
ABSTRACT
The Splendid Fairywren Malurus splendens has four distinct phenotypic forms currently recognised as subspecies, although three species have been recognised within the group in the past. Here, we use multilocus nuclear DNA to test the recent hypothesis that the Splendid Fairywren represents three allopatric taxa with little to no modern-day contact, despite phenotypic evidence of periods of past contact and gene flow. Our nuclear dataset is concordant with earlier mtDNA-based data in supporting three distinct genetic groups within M. splendens. These genetic groups align with the three phenotypically distinct forms recently hypothesised to be allopatric and previously described at species-level – western M. splendens, central M. callainus and eastern M. melanotus. Nuclear gene flow does not appear to be rampant between the three forms; however, several individuals show signals of admixed nuclear ancestry, which we cannot distinguish as having resulted from incomplete lineage sorting, poor phylogenetic signal, or gene flow. Given changing modern taxonomic interpretations of the significance of gene flow in speciation, we argue that recognising three distinct species within the M. splendens complex might better represent their evolutionary distinctiveness, rather than continuing to treat these genetically and morphologically distinct forms as subspecies of a widespread polytypic species. Our study highlights the interesting complexity of determining species status when taxa are in the ‘grey zone’ near the subspecies/species continuum, and the value gained from adding perspectives from nuclear DNA.
Introduction
The Splendid Fairywren Malurus splendens (Quoy & Gaimard, 1832) is a widespread passerine of the Australian arid and semi-arid zones (). Four subspecies are currently recognised based on their distinct male breeding plumages (; Figure. S1) (Schodde and Mason Citation1999; Gill et al. Citation2021 IOC v 14.1). The westernmost form, M. s. splendens (Quoy & Gaimard, 1832), is distributed from humid south-western Australia north to the arid zone in inland central Western Australia. The central form, M. s. callainus Gould, 1867, is distributed through the Gibson and Great Victoria Deserts through to the southern and central Northern Territory and to the northern Eyre Peninsula in South Australia. The eastern form, M. s. melanotus Gould, 1841, is distributed east of the Flinders Ranges through to southern Queensland where it meets M. s. emmottorum Schodde & Mason, IJ, 1999 in inland central Queensland. Hereafter, we refer to these forms by their epithets for brevity.
Figure 1. Taxonomic boundaries in the Splendid Fairywren complex. (a) Distribution of -the Splendid Fairywren complex with colours indicating the range of the four currently recognised subspecies and approximate locations of hybrid zones between each of them—M. s. splendens (purple), M. s. callainus (turquoise), M. s. melanotus (dark blue), M. s. emmottorum (light blue). Localities of specimens examined in this study are indicated by black circles. Photos were obtained from the Macaulay Library at the Cornell Lab of Ornithology—M. s. splendens (ML611751385; Gosnells, Western Australia), M. s. callainus (ML519296141; Gawler Ranges, South Australia), M. s. melanotus (ML481245791; Mildura, Victoria), M. s. emmottorum (ML246987571; Barcoo, Queensland). (b) Population structuring of eight autosomal nuclear loci sampled across 36 Splendid Fairywrens with missing data for no more than 2 loci. Plots show results of STRUCTURE analyses for 2, 3, and 4 populations (K).
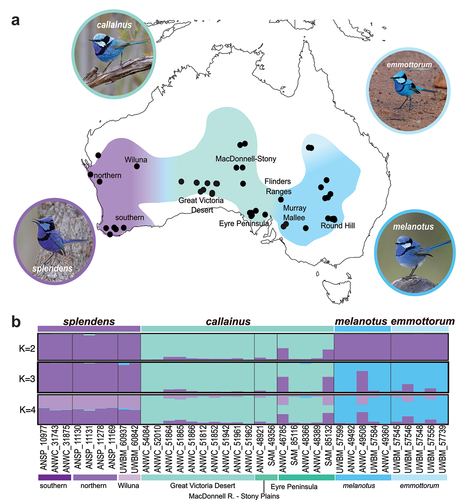
In earlier literature, western splendens, central callainus and eastern melanotus (including then undescribed emmottorum) were considered separate species (RAOU Citation1926; Condon Citation1968) until putative hybrids with intermediate plumage were identified where splendens and callainus, and callainus and melanotus meet (Ford Citation1975; Parker in Schodde Citation1975) (). For details of locations of intergradient zones see Black et al. (Citation2023, Citation2024). Johnstone and Storr (Citation2004) and Schodde and Mason (Citation1999). Birds from Wiluna, Western Australia were particularly identified as having lighter plumage than typical for splendens (Figure. S1), which was hypothesised to reflect gene flow from callainus into splendens (Ford Citation1975). The apparent lack of reproductive isolation between splendens and callainus, and between callainus and melanotus, prompted their subsequent treatment as subspecies within a single species, M. splendens under the biological species concept (Ford Citation1975; Parker in Schodde Citation1975; Schodde and Mason Citation1999). Yet, analysis of mtDNA showed no evidence of hybridisation between western splendens, central callainus and eastern melanotus/emmottorum, which formed three distinct genetic groups with approximately 1.2–1.4% net-divergence (; Kearns et al. Citation2009; Dolman and Joseph Citation2012). This included populations in and near the putative zones of intergradation, such as Wiluna (Kearns et al. Citation2009). Furthermore, narrow distributional gaps have been documented between splendens and callainus (100 km) (Black et al. Citation2024), and callainus and melanotus (80–150 km) (Reid et al. Citation1977; Black et al. Citation2023) leading Black et al (Citation2023, Citation2024). to hypothesise that there might be little or no present-day contact between splendens and callainus, and between callainus and melanotus. These distributional gaps occur across regions known to have represented vicariant biogeographic barriers during the Pleistocene – Nullarbor Plain, Great Victoria Desert and Gibson Desert for splendens and callainus, and the Eyrean Barrier for callainus and melanotus (Ford Citation1974, Citation1987; Schodde and Mason Citation1999). Consequently, putative hybrid birds with phenotypically intermediate male breeding plumage were hypothesised to result from ancient gene flow during periods of earlier contact rather than ongoing gene flow between splendens and callainus, and between callainus and melanotus (Black et al. Citation2023, Citation2024). If this is the case, it may be argued that western splendens, central callainus and eastern melanotus/emmottorum might be better treated as distinct species rather than as a single polytypic species. Critically, given the prevalence of mitonuclear discordance in nature (Funk and Omland Citation2003; Toews and Brelsford Citation2012; Winker Citation2021), including in Australian birds (Kearns et al. Citation2014; Morales et al. Citation2017; Shipham et al. Citation2017; Joseph Citation2021), nuclear DNA data are ultimately necessary to test this hypothesis.
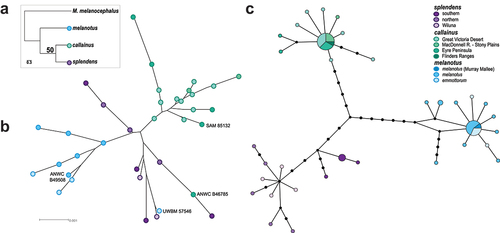
Figure 2. Nuclear and mtDNA variation across the Splendid Fairywren complex. (a) Species tree estimated from all concatenated data. (b) Unrooted Neighbor-joining tree estimated for the unphased concatenated 8-locus dataset. The museum accession number of specimens identified with admixed ancestry in STRUCTURE analyses are included for reference. (c) MtDNA ND2 haplotype network combined from specimens in the studies of Kearns et al. (Citation2009) and Dolman and Joseph (Citation2012).
Here, we use multilocus nuclear DNA (nuDNA) to test whether western, central, and eastern forms of the M. splendens complex represent independent evolutionary lineages between which gene flow is restricted. We use the nuDNA alongside multiple lines of other evidence from mtDNA, phenotype and distribution to offer a much needed reassessment of taxonomic rank for taxa within M. splendens sensu lato.
Methods
DNA extraction and sequencing of nuclear loci
DNA extraction was performed following the salting-out method (Miller et al. Citation1988). Primers for all nuclear autosomal loci in Lee and Edwards (Citation2008) were tested on tissue samples from seven specimens representing the four subspecies and mtDNA clades in M. splendens, as well as an outgroup, M. pulcherrimus, and positive control, M. melanocephalus. Of these, we selected nine loci (see Table S1) that amplified all target taxa. Tissues from a total of 57 specimens from splendens (n = 14), callainus (n = 24), melanotus (n = 11) and emmottorum (n = 8) were amplified, however, not all individuals were successfully sequenced for all loci (see Table S2). Polymerase chain reactions (PCR) and sequencing were performed following Dolman and Joseph (Citation2016) using primers presented in Table S1. Phasing of sequences with multiple heterozygous sites was performed following methods detailed in Dolman and Moritz (Citation2006) and Dolman and Joseph (Citation2016).
Population structure
Unrooted allele networks were estimated for each phased intron using TCS (Templeton, Crandall and Sing’s) in popart v1.7 (Leigh et al. Citation2015). PGDSpider (Lischer and Excoffier Citation2011) was used to convert aligned sequence data to structure format using unique alleles within each locus. We used STRUCTURE v2.3.4 (Pritchard et al. Citation2000) to estimate population structure under an admixture model with correlated frequencies. STRUCTURE was run 10 times for 1 million generations with a 500,000 generation burnin for K 1–8 on two datasets with different filtering approaches to minimise the impact of missing data. The first used all 9 nuclear loci and filtered the dataset to sample 52 individuals with missing data for no more than 4 loci. The second excluded locus TGFb2 owing to excess missing data (>40%) and sampled 36 individuals with missing data for no more than 2 loci (see Table S1). StructureSelector webserver (Li and Liu Citation2018) was used to summarise and plot K across the 10 replicate runs using CLUMPAK (Kopelman et al. Citation2015) and to evaluate the best value of K using DeltaK and mean LnP(K) (Pritchard et al. Citation2000; Evanno et al. Citation2005). We used Splitstree v1.49.0 (Huson Citation1998) to estimate an unrooted Neighbour-Joining tree using a concatenated unphased sequence matrix for the same 36 individuals and 8 loci used for STRUCTURE analyses. Species trees were estimated from all concatenated loci and individuals using SVDQuartets based on all quartets and 1000 bootstrap replicates (Chifman and Kubatko Citation2014).
Results
Nuclear loci mirror mtDNA and phenotype in supporting the distinctiveness of western splendens, central callainus and eastern melanotus/emmottorum (). Unrooted allele networks for all nine nuclear loci exhibited a lack of reciprocal monophyly between the four taxa, as well as between the three mtDNA groups identified earlier (Figure. S2). Yet, most alleles shared between the western, eastern and central forms are internal in the networks and differentiated alleles that are unique to either splendens, callainus or melanotus/emmottorum are present in almost every locus (Figure. S2).
STRUCTURE analyses run with no specimen locality data identified K = 3 as the best K in both the 8-locus and 9-locus (; Figure. S3–S5). The three genetic clusters corresponded to the western splendens, central callainus and eastern melanotus/emmottorum forms. However, not all individuals were unambiguously assigned to clusters corresponding to their taxonomic identifications at K = 2 or 3 (; Figure. S3–S5). Signals of mixed ancestry within specimens was most evident in the lower quality 9-locus dataset (Figure. S4); however, it was also present in individuals without missing data in the 8-locus dataset (). Thus, while noise owing to missing data (Table S2) may contribute to this signal, we also cannot rule out the impact of poor phylogenetic signal in the dataset or incomplete lineage sorting (ILS) or gene flow, especially for individuals sampled near putative zones of intergradation ().
The unrooted network estimated from the unphased concatenated dataset mirrored the STRUCTURE analyses in reconstructing three genetic groups corresponding to callainus, splendens and melanotus/emmottorum (). However, the nine splendens specimens formed two separate clusters. Notably, one callainus specimen from Eyre Peninsula, South Australia and one emmottorum specimen from central Queensland fell out with splendens (). These two specimens were both identified in STRUCTURE as having admixed splendens ancestry (), and both share many internal alleles with splendens in the phased allele networks (Figure. S2). Species trees estimated by SVDQuartets were unresolved (). The species tree had a sister relationship between callainus and splendens but with an equivocal bootstrap value (50) and a near-even split between incompatible (59.9%) and compatible (40.1%) quartets.
Discussion
Here, we used multilocus nuclear DNA from the Splendid Fairywren Malurus splendens complex to test the hypothesis based on earlier phenotypic and mtDNA studies that little to no present-day gene flow is occurring between western splendens, central callainus and eastern melanotus/emmottorum (Kearns et al. Citation2009; Dolman and Joseph Citation2012; Black et al. Citation2022, Citation2024) and that they therefore represent distinct evolutionary lineages better treated as species. We found that nuclear structuring mirrors mtDNA and phenotypic structuring in showing that splendens, callainus and melanotus/emmottorum have differentiated into three distinct lineages with clear spatial boundaries ().
Ultimately, more extensive sampling of the nuclear genome will be required to test conclusively whether gene flow is ongoing or whether the three forms are reproductively isolated. Acknowledging the limited power of 9 autosomal nuclear loci to detect gene flow, we do note that our nuDNA dataset does not fit with expectations if gene flow were rampantly occurring between splendens and callainus, and between callainus and melanotus (). Instead, we see clear genetic differentiation of the three forms with spatial boundaries coinciding with phenotype. We do, however, detect some signals, which could stem from gene flow – namely, individuals with mixed population assignments in STRUCTURE analyses (, S3, S4), placement of one callainus and one emmottorum specimen within an otherwise exclusively splendens lineage in the concatenated unrooted phylogenetic networks (), and numerous shared alleles amongst all four subspecies in the phased allele networks (Figure. S2). Whether these reflect the impact of ongoing gene flow – perhaps sporadic (‘leaky’) gene flow as per Variegated M. lamberti and Purple-backed M. assimilis Fairywrens (McLean et al. Citation2017) – or are indicative of incomplete lineage sorting (ILS) or the impact of missing data is unclear. We note here that the pattern of sharing in the phased allele networks for each locus (sharing of some alleles, uniqueness of others and the magnitude of divergences among the forms) is a typical hallmark of ILS rather than gene flow (Omland et al. Citation2006), suggesting that many shared alleles could stem from ILS (Figure. S2). Such shallow patterns of divergence and ILS are also typical of species-level divergences in other groups of closely related species sequenced for similar sets of loci—e.g. Red-backed Malurus melanocephalus and White-winged M. leucopterus Fairywrens (Lee and Edwards Citation2008), as well as the Pacific Robin Petroica multicolor species complex (Kearns et al. Citation2019). Our failure to reconstruct a robustly supported species tree () could reflect the impact of ILS, missing data, lack of phylogenetic signal or gene flow. However, it is possible that ambiguity in both the unrooted phylogenetic network and the species tree could reflect a truly rapid, near-simultaneous, divergence of ancestral populations into present-day splendens, callainus and melanotus/emmottorum. High-coverage SNP data will be needed for robust estimates of phylogenetic relationships, speciation history and gene flow in this complex.
Phenotypic analyses detected spatially patchy evidence of hybridisation between splendens and callainus, and between callainus and melanotus (Black et al. Citation2023, Citation2024). In those studies, some birds from putative intergradient zones were scored with intermediate plumage, while others were indistinguishable from their parental forms (Black et al. Citation2023, Citation2024). Such patchy phenotypic signals of hybridisation could stem from sporadic hybridisation between the forms, ecophenotypic responses or historical gene flow when splendens, callainus and melanotus were last in contact (sensu Black et al. Citation2023, Citation2024). This was potentially most recently in the Pleistocene based on dating of mtDNA ND2 sequences (Kearns et al. Citation2009). Our sampling unfortunately does not overlap extensively with the specimens examined by Black et al. (Citation2023, Citation2024). and thus, we cannot directly compare whether birds with intermediate plumage also had mixed genetic ancestry. However, we note the following for the potentially admixed specimens in our dataset. First, the placements of emmottorum UWBM 57,546 amongst splendens in the concatenated unrooted phylogenetic network and both emmottorum UWBM 57,546 and melanotus ANWC B49508 as admixed in the STRUCTURE analysis () seem unlikely to be caused by gene flow given the geographic separation between emmottorum and splendens, and melanotus and splendens (). These specimens shared many alleles with splendens specimens in the 9 nuclear loci sampled (Figure. S2), and as such, these patterns more likely reflect ILS and/or the limited phylogenetic power of our dataset (). The melanotus specimen, ANWC B49508, has the typical phenotype of melanotus (Figure. S1), and was collected from Round Hill, New South Wales, a straight-line distance of ~ 650 kilometres from the range of callainus (). The potentially admixed callainus specimens, ANWC B46785 and SAM ABTC 85132, from the Eyre Peninsula and ANWC B54202 and ANWC B54095 from the Great Victoria Desert, also shared many alleles with splendens in the 9 nuclear loci sampled (Figure. S2). Here, ILS and gene flow could both be likely given that callainus and splendens do potentially have opportunity for gene flow (). No comparisons of male breeding plumage are possible in these cases since ANWC B46785 is a subadult male and both ANWC B54202 and ANWC B54095 are females – see Figure. S1 for photos of male breeding plumage phenotypes from these regions. Finally, we note that the potentially intergradient splendens population in the Wiluna area aligns with splendens in both mtDNA and nuDNA datasets (), despite their lighter male breeding plumage (Figure. S1), which has long been hypothesised to result from introgression with callainus (Ford Citation1975).
Our dataset cannot fully reject that gene flow, long taken to indicate conspecificity in this group (Ford Citation1975; Parker in Schodde Citation1975; Schodde and Mason Citation1999), still occurs. Yet, taxonomic interpretations of hybridisation and gene flow have changed substantially over the last ~70 years. There has been a shift away from a need for complete reproductive isolation under the biological species concept, towards concepts like the phylogenetic or evolutionary species concepts wherein reproductive isolation is no longer a necessary criterion for naming new species. This shift has been fuelled by growing evidence from genome-scale datasets that reproductive isolation between species need not be total or genome-wide. As such, there is a growing sentiment that it may not be useful taxonomically to treat taxa that rarely interbreed as subspecies of a single species. More cases are being identified of species having diverged with continuous gene flow or with sporadic episodes of introgressive hybridisation, which are sometimes confined to only mtDNA or only certain chromosomes on the nuclear genome (Poelstra et al. Citation2014; Hejase et al. Citation2020; Winker Citation2021; Burley et al. Citation2023; Musher et al. Citation2023). This includes some notable examples from Australo-Papua (Joseph Citation2021), such as masked (Artamus personatus) and white-browed (A. superciliosus) woodswallows that display little divergence across their genome except for specific loci involved in plumage formation (Peñalba et al. Citation2022). Discussions of how to taxonomically interpret such complexities continue (Dufresnes et al. Citation2023) generating ongoing interest in speciation research.
Whether the degree of phenotypic and genetic divergence present within the M. splendens complex warrants recognition at species-level is debatable and serves a case study of the inadequacies of the Linnaean taxonomic system for taxa that fall within the ‘grey zone of speciation’ – where differentiation is greater than that seen between most subspecies, but shallower than that between most undisputed species (Roux et al. Citation2016; Winker Citation2021). Black et al. (Citation2023, Citation2024) reached several conclusions concerning the relationship between melanotus and callainus, and splendens and callainus that are relevant here: limited, past hybridisation has occurred; speciation was incomplete when they were in secondary contact (likely last in the Pleistocene); the lineages are now narrowly allopatric; if gene flow between them has indeed ceased then speciation may continue; gene flow is likely restricted (although their reproductive isolation is moot). These lines of evidence were used to argue that splendens, callainus and melanotus (including emmottorum) represent ‘semispecies’ or ‘almost-species’, falling within the continuum of divergence between typical subspecies and species (Black et al. Citation2023). Unfortunately, however, the Linnaean system and the rules of zoological nomenclature offer no mechanism for recognising such nuance, allowing only species or subspecies. Considering splendens, callainus and melanotus (including emmottorum) as subspecies of M. splendens (Quoy and Gaimard 1830), which Black et al. (Citation2023: 65) did ‘on the basis of present evidence’, clearly gives no indication that these forms are ‘almost species’. Here we have added another line of evidence, in the form of the first nuclear perspectives for the species complex. We show that nuclear loci also support the distinctiveness of splendens, callainus and melanotus (including emmottorum) despite their geographic ranges being either in contact or narrowly allopatric. We argue that recognising these three distinct lineages at species rank, rather than continuing to treat the three at subspecies rank, makes more biological sense. It better reflects the data now available on levels of divergence, relationships and gene flow and it better reflects Black et al. (Citation2023) indication that speciation is set to continue and not be arrested.
Conclusion
We have demonstrated here that there are multiple concordant lines of evidence (phenotype, mtDNA, nuDNA, putative allopatry) supporting the recognition of three independent evolutionary lineages in the M. splendens complex. Coupled with changing approaches for defining species boundaries with gene flow, we argue that current evidence justifies restoring the western splendens, central callainus and eastern melanotus/emmottorum forms to the status of distinct species, and in accordance with the phylogenetic species concept. We argue that this better represents the weight of evidence currently available for this species complex, which is right on the cusp of the species/subspecies continuum. As with any proposed taxonomic treatment, this represents a hypothesis that lays a foundation for future studies to test. The following nomenclature applies: Splendid Fairywren M. splendens (Quoy & Gaimard, 1832) for the western form, Turquoise Fairywren M. callainus Gould, 1867 for the central form and Black-backed Fairywren M. melanotus Gould, 1841 for the eastern form, which includes two subspecies, M. m. emmottorum Schodde & Mason, IJ, 1999 and M. m. melanotus Gould, 1841.
Supplemental Material
Download PDF (2.5 MB)Acknowledgments
This study would not have been possible without the contribution of specimens from the Australian National Wildlife Collection (ANWC), South Australian Museum (SAM), Academy of Natural Sciences, Philadelphia (ANSP), and Burke Museum (UWBM). We thank all collectors, especially Alex Drew and Ian Mason at the ANWC, funding- and permit-granting agencies and collection management staff involved in the collection and curation of these specimens over the years, and for promoting their accessibility. We are grateful to The Macaulay Library at the Cornell Lab of Ornithology for the use of photographs used in . This work was supported by a CSIRO postdoctoral fellowship to GD. We particularly acknowledge that field work for GD’s research was made possible by the Maralinga-Tjarutja People and Community and the Tjuntjuntjara People and Community. Collecting was done under CSIRO Ethics permits 713-08/08 and 07-04 and by scientific collecting permits kindly granted by the South Australian Department of Environment and Heritage (Y 25496 1) and the Western Australian Department of Environment and Conservation (CE 001990, SF 006336). We thank Andrew Black and three anonymous reviewers for their constructive comments.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Nuclear DNA sequences generated for this study are lodged with GenBank under the following accessions (PP412882 – PP413260). MtDNA sequences generated by previous studies can be found on GenBank under the following accessions (Kearns et al. (Citation2009): EU144234- EU144303 and EU534188-EU534192; Dolman and Joseph (Citation2012): JQ027448-JQ027433).
Supplementary material
Supplemental data for this article can be accessed at https://doi.org/10.1080/01584197.2024.2352400
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
References
- Black, A., Horton, P., Johnston, G., and Blaylock, B. (2022). Phenotypic separation of three divergent taxa within the Splendid Fairywren Malurus splendens. South Australian Ornithologist 47(1), 38–43.
- Black, A., Horton, P., Johnston, G., and Blaylock, B. (2023). What is the evidence of contact and interaction between the two divergent lineages of Splendid Fairywren Malurus splendens in South Australia? South Australian Ornithologist 47(2), 49–68.
- Black, A., Johnstone, R., Johnston, G., and Hamilton, N. (2024). What is the evidence of contact and interaction between three lineages of Splendid Fairywren Malurus splendens in Western Australia? South Australian Ornithologist 48(1 & 2), 24–36.
- Burley, J. T., Orzechowski, S. C. M., Sin, S. Y. W., and Edwards, S. V. (2023). Whole-genome phylogeography of the blue-faced honeyeater (Entomyzon cyanotis) and discovery and characterization of a neo-Z chromosome. Molecular Ecology 32(6), 1248–1270. doi:10.1111/mec.16604
- Chifman, J., and Kubatko, L. (2014). Quartet inference from SNP data under the coalescent model. Bioinformatics 30(23), 3317–3324. doi:10.1093/bioinformatics/btu530
- Condon, H. T. (1968). ‘A Handlist of the Birds of South Australia.’ (South Australian Ornithological Association: Adelaide).
- Dolman, G., and Joseph, L. (2012). A species assemblage approach to comparative phylogeography of birds in southern Australia. Ecology and Evolution 2(2), 354–369. doi:10.1002/ece3.87
- Dolman, G., and Joseph, L. (2016). Multi-locus sequence data illuminate demographic drivers of pleistocene speciation in semi-arid southern Australian birds (Cinclosoma spp.). BMC Evolutionary Biology 16(1), 1–14. doi:10.1186/s12862-016-0798-6
- Dolman, G., and Moritz, C. (2006). A multi-locus perspective on refugial isolation and divergence in rainforest skinks (Carlia). Evolution 60(3), 573–82. doi:10.1111/j.0014-3820.2006.tb01138.x
- Dufresnes, C., Poyarkov, N., and Jablonski, D. (2023). Acknowledging more biodiversity without more species. Proceedings of the National Academy of Sciences USA 120(40), e2302424120. doi:10.1073/pnas.2302424120
- Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology 14(8), 2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
- Ford, J. (1974). Speciation in Australian birds adapted to arid habitats. Emu 74(3), 161–168. doi:10.1071/MU974161
- Ford, J. (1975). Hybridization of Splendid and Turquoise Wrens. Emu 75(3), 153–154. doi:10.1071/MU9750153
- Ford, J. (1987). Hybrid zones in Australian birds. Emu 87(3), 158–178. doi:10.1071/MU9870158
- Funk, D., and Omland, K. E. (2003). Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annual Review of Ecology, Evolution, and Systematics 34(1), 397–423. doi:10.1146/annurev.ecolsys.34.011802.132421
- Gill, F., Donsker, D., and Rasmussen, P. (Eds). (2021). IOC World Bird List (V11.2). doi:10.14344/IOC.ML.11.2
- Hejase, H. A., Salman-Minkov, A., Campagna, L., Hubisz, M. J., Lovette, I. J., Gronau, I., and Siepel, A. I. (2020). Genomic islands of differentiation in a rapid avian radiation have been driven by recent selective sweeps. PNAS 117(48), 30554–30565. doi:10.1073/pnas.2015987117
- Huson, D. H. (1998). SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14(1), 68–73. doi:10.1093/bioinformatics/14.1.68
- Johnstone, R. E., and Storr, G. M. (2004). ‘Handbook of Western Australian Birds. Volume II. Passerines (Blue-Winged Pitta to Goldfinch).’ (Western Australian Museum: Perth).
- Joseph, L. (2021). Species limits in birds: Australian perspectives on inter-related challenges of allopatry, introgression of mitochondrial DNA, recent speciation, and selection. Ornithology 138(2). doi:10.1093/ornithology/ukab012
- Kearns, A. M., Joseph, L., Edwards, S. V., and Double, M. C. (2009). Inferring the phylogeography and evolutionary history of the splendid fairy-wren Malurus splendens from mitochondrial DNA and spectrophotometry. Journal of Avian Biology 40(1), 7–17. doi:10.1111/j.1600-048X.2008.04383.x
- Kearns, A. M., Joseph, L., Toon, A., and Cook, L. G. (2014). Australia’s arid-adapted butcherbirds experienced range expansions during pleistocene glacial maxima. Nature Communications 5(1), 5. doi:10.1038/ncomms4994
- Kearns, A. M., Malloy, J. F., Gobbert, M. K., Thierry, A., Joseph, L., Driskell, A. C., and Omland, K. E. (2019). Nuclear introns help unravel the diversification history of the australo-pacific Petroica robins. Molecular Phylogenetics and Evolution 131, 48–54. doi:10.1016/j.ympev.2018.10.024
- Kopelman, Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A., and Mayrose, I. (2015). Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Molecular Ecology Resources 15(5), 1179–1191. doi:10.1111/1755-0998.12387
- Lee, J., and Edwards, S. (2008). Divergence across Australia’s Carpentarian barrier: statistical phylogeography of the red-backed fairy wren (Malurus melanocephalus). Evolution 62(12), 3117–3134. doi:10.1111/j.1558-5646.2008.00543.x
- Leigh, J. W., Bryant, D., and Nakagawa, S. (2015). PopART: full-feature software for haplotype network construction. Methods in Ecology and Evolution 6(9), 1110–1116.
- Li, Y. L., and Liu, J. X. (2018). StructureSelector a web-based software to select and visualize the optimal number of clusters using multiple methods. Molecular Ecology Resources 18(1), 176–177. doi:10.1111/1755-0998.12719
- Lischer, H. E. L., and Excoffier, L. (2011). Pgdspider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28(2), 298–299. doi:10.1093/bioinformatics/btr642
- McLean, A., Joseph, L., Toon, A., Schmidt, D., Drew, A., Mason, I. J., and Hughes, J. (2017). Reassessment of a possible case of intraspecific gene flow across Australia’s great dividing range in the variegated fairy wren, Malurus lamberti (aves: Maluridae), and its systematic consequences. Biological Journal of the Linnean Society 122(1), 210–223. doi:10.1093/biolinnean/blx054
- Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research 16(3), 1215. doi:10.1093/nar/16.3.1215
- Morales, H. N. E., Sunnucks, P., Joseph, L., and Pavlova, A. (2017). Perpendicular axes of differentiation generated by mitochondrial introgression. Molecular Ecology 26(12), 3241–3255. doi:10.1111/mec.14114
- Musher, L. J., Del-Rio, G., Marcondes, R. S., Brumfield, R. T., Bravo, G. A., and Thom, G. (2023). Geogenomic predictors of genetree heterogeneity explain phylogeographic and introgression history: a case study in an Amazonian bird (Thamnophilus aethiops). Systematic Biology syad061. doi:10.1093/sysbio/syad061
- Omland, K. E., Baker, J. M., and Peters, J. L. (2006). Genetic signatures of intermediate divergence: Population history of old and new world holarctic ravens (Corvus corax). Molecular Ecology 15(3), 795–808. doi:10.1111/j.1365-294X.2005.02827.x
- Peñalba, J., Peters, J., and Joseph, L. (2022). Sustained plumage divergence despite weak genomic differentiation and broad sympatry in sister species of Australian woodswallows (Artamus spp.). Molecular Ecology 31(19), 5060–5073. doi:10.1111/mec.16637
- Poelstra, J. W., Vijay, N., Bossu, C. M., Lantz, H., Ryll, B., and Müller, I., et al. (2014). The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 344(6190), 1410–1414. doi:10.1126/science.1253226
- Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155(2), 945–959. doi:10.1093/genetics/155.2.945
- RAOU. (1926). ‘The Official Checklist of the Birds of Australia.’ (Royal Australasian Ornithologists Union: Melbourne).
- Reid, N., Paton, J. B., and Paton, D. C. (1977). Critical range limits of the turquoise and black-backed wrens in South Australia. South Australian Ornithologist 27, 216–221.
- Roux, C., Fraïsse, C., Romiguier, J., Anciaux, Y., Galtier, N., Bierne, N., and Moritz, C. (2016). Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLOS Biology 14(12), e2000234. doi:10.1371/journal.pbio.2000234
- Schodde, R. (1975). ‘Interim List of Australian Songbirds. Passerines.’ (Royal Australasian Ornithologists Union: Melbourne).
- Schodde, R., and Mason, I. J. (1999). The Directory of Australian Birds: Passerines. (CSIRO Publishing: Collingwood).
- Shipham, A., Schmidt, D. J., Joseph, L., and Hughes, J. M. (2017). A genomic approach reinforces a hypothesis of mitochondrial capture in eastern Australian rosellas. The Auk 134(1), 181–192. doi:10.1642/AUK-16-31.1
- Toews, D. P. L., and Brelsford, A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology 21(16), 3907–3930. doi:10.1111/j.1365-294X.2012.05664.x
- Winker, K. (2021). An overview of speciation and species limits in birds. Ornithology 138(2), 1–27. doi:10.1093/ornithology/ukab006