the microbial culture from contamination, large glass beakers containing microbial suspension and Mn(II) were covered by caps. The beakers were uncovered once a day for a few minutes to take small samples (for measurements of Mn(II) and proteins in solution), to check the pH and were quickly covered again. At the end of the experiments, ‘adsorbent’ (microbial cells) was separated from aqueous solutions by centrifugation, filtered using a 0.2-µm pore diameter membrane filter (Advance Microdevices P. LTD), dried at ambient temperature and used for spectroscopic analyses. Frequent sampling of bacterial suspension was avoided to maintain the same experimental conditions during long-term experiments (Chubar et al. Citation2013, Citation2015).
Adsorption was calculated using standard empirical formula:
(1)
(1)
where q (mg/gdw) is the amount of Mn2+ sorbed per gram of adsorbent/bacteria (dry weight), Co (mg/L) is the initial concentration of Mn(II), Ceq (mg/L) is the final/equilibrium concentration of Mn(II) (or measured at a particular contact time), V (mL) is the volume of solution, and m (mg) is the adsorbent weight.
Spectroscopy analysis: Fourier transform infrared (FTIR) and X-ray photoelectron (XPS)
FTIR spectra of the air-dried samples of S. putrefaciens were recorded using KBr pellets at ambient conditions with a Perkin-Elmer 2000 FTIR spectrometer equipped with a DTGS detector. The optical resolution of the spectra was 4 cm−1; 25 scans were collected for each spectrum.
XPS spectra were collected with Kratos Axis Ultra electron spectrometer using a mono Al Kα source operated at 150 W and a low-energy electron gun for charge compensation. Survey scan spectra were written in 5 min using pass energy of 160 eV and a 1 eV step. The hybrid lens mode was used for all measurements. The samples were hand-pressed in a molybdenum holder with a 5 mm diameter through of variable volume. The spectra were processed using Kratos software. The binding energy (BE) scale was referenced to the C 1 s line of aliphatic carbon contamination, set at 285.0 eV.
Analytical chemistry measurements
Concentration of Mn2+ was analyzed using inductively coupled plasma mass spectrometry (ICP-MS) and spectrophotometry method (formaldoxime colorimetric method (Burle and Kirby-Smith Citation1979). The proteins concentration was determined by the Bradford reagent using bovine serum albumin as standard (Bradford Citation1976). These measurements were conducted in the same sample taken once a day and filtered using 0.2 µm pore size membrane filter which ensures that we analyzed the substances in fully dissolved state. Bradford method is well-known, widely accepted and approved tool for measurements of total protein-like substances.
Results and discussion
Long-term interaction of viable S. putrefaciens (0.5 gdw/L) with aqueous Mn(II) in absence/presence of equal concentration of dissolved fulvic acids of 0.5 g/L
In the first series of experiments, we were answering the fundamental science question which tested the competitiveness of complexing ability of dissolved fulvic acids and the sorptive (cation binding) power of S. putrefaciens at their equal cation exchange capacities. An average cation exchange capacity of fulvic acids was estimated from the literature which showed that total acidity (from proton titration) of humic and fulvic acids ranges 9.0–11.6 meq/gC (Ma et al. Citation2001); whereas the carbon (C) content in the same substances constitutes 30–50%. These data indicated that the cation exchange capacities of viable S. putrefaciens (5.3 meq/gdw calculated from batch potentiometric titration (Chubar et al. Citation2008) and fulvic acids were roughly equal. Since we decided to use an adsorbent dose of 0.5 gdw/L in this batch study, the concentration of fulvic acids was also fixed at 0.5 mg/L. Results of Mn2+ sorption by S. putrefaciens in absence/presence of the same concentration of fulvic acids are shown in and for short-term and long-term contact time, respectively.
Figure 1. Short-term removal of aqueous Mn(II) by viable S. putrefaciens in absence/presence of equal concentration of fulvic acids (A) and alteration of pH in the same experimental batches (B).
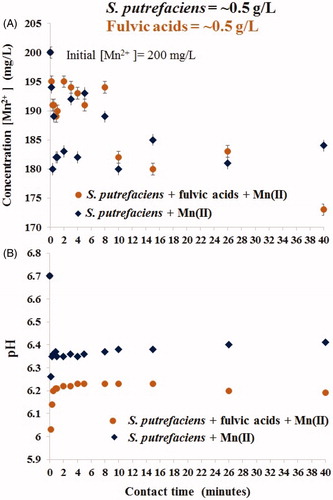
Figure 2. Long-term uptake of aqueous Mn(II) by S. putrefaciens in absence/presence of equal concentration of fulvic acids (A) and drift of pH in the same experiments (B).
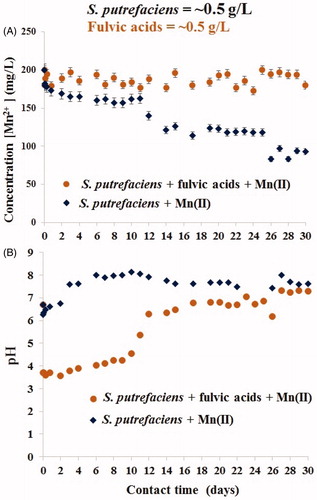
reflects the Mn(II) removal data (A) and pH drift in the same batches (B) during the first 40 min. presents results from continuation of the same experiments over 30 days. demonstrates that despite the lower pH values caused by the presence of (highly acidic) fulvic acids (B), over the first 40 min the rates of Mn2+ removal was close in value in both fulvic-free and fulvic-loaded studies (A). In both experiments, Mn(II) concentration was reduced from 200 to ∼180 mg/L. Such fluctuation of metal cation concentration (due to adsorption/desorption processes) is very typical phenomenon at the interface with viable microbial cells due to their viability. The cells were strong (which is indirectly shown by stable pH without fluctuation) and were trying to desorb Mn2+ from their surface (reflected by change of aqueous Mn(II) concentration). Also, due to viability of the ‘sorbent’ there was no traditional pH effect of metal cation sorption: the higher pH the greater uptake, which also proves a leading role of microbial viability in Mn2+ removal. During the first 24 h, Mn2+ was removed with the same rate from both batches after which its uptake continued from fulvic-free experiment only, see . In fulvic-present study, we observed a partial desorption (by ∼50% of just sorbed cations), from 180 to 190 mg/L on days 2–3, and thereafter nearly the same Mn2+ concentration of ∼190 mg/L was measured on from the 2nd to 24th days. Full desorption was recorded at 24–30 days as confirmed by the release of Mn(II) concentration in solution to the initially introduced ∼200 mg/L. Interestingly, the presence of fulvic acids resulted in greater fluctuation of adsorption/desorption reactions compared with fulvic-free systems, see . The alignment of pH values in both experiments at >14 days did not change the previously established trends of Mn(II) removal in each batch.
Although the solid phases were not collected from these studies, we can conclude from the curves in that bio-precipitation of Mn-containing compounds also took place in fulvic-free system at bacterial density of 0.5 gdw/L. The previous results revealed that exclusively adsorptive capacity of dead S. putrefaciens to Mn(II) (without contribution from biomineralization) cannot be higher than approximately 20 mg[Mn]/gdw (Chubar et al. Citation2013). It was shown that for short contact time (24 h), Mn2+ removal by both live and dead S. putrefaciens was nearly the same. We also defined in the earlier works that biomineralization of Mn2+ by S. putrefaciens begins at >4 days. Consequently, we can conclude (roughly estimate) that deeper (than approximately 20 mg/gdw) aqueous Mn(II) removal from water solution by viable cells was always caused by an additional interfacial process, in particular, bio-precipitation of Mn-containing minerals. presents the precisely calculated data on Mn2+ removal in fulvic-free and fulvic-affected batches using two units of measurements, % removal and sorption (mg[Mn]/gdw). We realize that the value ‘sorption’ (q, mg/gdw) measured is inaccurate (conditional) because this uptake includes also precipitated minerals attracted to microbial surface, however, it is always useful to treat experimental data using both units of measurements (traditional in adsorption science).
Table 1. Adsorptive (q, mg/gdw) and removal (%) characteristics of viable S. putrefaciens of the density (adsorbent dose) of 0.5 and 1.0 gdw/L at Mn2+ initial concentration of 200 mg/L.
Long-term uptake of Mn2+ by viable S. putrefaciens (1 gdw/L) in absence/presence of the environmentally relevant concentrations of dissolved fulvic acids of 45 mg/L
To estimate the influence of these major natural organic matter on the processes taking place in natural ecosystems at the interface with microbial cells, in the second series of the experiments we applied much smaller concentration of fulvic acids of 45 mg/L. It is a typical average concentration of fulvic acids occurred in natural waters. and show the rate of Mn2+ removal over 30 min and 38 days, respectively, by viable cells of S. putrefaciens (1 gdw/L) uninfluenced/affected by fulvic acids (45 mg/L) (A) and drift of pH in the same suspensions (B).
Figure 3. Kinetics of Mn(II) sorption by viable S. putrefaciens (1 gdw/L) for 30 min in absence/presence of fulvic acids (45 mg/L) at the initial Mn(II) concentration of 200 mg/L (A) and drift of pH in the same batches (B).
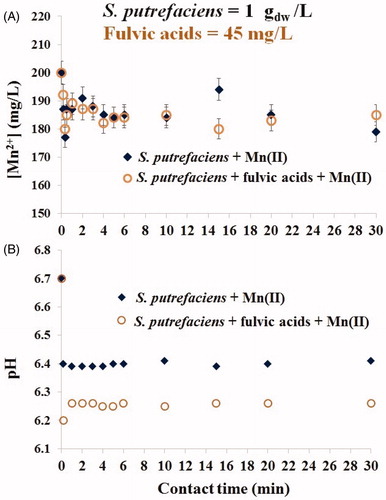
Figure 4. Kinetics of Mn(II) sorption by viable S. putrefaciens (1 gdw/L) for 38 days in absence/presence of fulvic acids (45 mg/L) at the initial Mn(II) concentration of 200 mg/L (A) and change of pH in the same experiments (B).
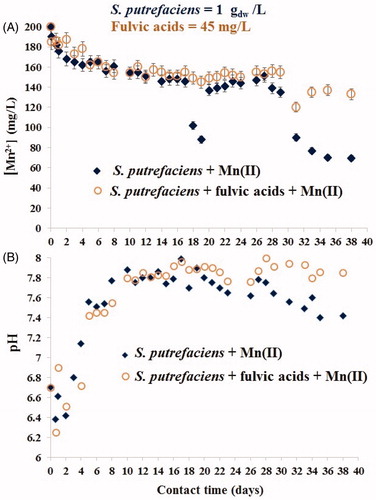
We observed that in the beginning of solid-liquid interactions () the environmentally relevant concentration of fulvic acids did not affect the adsorptive capability of living cells (A), however, pH in the same suspensions was lowered (B). Notably: after 24 h, viable cells were able to align the pH values in fulvic-present batches to an approximately the same level as in the fulvic-free systems, and this trend was preserved over the next 20 days (). demonstrates that during the first 26 days, fulvic acids did not alter the Mn(II) removal of viable S. putrefaciens (A) which correlated with the unchanged pH values in fulvic-free and fulvic-present batches (B). However, extension of contact time until longer than 26-day period caused different sorption results. The removal of Mn2+ from fulvic-free batches continued and even intensified for additional 10 days, but it was highly inhibited in fulvic-present systems. These data suggest that the physico-chemical conditions, established in the experimental batches on 26th day and thereafter, became a dominating factor (which directed the interfacial processes) over bacterial viability. Both the shape of the curves in and the values of the uptake capacities indicated that bio-precipitation was a major mechanism of aqueous Mn(II) removal by the twice smaller number of S. putrefaciens cells, 1 gdw/L, compared with the previously used 2 gdw/L (Chubar et al. Citation2013, Citation2015). Two spectroscopy methods, FTIR and XPS, confirmed the fact of formation of the same two major precipitates (manganese phosphate and manganese carbonate) in both fulvic-free and fulvic-present systems which is the topic of the next sections.
We would like to remind that the interfacial processes in the system {viable S. putrefaciens + Mn2+ aqueous} always start from sorption/cation exchange and continue (from >4th day) by biomineralization which also include periods of desorption and, if having different background electrolyte, may be accompanied by Mn(II) oxidation (there was no Mn2+ oxidation in the previous works, without humic substances (Chubar et al. Citation2015)). So, the term ‘removal’ here includes several reactions which extract aqueous Mn2+ from water solutions: sorption/cation exchange (whereas Mn(II) is pulled out by binding to the surface of S. putrefaciens) and Mn2+ bio-precipitation (when dissolved Mn2+ is eliminated from aqueous phase via the formation of new, solid, phases).
Removal of aqueous Mn2+ by viable S. putrefacient influenced by fulvic acids under the experimental conditions described in two previous subsections
Since it is (visually) difficult to assess Mn2+ removal itself from and , has been prepared to gather and present the calculated data on Mn(II) uptake from both experiments (shown in ) using two (standard for sorption science) units of measurements: % of Mn(II) removal and sorption (q, mg[Mn]/gdw). (along with and ) shows that in fulvic-free batches the Mn2+ uptake efficiency (% removal) was nearly the same for higher (1.0 gdw/L) and lower (0.5 gdw/L) density of viable microbial cells. This indicates that in long-term process, the rate of Mn2+ biomineralization by both quantities of bacterial cells (0.5 and 1.0 gdw/L) was unaffected by adsorbent dose (within this range), and bio-precipitation was a function of Mn(II) concentration and microbial viability. The formation of inorganic compounds in such systems was initiated by living microorganisms; biomineralization could not have taken place without viable bacterial cells as demonstrated in the previous works (Chubar et al. Citation2013, Citation2015). It is known that large microbial community assembles stronger each cell making them much ‘greater than the sum of parts’ (as written in any reference book of microbiology or water disinfection). However, from these data it seems to us that at microbial density ranged 0.5–1.0 gdw/L, the contribution from bacterial viability to the interfacial processes was a constant value but only in the absence of fulvic acids. The environmentally relevant concentrations of fulvic acids did not prevent either sorption of Mn2+ or its bio-mineralization initiated by viable S. putrefaciens.
FTIR examination of S. putrefaciens samples (1 gdw/L) contacted to aqueous Mn2+ over 38 days in presence/absence of (45 mg/L) dissolved fulvic acids
FTIR spectroscopy is powerful tool to reveal mechanism of metal cation sorption by biomaterials and to detect (confirm the fact of) biomineralization. However, when biomineralization (of inorganic phases) has started by microorganisms this process becomes dominating and we cannot distinguish (observe) sorption/ion exchange anymore in FTIR. Therefore, before to move to analysis of FTIR spectra of S. putrefaciens samples dominated by new inorganic phases generated by these microbes for 30 days (), we first review the FTIR of the same bacterium which was in contact with aqueous Mn2+ for three days only, when bio-precipitation did not begin yet. shows FTIR spectra of two S. putrefaciens samples one of which was contacted to aqueous Mn2+ in 0.1 M NaCl background electrolyte for three days (sorption experiment) and the second was recorded from original (not contacted to aqueous Mn2+) bacterial cells, presented in region 400–2000 cm−1 in which all functional groups of microorganisms are reflected. We can easily conclude that IR-spectra of both samples are identical: there are exactly the same IR-features resulted from vibrations of the same functional groups. Mn2+ sorbed to S. putrefaciens for three days did not do any principal changes to this biosorbent. In region 400–1170 cm−1, there are IR-active bands generated due to vibrations of carbohydrates including polymeric sugars known as Extracellular polymeric substances (EPS). In the same region can be found also IR-reflections of inorganic materials, metal phosphates (see the following paragraphs), however, in there are no evidences of new inorganic phases precipitated over 3 days in bacterial samples due to the contact with Mn2+. To conclude on a particular new inorganic phase, in FTIR we must see distinguished sharp shoulders (or peaks) in the respective characteristic regions. Comparing the two FTIR spectra in we can only suggest that some (tiny amounts) of EPS might have been generated as evidenced by an increase of broad band at around 550 cm−1. Carbohydrates are abundant in OH– functional groups which are not classical cation exchange centers, so we did not expect these groups to be major cation removal sites. This anticipation was confirmed as there are no shifts of any bands assigned to carbohydrates in FTIR of Mn(II)-sorbed sample compared with the initial S. putrefaciens (1164/1165, 1070/1067 cm−1 and the region at around 550 cm−1). Complexation or cation exchange to sorbent binding sites are usually recognized from the shifts of characteristic bands for at least approximately 10 cm−1. Surprisingly, phosphate groups did not play a role in the removal of Mn2+ during three days as evidenced by the same position of characteristic features at 1235 cm−1 in original cells and those used in sorption study, respectively. However, as expected, carboxylic groups were actively involved in aqueous Mn2+ sorption as shown by band shift from 1400 to 1385 cm−1 in initial cells and those used in sorption experiment, respectively. Although coordination ability of amino groups to metal cations is well-known, we did not forecast these groups (in companionship with carboxylate and phosphate) to be major Mn2+ removing sites. And indeed, there was no shift of amide II characteristic bands at 1535 cm−1 (either amine I). Some difference in highest of these bands resulted from the spectra treatment (normalization to one band) and preparation (different amounts of S. putrefaciens samples might have been pressed in KBr pellets during FTIR recording) but the spectra contain reflections of the same chemical bonds of the same material (substances).
Figure 5. FTIR spectra of initial S. putrefaciens and contacted to aqueous Mn2+ in 0.1 M NaCl electrolyte for three days.
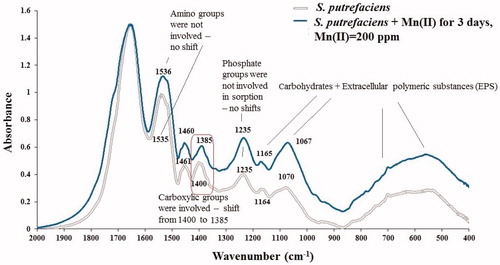
After consideration of FTIR spectra of S. putrefaciens with Mn(II) sorbed for 3 days () we can easily acknowledge the fact of biomineralization in and see the changes (‘damage’) made to organic material (S. purefaciens) by mineralized cations of Mn2+ transformed into new inorganic phases, Mn-containing precipitates. presents FTIR spectra of the experimental samples (shown in ), original (not used in the experiments) S. putrefaciens, and fulvic acids, as well as FTIR of two references, Mn3(PO4)2 (B) and MnCO3 (C). summarizes (interprets ) the main characteristic IR-active features in four samples shown in (we do not repeat all of them in the text); the literature references in are given to support the interpretation of FTIR spectra of .
Figure 6. FTIR spectra of initial S. putrefaciens (1 gdw/L), contacted to Mn2+ in absence/presence of fulvic acids (45 mg/L) for 38 days and fulvic acids used in this study (A), and of two references, Mn3(PO4)2 (B) and MnCO3 (C).
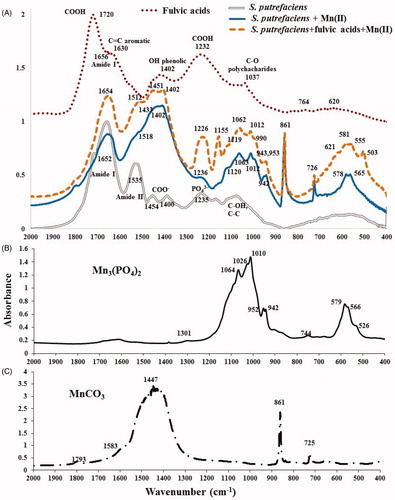
Table 2. Characteristic (400–2000 cm−1) IR-vibrations in experimental samples of original S. putrefaciens (1 gdw/L) and influenced by aqueous Mn2+ in absence/presence of fulvic acids (shown in ).
FTIR of fulvic acids (used in the experiments) displayed all characteristic vibrations of this class compounds ( and ), however, most of major bands (at 620, 1232 and 1402 cm−1) were smoother than those observed by other researchers (Naidja et al. Citation2002) as indicated by several overlapping peaks in each IR-active region.
A quick glance at FTIR-spectra of the experimental samples in (30 days contact time) and comparison with IR-spectra in (3-day experimental duration) clearly demonstrates that the spectra in are heavily dominated by vibrations of new inorganic phases; IR-active features of purely organic adsorbent (S. putrefaciens) is hardly visible anymore. Reviewing the main characteristic bands in together with FTIR of two references, Mn3(PO4)2 () and MnCO3 (), allows to conclude that these two compounds are major precipitates in both bio-composites, formed in fulvic-free and fulvic-affected batches, however, fulvic acids promoted the formation of an addition substance. suggests that biomineralization was a dominating process at the interface: S. putrefaciens-aqueous Mn2+, under both the fulvic-free and the fulvic-affected experimental conditions as reflected at 400–650 cm−1 (characteristic IR-region of phosphates, oxides and polysaccharides), 900–1300 cm−1 (inorganic phosphates), and {700–900 + 1300–1600 cm−1} (inorganic carbonates), see also . contains the explanation to with assignments to each band which is supported by the literature. Although the FTIR does not allow to quantitively define the ratio between Mn3(PO4)2 and MnCO3 (as done by XANES/EXAFS in (Chubar et al. Citation2013)) it qualitatively confirmed the presence of MnCO3, which is brightly shown by two characteristic bands at 861 and 726 cm−1 complemented by several others (at 1402–1518 cm−1), and of Mn3(PO4)2, whose characteristic features are easily recognizable at 942–1120 cm−1, see and . These reflections cannot belong to any organic compound in S. putrefacies, not contacted to Mn2+. Three IR-features at 1402–1518 cm−1 attributed to manganese carbonate (formed by S. putrefaciens) differed from IR-spectrum of pure compound MnCO3-reference () which usually shows one high peak at around 1447 cm−1 and weak shoulder on both sides of it which is expected feature due to complexity of bio-precipitate composites. Other researchers also observed similar phenomena of severe carbonate vibrations near 1400 cm−1 which were attributed to either CO32− replacing PO43− ions without an adjacent OH− ion (Vignoles-Montrejaud Citation1984) (at 1480 cm−1) or to the A-type carbonate substitution (Elliot Citation1994) (weak shoulders at 880, ∼1495 and ∼1530 cm−1). In the previous articles (Chubar et al. Citation2013, Citation2015) we evidenced that biomineralization of Mn-containing compounds by S. putrefaciens always starts from the formation of Mn phosphate (at ≥4 days) and, in larger batches, continues with precipitation of MnCO3 (at ≥8 days). Such regularity of stepwise biomineralization is also reflected in carbonate IR-region at 1402–1518 cm−1, which indicates either replacement of a part of Mn3(PO4)2 by MnCO3 or (most probably) the the formation of mixed/hybrid compounds Mn3(PO4)2-MnCO3. FTIR spectrum of fulvic-influenced sample also revealed both the presence of fulvic acids and their influence on the interfacial processes. Two new distinguishing IR-features at 1155 and 1226 cm−1 in fulvic-affected sample, which were not measured in fulvic-free systems, were attributed to vibrations of a new inorganic phase (an additional-different compound to Mn phosphate and Mn carbonate) and fulvic acids, respectively. Since the formation of Mn hydrous oxides in fulvic-present system was confirmed by XPS analysis, bands at 1155 and 503 cm−1 were attributed to this new inorganic phase. It was shown in the literature that metal oxides often produce IR-bands in the same regions (Parikh and Chorover Citation2005). Similar IR-active features were measured in the PANI functional MnO2 nanocomposite (1222 and 507 cm−1), see Yang et al. (Citation2011). The second peak at 1226 cm−1 was attributed to fulvic acids which was sorbed alongside with Mn2+. Overall, the intensively formed inorganic compounds strongly dominate most of the spectra in the same IR-region which is also characteristic of both organic phases (bacterial cells and fulvic acids). Generous presence of inorganic phases oppresses vibrations in organic substances making the later invisible (or less detectable) in IR-spectra. Region 500–600 cm−1 reflects an occurrence of both fulvic acids (at 621 cm−1) and Mn hydrous oxide (at 503 cm−1). However, typical sharp duplets at 656/578 and 555/581 cm−1 in fulvic-free and fulvic-affected samples, respectively, shall be attributed to Mn phosphate (Chubar et al. Citation2013) (see ); these peaks cannot be assigned to organic material.
Overall, FTIR examination already answered the major questions of the research tasks. Twice smaller number of living cells (1 gdw/L) produced the same two precipitates (Mn phosphate and Mn carbonate) as 2 gdw/L, discovered in the previous works (Chubar et al. Citation2015). Environmentally relevant quantities of fulvic acids did not prevent the formation of these two inorganic minerals, however, they affected the composition of bio-composites and most probably occasioned precipitation of an additional, third, mineral.
XPS surface analysis of raw viable S. putrefaciens (1.0 gdw/L) and contacted to Mn2+ for 38 days in presence/absence of fulvic acids
XPS data (shown in and ) confirmed the presence of Mn in both samples of S. putrefaciens contacted to aqueous Mn2+ in absence/presence of fulvic acids.
Table 3. C 1 s and N 1 s XPS features in four samples: uncontacted S. putrefaciens and with Mn(II) sorbed in absence/presence of fulvic acids over 38 days, and fulvic acids.
Table 4. XPS features (O 1 s, Mn 2p, Na 1 s, P 2p, Cl 2p and S 2p) in four samples: initial S. putrefaciens and with Mn(II) sorbed in absence/presence of fulvic acids over 38 days, and fulvic acids.
Concentration of Mn at.% on the surface measured by XPS was in correspondence with the removal data defined by Mn measurements in solution, see and . XPS analysis revealed that uptake of Mn2+ by this biosorbent from fulvic-free batch was almost twice higher than from fulvic-affected systems, 3.32 and 2.11 at.% of Mn, respectively. It is a good correlation with the removal measured in liquid phase considering very narrow surface layer studied by XPS. O 1 s XPS analysis demonstrated that Mn hydrous oxides are present in fulvic-influenced sample as shown by peak at 529.9 eV to be attributed to Mn-O (https://xpssimplified.com/). The XPS data suggest that an approximately 25% of Mn-containing precipitates (0.73 at%) in fulvic-affected sample was found in hydrolyzed (hydroxide) form. Positions of Mn 2p 3/2 XPS peaks (at 641.2 and 641.3 eV in fulvic-free and fulvic-present samples, respectively) demonstrated partial oxidation of Mn(II) to Mn(III) in fulvic-loaded sample (Nesbitt and Banerjee Citation1998; Huang et al. Citation2015). Although the available data confirmed the fact of Mn(II) oxidation, they did not allow precise estimation of Mn oxidation state.
Bio-precipitation of Mn3(PO4)2 was proved by an increase of phosphorus content (XPS feature P 2p 3/2 XPS at 133.3 eV attributed to PO43−) in bacterial sample contacted to aqueous Mn(II) (1.81 and 1.78 at.% in fulvic-free and fulvic-present samples, respectively) compared with raw (uncontacted) microbial cells (0.73 at.%), see . The formation of MnCO3 was evidenced by an increase of carbon (C) surface concentration (associated with carbonate-attributed band at 289.3 (in virgin biomass) and 289.6 (in used for experiment cells) eV (C 1 s)) from 1.0 to 1.70/2.41 at.% in original biomass and contacted to aqueous Mn(II) in absence/presence of fulvic acids, respectively. XPS databases refers BE of C1s at 288–290 eV to metal carbonates (https://xpssimplified.com/elements/carbon.php#inorganic). Even the fact of different BE of C 1 s in original cells (289.3 eV) compared with those used in sorption studies (289.6 eV) already demonstrates that something new, inorganic, has been originated, which, in this case, is Mn carbonate, as already proved (100%) by FTIR spectra. Moreover, XPS database, mentioned above, also indicates that greater BE of C 1 s in inorganic carbonate reveals an occurrence of ‘possible metal hydrocarbonate’ (in contrast to metal carbonate) as also written in the article (Shchukarev and Korolkov Citation2004) referred by this site. We know from Mn 2p XPS (and from the removal data) that amount of Mn-containing precipitates is smaller in fulvic-affected batch (see ) which shall be shown by lower At.% of both, inorganic phosphate (P 2p) and inorganic carbonate (C 1 s). The above estimation indeed was reflected in greater content of inorganic phosphate, P 2p (see ) in fulvic-free sample. However, instead, we measured larger At.% of carbonate/carboxylate-assigned C 1 s in fulvic-present batch (compared with fulvic-free sample) which can be referred to carboxylic groups of fulvic acids sorbed to S. putrefaciens, only.
The presence of fulvic acids in fulvic-affected sample in XPS data was indicated by: both higher concentration of C (assigned to carbonate + carboxylate) in fulvic-influenced biomass (2.41 at.%) compared with fulvic-free batches (1.70 at.%) due to greater content of carboxylic (-COOH) groups (featured at 289.1 eV), 10.69 at.%, in fulvic acids itself; greater content of C in amide (10.20 At.%) compared with fulvic-free study (9.91At.%); bigger quantity of unprotonated amide from N1s (8.89 vs 7.39 At.%) and greater amount of protonated amide from N1s (0.64 against 0.51 At.%), in fulvic-affected and fulvic free samples, respectively, see . All these increased XPS features of organic constituents in fulvic-affected sample could come from an additional organic matter only which is nothing else but fulvic acids. We rely on the reviewing of the entire XPS data. Moreover, we noted on FTIR that fulvic acids (highly acidic substance) were sorbed by S. putrefaciens along with Mn2+. Concentration of total O and total N on the surface of fulvic-affected cells was an approximately 15% higher than in fulvic-free batches which indicated the presence of larger portion of organic phases in this organic-inorganic composite compared with the fulvic-free experiment. Conclusions drawn from XPS measurements correlate with FTIR spectra () which also demonstrated that fulvic-affected sample contained a greater amount of organic substances than fulvic-free biomass.
Uptake of fulvic acids alongside with sorption of Mn2+ resulted in full elimination of Na (see Na 1 s XPS data at 1071.70 eV in ). However, concentration of Na+ in S. putrefaciens used to sorb Mn2+ from fulvic-free batches decreased from 2.60 at.% in uncontacted bacterium cells to 0.28 at.% . demonstrates that Na was not detected on the surface of S. putrefaciens contacted to Mn2+ in the presence of fulvic acids which were also sorbed by these microorganisms. We cannot provide (research-based) interpretation of this outcome on full Na+ removal in fulvic-affected sample as it was collected at the end of the experiment, at 38 days, after long-term (from 4th to 5th day onward) domination of bio-precipitation which was stabilized (not continued) in the last 6 days (see ). However, we can suggest a theoretical explanation based on the literature. As for the beginning of the interfacial interactions, when cation exchange (sorption) was the initiating step during the first three days, we suppose that cation exchange of either surface H+ or Na+ (of –COOH and COONa, respectively) for aqueous Mn2+ was more preferred in fulvic-free and fulvic-affected batches, respectively. Also, while fulvic acids was sorbed in parallel with Mn2+, it is possible that divalent ions Mn2+ could serve as a bridge (as described by Kawano and Tomita Citation2001) linking negatively charged carboxyls on the cell surface to negatively charged carboxyls of fulvic acids, and cations Na+ made it easier (energetically favorable) over H+.
For the last 6 days (at 32–38 days) (when the removal of Mn(II) continued in fulvic-free batch) there was a period of stagnation (stabilization) of Mn2+ uptake in fulvic-affected study, during which the interactions between all components prolonged which resulted in full wash out of Na+ from solid phase by dissolved fulvic acids. We suggest that in this latest period, Mn2+ was kept in dissolved state (and not being allowed to precipitate) due to its chelation by the strong complexing agent, fulvic acids.
Release of proteins by viable cells as reflection of their viability
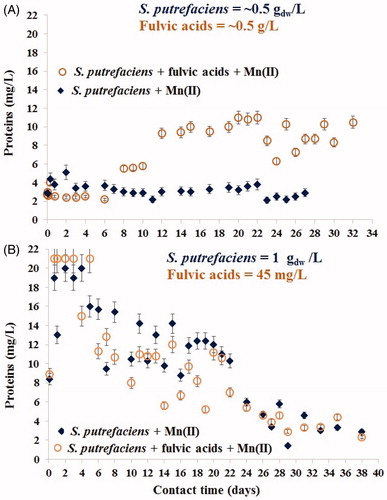
Protein-like compounds released by viable S. putrefaciens contacted to aqueous Mn2+ were measured (in the same filtered sample used to measure Mn which contained fully dissolved state of the matter – so only dissolved total protein-like substances were determined) to better interpret the adsorption/biomineralization processes as these data reflect the response of viable cells of S. putrefaciens (of densities of 0.5 gdw/L (A) and 1.0 gdw/L (B)) to the contact with aqueous Mn(II) in absence/presence of fulvic acids, see . In this figure, beginning of the interfacial processes is shown in the upper graphs (1). Although it is known that concentration of proteins released by microbes is a function of their density (quantity of cells), we could not predict that twice greater number of microorganisms can release an approximately sevenfold much of dissolved protein. Concentrations of these compounds measured over the first 6 days constituted 3–4 and 19–21 mg/L in fulvic-heavy/lightly loaded experiments, respectively, see . Fivefold increase of proteins on the 7th day in batches exposed to high content of fulvic acids (0.5 g/L) indicated more vigorously metabolizing cells (and their stronger viability) compared with the fewer-fulvic loaded experiment. These particularities of metabolism intensiveness correlated with desorption of Mn(II) in fulvic-heavily-loaded batch, see . In the system with the environmentally relevant content of fulvic acids (45 mg/L), biomineralization did not start during the first 4–6 days until the release of proteins was high: 19–21 mg/L, see (Chubar et al. Citation2015). Beginning of bio-precipitation in this study correlated with a decrease of dissolved proteins content over time which also indicated the inhibition of bacterial metabolism intensiveness, see . This observation demonstrated that bacterial biomineralization is a function of their viability. It also revealed that the biomineralization processes were initiated by viable-but-nonculturable bacterial cells which were true at least for the formation of Mn-containing minerals. There was no correlation, however, between the release of proteins () and the uptake of Mn(II) () at 28–38 days. This shows that although Mn2+ biomineralization was initiated by living-nonculturable microbial cells, at such a longer contact time this process was already ruled by physico-chemical conditions (not microbial viability). Most probably, the predominant part of microorganisms was already dead. That was why dissolved fulvic acids (as a strong chelating agent) was capable to keep the rest of Mn2+ cations (not precipitated yet) in dissolved state via complexation.
Figure 7. Concentrations of dissolved protein-like substances released by viable S. putrefaciens for one month contact with aqueous Mn2+ in absence/presence of very high (A) and low (environmentally relevant) (B) contents of fulvic acids.
Mn2+ removal mechanism, and implication to biomineralization and environmental cycling
Despite the well-known complexity of natural environmental systems, laboratory-based setups aiming to investigate the fate and speciation of chemical elements in the environment (such as metal-microbe interaction) usually involve two major participants. Although the knowledge on the simplified systems (microbes + aqueous metal cation; or humic compounds + metal cation) are utmost important to reveal the mechanisms of the respective interactions, we must increase experimental complexity to compare results obtained from both the simplified and the complex studies. This is the only way to explain many phenomena in the Earth’s history.
Numerous articles demonstrated that microorganisms played a leading role in the most mineralization processes taking place over centuries and millennia. However, understanding of the molecular-level mechanisms of the mineral’s formation is presently in the embryonic stage. In the previous works (Chubar et al. Citation2013, Citation2015), we discovered two biomineralization processes resulted in precipitation of Mn3(PO4)2 and MnCO3 at the interface of S. putrefaciens and aqueous Mn2+ which was due to optimization of the experimental conditions. Among the rarely explored experimental set-ups were: (a) an extended contact time (until 30 days) which closed the laboratory-based experimental conditions to those in real natural environment; (b) application of a typical (in adsorption science) bacteria density (called an adsorbent dose) of 2 gdw/L; (c) large batch volumes of 1000 mL (with high microbial density); and (d) long-term temperature effect investigations.
The first series of our experiments on Mn(II) sorption by S. putrefaciens (Chubar et al. Citation2013, Citation2015) did not contain any organic substances (which could be a carbon source for viable microorganisms) as they could strongly affect the sorption processes (organic substances are binding ligand for metal cations). However, within the time allotted by the project we conducted several additional studies which included dissolved fulvic acids in the system: S. putrefaciens+ aqueous Mn2+. Humic substances (especially, fulvic acids) and microorganisms are both abundant in carboxylic groups materials (-COOH) which serve as perfect scavengers of metal cations in any liquid or solid states. The presence of both of them in the same environment (experiments) will affect the interfacial processes. Any mineralization always depends on experimental conditions including background electrolyte (Webb et al. Citation2005). The presence of fulvic acids in the system: S. putrefaciens + aqueous Mn2+, might alter the composition of bio-precipitates, can result in the formation of different minerals or even might prevent Mn2+ sorption or its bio-precipitation. Humic substances can also desorb the initially sorbed Mn(II) by microorganisms at an extended contact time in the same manner as the humic acids were the washing agents for Cd-contaminated soils (Meng et al. Citation2017). Due to microbial viability, we cannot predict the influence of fulvic acids on such processes which requires empirical evidences.
In this work, we give the answers for several research questions (obtained experimentally) regarding S. putrefaciens-versus-fulvic acids competitive interactions with aqueous Mn2+. It has been demonstrated that over the first 3–4 weeks, environmentally relevant concentrations of dissolved fulvic acids did not influence a capability of viable S. putrefaciens to sorb aqueous Mn2+. However, at longer than 4-week contact time, fulvic acids prevented a continuation of Mn(II) removal.
Although environmentally relevant concentrations of dissolved fulvic acids did not stop bio-precipitation of aqueous Mn(II) at the interface with viable cells of S. putrefaciens, they changed chemical compositions of adsorbate (as reflected by the concentrations of dissolved proteins), fortified the viability of microbial cells and were also sorbed by bacterial cells alongside with Mn2+. Such multi-influence of fulvic acids can result in the formation of either more precipitates or/and their different structures compared with the fulvic-free systems. We found out that the same two major compounds (Mn3(PO4)2 and MnCO3), which were reported in the previous paper (Chubar et al. Citation2015), were also formed in both fulvic-free and fulvic-loaded batches by smaller quantity of bacterial cells, however, dissolved fulvic acids caused precipitation of an additional (third) mineral (Mn hydrous oxides) and partial oxidation of Mn(II). Both of the later processes were at the initial stage.
The earlier discovered bio-precipitation of Mn3(PO4)2 and MnCO3 initiated by viable S. putrefaciens at the interface with aqueous Mn2+ did not perfectly correlate with the overall data on minerology of Mn. It is known that most of Mn-based minerals are either hydrous (mixed/double) oxides or partially hydrolyzed (hydroxy) Mn salts (phosphates, carbonates, silicates) (http://webmineral.com/). However, introduction of fulvic acids to the experiments put these results on Mn2+ biomineralization regularities in the systems {S. putrefaciens - aqueous Mn2+} in agreement with the mineralogy of Mn. As a matter of fact, whatever the natural environment (soils or waters including groundwaters, thermal waters and wastewaters), humic substances are always a part of its chemical composition. In this work, we demonstrated that humic substances (widely present everywhere in natural environment) shifted the biomineralization processes toward the formation of Mn hydrous oxides which is the reason of their abundance among Mn-based minerals.
Unlike the environmentally relevant contents of fulvic acids, very high concentrations of fulvic acids, nearly equal to the dose of bacterial cells referring to cation exchange capacity, indeed prevented the sorption of aqueous Mn2+ by viable cells of S. putrefaciens. The initially sorbed (in small quantities) cations of Mn(II) were fully desorbed at the end of the long-term experiment. This was a purely fundamental science research study, however, it resulted in a practical discovery. It was noted that Mn(II) sorption/desorption reactions did not correlate with a change of pH in the same batches. However, the data agreed with the concentrations of dissolved protein-like substances released by bacterial cells which reflected their viability. Very high concentrations of fulvic acids served as organic food for these cells keeping them viable for longer time which in turn prevented Mn2+ biomineralization and finally resulted in full desorption of Mn(II). It is the first observation which demonstrated that biomineralization processes at the interface with bacterial cells are associated with the intermediate state of microbial viability, called viable but nonculturable anymore (which differ them from fully viable and completely dead microorganisms). This information can be useful in a variety of applications in which biomineralization play a key role.
The idea of the role of bacterial viability in biomineralization is supported by both (fulvic-lightly/heavy loaded experiments). At equal binding powers of fulvic acids and microbes (0.5 q/L), the former was the winner in the competition with viable cells for aqueous Mn2+ (which prevented biomineralization). At the environmentally relevant content of fulvic acids (45 mg/L), living bacteria was the winner in the same competition, however, only during the period when the cells were still in principle alive (even nonculturable). From the moment, when most cells became dead (at the end of the experiment), fulvic acids turned into the winners again, keeping the remaining cations Mn2+ in dissolved state via complexation and preventing the continuation of further biomineralization. All results presented in this work correlate well with each other.
Gross and Harrison (Citation2019) proposed a model of SOC cycling in root system, and raised a question of how much do the extracted portions of soil organic matter reflect the behavior of organic matter in the environment. These authors also reviewed a number of interaction reactions among which is a multiple stage microbial processing which either increases a solubility of SOC or assimilate it with microbial cells. In this work we showed possible mechanisms for both of such processes (an increase of solubility ruled by the presence of fulvic acids and assimilation with microbial cells via biomineralization) that cycle the soil organic carbon in the environment.
Conclusions
The presence of fulvic acids in the system: S. putrefaciens – aqueous Mn(II) indeed affected the long-term interfacial processes including bio-precipitation. The influence of fulvic acids was dependent on their concentration and was realized via the two interdependent pathways: the change of chemical compositions of the formed precipitates and the alteration of microbial viability.
An occurrence of smaller (environmentally relevant) concentrations of fulvic acids (45 mg/L) resulted in the formation of Mn hydrous oxides, an additional precipitate to the earlier discovered Mn3(PO4)2 and MnCO3. This finding put in agreement the information on Mn-containing minerals most of which are composite materials containing Mn hydrous oxides.
Very high concentration of fulvic acids (500 mg/L) which was roughly the same as the concentration of S. putrefaciens prevented both the sorption of Mn2+ and its biomineralization. However, this fundamental science research question helped us to reveal an additional regularity on the molecular-level mechanisms of the interfacial processes taking place on the surface of viable cells. It was observed that microbially mediated mineralization was initiated by the intermediate bacterial metabolic state called viable but nonculturable.
Acknowledgments
We thank Dr. Andrey Shchukarev (Umea University, Sweden) and Dr. Tom Visser (Utrecht University) for XPS analysis and FTIR measurements, respectively. Ms. Cristina Avramut (Leiden University, Netherlands) is gratefully acknowledged for her participation in the experiments. We are much obliged to anonymous Reviewers and Editors who helped us to improve this paper considerably.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bahafid W, Joutey NT, Sayel H, Iraqui-Houssaini M, El Ghachtouli N. 2013. Chromium adsorption by three yeast strains isolated from sediments in Morocco. Geomicrobiol J 30(5):422–429.
- Barnie S, Zhang J, Wang H, Yin H, Chen H. 2018. The influence of pH, co-existing ions, ionic strength, and temperature on the adsorption and reduction of hexavalent chromium by undissolved humic acid. Chemosphere 212:209–218.
- Biswas A, Besold J, Sjöstedt C, Gustafsson JP, Scheinost AC, Planer-Friedrich B. 2019. Complexation of arsenite, arsenate, and monothioarsenate with oxygen-containing functional groups of natural organic matter: an XAS study. Environ Sci Technol 53(18):10723–10731.
- Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye vinding. Anal Biochem 72(1–2):248–254.
- Burle E, Kirby-Smith WW. 1979. Application of formaldoxime colorimetric method for the determination of manganese in the pore water of anoxic estuarine sediments. Estuaries 2:198–201.
- Calvert SE, Pedersen TF. 1996. Sedimentary geochemistry of manganese: implications for the environment of formation of manganiferous black shales. Econom Geol 91(1):36–47.
- Chowdhury I, Cwiertny DM, Walker SL. 2012. Combined factors influencing the aggregation and deposition of nano-TiO2 in the presence of humic acid and bacteria. Environ Sci Technol 46(13):6968–6976.
- Chubar N, Avramut C, Visser T. 2015. Formation of manganese phosphate and manganese carbonate during long-term sorption of Mn(2+) by viable Shewanella putrefaciens: effects of contact time and temperature. Environ Sci Process Impacts 17:780–790.
- Chubar N, Behrends T, Van Cappellen P. 2008. Biosorption of metals (Cu(2+), Zn(2+)) and anions (F(-), H(2)PO(4)(-)) by viable and autoclaved cells of the Gram-negative bacterium Shewanella putrefaciens . Colloids Surf B Biointerfaces 65(1):126–133.
- Chubar N, Visser T, Avramut C, de Waard H. 2013. Sorption and precipitation of Mn2+ by viable and inactivated Shewanella putrefaciens: effect of contact time. Geochim Cosmochim Acta 100:232–250.
- Deonarine A, Lau BL, Aiken GR, Ryan JN, Hsu-Kim H. 2011. Effects of humic substances on precipitation and aggregation of zinc sulfide nanoparticles. Environ Sci Technol 45(8):3217–3223.
- Elliot JC. 1994. Structure and Chemistry of the Apatites and other Calcium Orthophosphates. Amsterdam: Elsevier.
- Gross CD, Harrison RB. 2019. The case for digging deeper: soil organic carbon storage, dynamics, and controls in our changing. World Soil Syst 3(28):1–24.
- Grunenwald A, Keyser S, Sautereau AM, Crubezy E, Ludes B, Drouet C. 2014. Revising carbonate quantification in carbonate (bio)minerals: a validated FTIR methodology. J Archaeological Sci 49:134–141.
- Haas JR. 2004. Effects of cultivation conditions on acid–base titration properties of Shewanella putrefaciens. Chem Geol 209(1-2):67–81.
- Huang Z, Zhou W, Ouyang C, Wu J, Zhang F, Huang J, Gao Y, Chu J. 2015. High performance of Mn-Co-Ni-O spinel nanofilms sputtered from acetate precursors. Sci Rep 5:10899.
- Jiang W, Saxena A, Song B, Ward BB, Beveridge TJ, Myneni SCB. 2004. Elucidation of functional groups on Gram-positive and Gram-negative Bacterial surfaces using infrared spectroscopy. Langmuir 20(26):11433–11442.
- Kawano M, Tomita K. 2001. Microbial biomineralization in weathered volcanic ash deposit and formation of biogenic minerals by experimental incubation. Am Mineral 86(4):400–410..
- Kouhail YZ, Benedetti MF, Reiller PE. 2016. Eu(III)-fulvic acid complexation: Evidence of fulvic acid concentration dependent interactions by time-resolved luminescence spectroscopy. Environ Sci Technol 50(7):3706–3713.
- Kulczycki E, Ferris FG, Fortin D. 2002. Impact of cell wall structure on the behavior of bacterial cells as sorbents of cadmium and lead. Geomicrobiol J 19(6):553–565.
- Linnik PN, Ivanechko YS, Linnik RP, Zhezherya VA. 2013. Humic Substances in Surface Waters of the Ukraine. Russ J Gen Chem 83(13):2715–2730.
- Liu D, Zhang Q, Wu L, Zeng Q, Dong H, Bishop M, Wang H. 2016. Humic acid-enhanced illite and talc formation associated with microbial reduction of Fe(III) in nontronite. Chem Geol 447:199–207.
- Ma H, Allen HE, Yin Y. 2001. Characterization of isolated fractions of dissolved organic matter from natural water and a waste water effluent. Wat Res 35(4):985–996.
- Maurice PM, Manecki M, Fein JB, Schaefer J. 2004. Fractionation of an aquatic fulvic acid upon adsorption to the bacterium, Bacillus subtilis. Geomicrobiol J 21(2):69–78.
- Mayanna S, Peacock CL, Schäffner F, Grawunder A, Merten D, Kothe E, Büchel G. 2015. Biogenic precipitation of manganese oxides and enrichment of heavy metals at acidic soil pH. Chem Geol 402:6–17.
- Meng F, Yuan G, Wei J, Bi D, Ok YS, Wang H. 2017. Humic substances as a washing agent for Cd-contaminated soils. Chemosphere 181:461–467.
- Merroun ML, Omar NB, Alonso E, Arias JM, GonzÁlez-MuÑoz MT. 2001. Silver sorption to Myxococcus xanthus biomass. Geomicrobiol J 18(2):183–192.
- Nachtegaal N, Sparks DL. 2003. Nickel sequestration in a kaolinite-humic acid complex. Environ Sci Technol 37(3):529–534.
- Naidja A, Huang PM, Anderson DW, Van Kessel C. 2002. Fourier transform infrared, UV-visible, and X-ray diffraction analyses of organic matter in humic, humic acid and fulvic acid fractions in soil exposed to elevated CO2 and N fertilisation. Appl Spectrosc 56(3):318–324.
- Nesbitt HW, Banerjee D. 1998. Interpretation of XPS Mn(2p) spectra of Mn oxyhydroxides and constraints on the mechanism of MnO2 precipitation. Am Mineral 83(3–4):305–315.
- Parikh SJ, Chorover J. 2005. FTIR spectroscopic study of biogenic Mn-oxide formation by Pseudomonas putida GB-1. Geomicrobiol J 22(5):207–218.
- Shchukarev AV, Korolkov DV. 2004. XPS study of group IA carbonates. Central Eur J Chem 2(2):347–362..
- Takenaka Y, Saito T, Nagasaki S, Tanaka S, Kozai N, Ohnuki T. 2007. Metal sorption to Pseudomonas fluorescens: influence of pH, ionic strength and metal concentrations. Geomicrobiol J 24(3–4):205–210.
- Tang T, Lu G, Wang W, Wang R, Huang R, Qiu Z, Dang Z. 2018. Photocatalytic removal of organic phosphate esters by TiO2: effect of inorganic ions and humic acid. Chemosphere 206:26–32.
- Town RM, Duval JSL, Buffle J, van Leeuwen HP. 2012. Chemodynamics of metal complexation by natural soft colloids: Cu(II) binding by humic acid. J Phys Chem A 116(25):6489–6496.
- Vignoles-Montrejaud M. 1984. Contibution a 1'etude des apatites carbonate es de type B. These d'Etat, Institut National Polytechnique de Toulouse.
- Wang H, Tang Z-Y, Liu Y-G, Lee C-S. 2009. Synthesis and behavior of Al-stabilized α-Ni(OH)2. Trans Nonferrous Met Soc China 19(1):170–175.
- Webb SM, Tebo BM, Barger JR. 2005. Structural influences of sodium and calcium ions on the biogenic manganese oxides produced by the marine Bacillus sp., strain SG-1. Geomicrobiol J 22(3–4):181–193.
- Yang S, Yang H, Ma H, Guo A, Cao F, Gong J, Deng Y. 2011. Manganese oxide nanocomposite fabricated by a simple solid-state reaction and its ultraviolet photoresponse property. Chem Commun (Camb) 47:2619–2621.
- Zhou Z, Hu D, Ren W, Zhao Y, Jiang L-M, Wang L. 2015. Effect of humic substances on phosphorus removal by struvite precipitation. Chemosphere 141:94–99.
Websites
- http://webmineral.com/. Accessed November 16, 2019.
- http://webmineral.com/chem/Chem-Mn.shtml#.WrOw7i5uapp. Accessed November 16, 2019.
- http://www.supremefulvic.com/documents/html/fulvic_acid.php. Accessed February 8, 2019.
- https://xpssimplified.com/. Accessed February 7, 2020.
- https://xpssimplified.com/elements/carbon.php#inorganic. Accessed February 7, 2020.
- https://xpssimplified.com/elements/oxygen.php. Accessed February 8, 2019.