
Abstract
Objectives: Previous studies have investigated the association between MTHFR A1298C (rs1801131) polymorphism and susceptibility to Alzheimer’s disease (AD). Nevertheless, an ultimate conclusion remains obscure. We then executed this meta-analysis to estimate this association more precisely.
Methods: Related studies were systematically searched on PubMed, Embase, China National Knowledge Infrastructure, Google scholar, and AlzGene databases. The association was evaluated by reviewing the odds ratios (ORs) with corresponding 95% confidence intervals (CIs). Publication bias, sensitivity analysis, and cumulative meta-analysis were performed to help draw a more definite conclusion.
Results: Ten eligible studies were finally enrolled in this meta-analysis. Lack of association between MTHFR A1298C polymorphism and AD risk was observed in five genetic models (allelic: OR = 1.17, 95% CI: 0.88–1.56; homozygous: OR = 1.15, 95% CI: 0.87–1.53; heterozygous: OR = 1.19, 95% CI: 0.76–1.86; dominant: OR = 1.23, 95% CI: 0.81–1.87; recessive: OR = 1.16, 95% CI: 0.89–1.52). The result of cumulative meta-analysis sorted by publication year was also detected a dynamic tendency of no correlation between MTHFR A1298C polymorphism and AD.
Conclusion: This meta-analysis reveals that MTHFR A1298C polymorphism may not be associated with AD risk.
Introduction
Alzheimer’s disease (AD) is a fatal neurodegenerative disease and the most frequent type of dementia in the elderly individuals (>65 years old) [Citation1,2]. Dementia is a general term for conditions and diseases characterized by a decay in memory and cognitive deterioration that influences a person’s ability to perform daily activities [Citation3]. The International Alzheimer’s Disease Report estimated that 47 million people worldwide are suffering from AD in 2015, and the number of AD patients will grow to 131 million by 2050 [Citation4]. Neuropathological characteristics of AD are the deposition of hyperphosphorylated tau proteins that form the paired helical filaments in neurofibrillary tangles, and the extracellular buildup of amyloid beta in plaques [Citation5,6]. There is no effective treatment for AD and no biomarkers for early diagnosis of AD [Citation7]. One challenge to transform disease-associated polymorphisms into clinical applications is deficient in knowledge about the biomarkers [Citation8]. If biomarkers that were linked to development and progression of AD could be ascertained, they would greatly contribute to early diagnosis of AD and exploring special prophylactics [Citation9].
The development of AD is probably associated with the interaction of multiple genetic and environmental factors [Citation10]. Approximately 70% of AD risk is attributed to genetic factors [Citation11]. It has been repeatedly demonstrated that the apolipoprotein E ε4 (ApoE ε4) allele is related to AD susceptibility. However, the ApoE ε4 gene polymorphisms only make up 20% of the genetic predisposition to AD [Citation12], which suggests a possible role of non-ApoE genetic factors in disease development. Several investigations have suggested the correlation with AD risk and some non-ApoE genetic loci, including BACE1, CR1, ZCWPW1, MTHFR gene [Citation1,13–16]. Among these genetic risk loci, the MTHFR gene polymorphisms and their correlations with susceptibility to AD are the center of our attention in this paper.
The human MTHFR gene (MIM *607093) is localized to chromosome 1p36.3 and is composed of 11 exons [Citation17]. Methylene-tetrahydrofolate reductase (MTHFR) is a pivotal enzyme to modulate folate metabolism and also affects DNA methylation and nucleic acid synthesis [Citation16,18]. MTHFR irreversibly converts 5,10-metilenetetraidrofolate (5,10-mTHFR) to 5-metiltetraidrofolate (5-mTHF) [Citation16], which is an important co-factor in homocysteine (Hcy) methylation. Mutations in MTHFR gene (C677T and A1298C) might cause amino acid substitutions that result in decreased enzyme activity with a reduced 5-mTHF availability. As a consequence of the MTHFR dysfunctions, Hcy level was increased in plasma [Citation19]. High levels of plasma homocysteine can produce a cytotoxic effect [Citation20] and compromise normal cell function in several different tissues [Citation16] (e.g. neurodegenerative disorders, including AD).
Several studies [Citation16,19,21–28] tried to find out any association between MTHFR A1298C polymorphism and the risk of AD; due to different populations, case selections, and sample sizes, the association remained inconclusive. Two articles [Citation16,25] revealed the genetic association of MTHFR A1298C polymorphism with AD risk, but others indicated lack of this association. Therefore, to provide a more precise estimation, we carried out a meta-analysis to clarify the association between MTHFR A1298C polymorphism and the susceptibility to AD.
Materials and methods
Identification and eligibility of relevant studies
To find all publications related to the risk of AD associated with MTHFR A1298C polymorphism, we performed a thorough literature search of electronic databases (up to 5 July 2016). We first searched Chinese databases, including China National Knowledge Infrastructure and Wanfang, but no eligible studies were found (data not shown in Figure ). Then, we performed a systematic literature search in PubMed, Embase, Google scholar, and AlzGene databases. The initial search terms were ‘MTHFR’, ‘methylene-tetrahydrofolate reductase’, ‘Alzheimer’s disease’, ‘polymorphism’, ‘mutation’, and ‘variant’. The eligible articles identified were restricted to case-control studies involving human subjects but without limitation on sample size, population, or time period. The eligible studies in this meta-analysis were considered if they: (1) assessed the association between MTHFR A1298C polymorphism and the risk of AD; and (2) were original research papers with a case-control design and with sufficient genotype data (such as AA, AC, and CC genotypes).
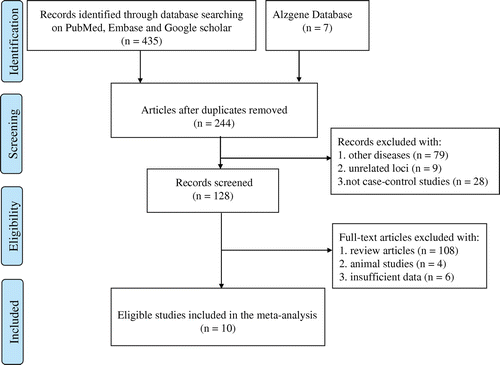
Figure 1. Flow diagram of the process used to select eligible studies.
Data extraction
Data from all eligible studies were mined independently by two authors on paper data collection forms according to the above inclusion criteria. Disparities were resolved by discussion between the authors. The extracted data included first author’s surname, publication year, and country of origin, ethnicity of participants (categorized as Caucasian, Asian, and Mixed); numbers of genotyped cases and controls, sources of control, genotyping method, diagnosis criteria, and Hardy–Weinberg equilibrium (HWE).
Statistical analysis
In this retrospective analysis, the pooled odds ratios (ORs) with corresponding 95% confidence intervals (CIs) of association between MTHFR A1298C polymorphism and AD risk were estimated for each study. Five genetic models, including the allelic (C vs. A), homozygous (CC vs. AA), heterozygous (CA vs. AA), dominant (CC + CA vs. AA), and recessive (CC vs. CA + AA) model were applied, respectively, to dissect the association patterns. The Z-test was used to estimate the significance of pooled ORs and a P ≤ 0.05 was judged to be statistically significant. We conducted Q-test and I2 test to assess between-study heterogeneity. When heterogeneity was present or not, the random-effect or fixed-effect model would be employed to compute the pooled ORs with 95% confidence intervals. If PQ > 0.10 and/or I2 < 50% (denoting an absence of obvious heterogeneity among included studies), then the fixed-effect model (Mantel–Haensze method) was applied to calculate the pooled ORs with 95% CIs. On the contrary, a PQ ≤ 0.10 and I2 ≥ 50% indicated a high degree of between-study heterogeneity, and the random-effect model (DerSimonian and Laird method) was utilized.
The HWE [Citation29,30] was tested by chi-square (χ2) to estimate whether the genotype frequencies and population genetic data in each study were balanced. A P < 0.05 was considered statistically significantly unbalanced, indicating that the study samples were not representative of the community in the corresponding area. The overall quality of included case-control studies was assessed through Newcastle–Ottawa Scale (NOS) criteria [Citation31]. The estimation of content in the NOS was categorized into three aspects: object selection, comparability, and exposure assessment. The overall quantitative evaluation of each study quality was accumulated from the scores estimated based on the aforementioned three aspects. An eligible study with high quality should obtain at least six points in this meta-analysis. Sensitivity analysis was carried out by excluding individual studies in sequence to observe the changes in pooled effect-size estimates [Citation32]. In this way, any individual study notably affecting the overall results could be discovered and the robustness of the meta-analysis could be examined. Cumulative meta-analysis according to publication year was conducted, where the pooled OR and 95%CI were updated every time when a new publication was accumulated. This statistical method could be used to detect the dynamic trend of the association result or further stabilize the meta-analysis conclusion [Citation33,34]. Publication bias was estimated by Egger’s and Begg’s tests among the eligible studies to avoid unreliable conclusions caused by the preference for positive results or other factors. A P value of more than 0.05 and the funnel plot symmetry suggested no apparent bias. In this meta-analysis, all statistical analyses were performed by the software STATA 14.0 (Stata Corporation College Station, Texas, U.S.A.) and Review manager 5.3 (The Cochrane Collaboration, Oxford, U.K.).
Results
Study characteristics
Figure displays a flow chart to describe the selection process of eligible studies included in our analysis. The original search strategy generated a total of 442 potential articles. Among them, 198 records were duplicated. We removed these duplications and obtained 244 papers. Then, we carefully checked the abstracts and/or full-texts, and discarded an additional 116 articles because they either studied other diseases/unrelated loci or were not in case-control design. Among the remaining 128 articles, 108 were reviews, four studied animal experiments, and six lacked the sufficient genotype data. Thus, 118 publications were futher excluded. Finally, 10 eligible studies [Citation16,19,21–28] were recognized in this meta-analysis. The main characteristics of all these studies are listed in Table . Among the ten included studies, eight articles had Caucasian study participants, one involved Asians, and one investigated the Mixed type (participants originated from Brazil and had different proportions of white, black, and indigenous blood [Citation19]). Besides, control groups of nine selected studies were from population-based settings and one study [Citation25] was from hospital-based settings. The genotype distributions in control groups of all the studies conformed to the HWE equilibrium except one reported by Ravaglia et al. [Citation23] (Table ). This study was also included in our meta-analysis but excluded from sensitivity analysis. Additionally, the NOS result indicated that all the included studies in the retrospective analysis were of high quality with scores above six points or more (Table Citation2).
Table 1. The baseline characteristics of all the included studies in this meta-analysis.
Meta-analysis results
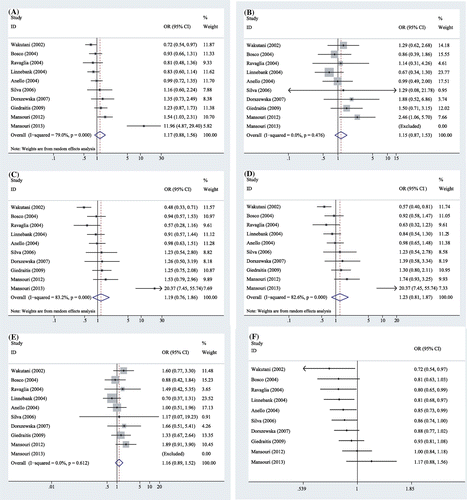
In this meta-analysis, a total of 10 studies involving 1067 AD cases and 1679 controls were included to examine the association between MTHFR A1298C polymorphism and AD risks. This meta-analysis showed a great heterogeneity among related studies in three genetic models (C vs. A, I2 = 79%, PQ < 0.001; CA vs. AA, I2 = 83%, PQ < 0.001; CC + CA vs. AA, I2 = 83%, PQ < 0.001, Table ), thus, the random-effect model was applied to compute the pooled ORs for these three models. In contrast, there was no heterogeneity in the other two genetic models (CC vs. AA, I2 = 0%, PQ = 0.48, CC vs. CA + AA, I2 = 0%, PQ = 0.61, Table ); the combined ORs were estimated for these two models using a fixed-effect model. Overall, the results illustrated that no significant association between MTHFR A1298C polymorphism and AD risk was observed in all five genetic models (allelic C vs. A: OR = 1.17, 95% CI: 0.88–1.56, P = 0.27; homozygous CC vs. AA: OR = 1.15, 95% CI: 0.87–1.53, P = 0.33; heterozygous CA vs. AA: OR = 1.19, 95% CI: 0.76–1.86, P = 0.44; dominant CC + CA vs. AA: OR = 1.23, 95% CI: 0.81–1.87, P = 0.33; and recessive CC vs. CA + AA: OR = 1.16, 95% CI: 0.89–1.52, P = 0.26, Table , Figure ).
Table 2. Quality assessment scheme for included studies (Newcastle–Ottawa Scale).
Figure 2. Forest plots of MTHFR A1298C polymorphism and the risk of Alzheimer’s disease in five genetic models: A: the allelic model (C vs. A), B: the homozygous model (CC vs. AA), C: the heterozygous model (CA vs. AA), D: the dominant model (CC + CA vs. AA), E: the recessive model (CC vs. CA + AA), and F: the allelic model (C vs. A) in cumulative meta-analysis by publication year.
Sensitivity analysis and cumulative meta-analysis
The leave-one-out sensitivity analysis was conducted to explore the different contributions of individual studies to entire pooled results because high heterogeneity existed in three genetic models. After the exclusion of the study conducted by Mansouri et al. in 2013 [Citation16], the heterogeneity calculated in clinical AD samples was all reduced from high to moderate under those three genetic models (C vs. A: I2 = 41%; CA vs. AA: I2 = 52%; CC + CA vs. AA: I2 = 49%). When any other study excluded, the between-study heterogeneity, however, was not substantially decreased. The pooled estimate became negative after the seventh study was added in cumulative meta-analysis. In addition, there have been an obvious trend in the combined ORs that clearly indicated no correlation between this polymorphism and AD risk when more publications were accumulated from 2007 to 2013 (Figure (F)).
Publication bias
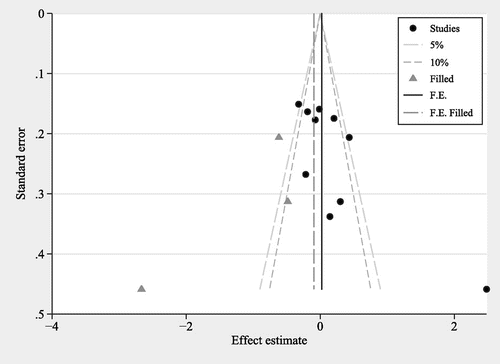
Publication bias was estimated by Egger’s quantitative test and Begg’s funnel plot. Egger’s quantitative test revealed that the results in the allelic model showed the existence of publication bias (C vs. A: P = 0.04). Thus, we conducted a funnel plot using trim and fill method. This method could mirror the positive studies that cause funnel plot asymmetry by conservatively imputing hypothetical negative studies as unpublished [Citation35]. The imputed studies generated a symmetrical funnel plot (Figure ). When the study performed by Mansouri et al. in 2013 [Citation16] was excluded, Egger’s test showed that no obvious publication bias was detected in the allelic model (C vs. A: P = 0.27).
Figure 3. Funnel plot for the allelic model (C vs. A) to analyze publication bias between MTHFR A1298C polymorphism and AD risk.
Discussion
The current study is a meta-analysis concerning MTHFR A1298C polymorphism and AD risk in the general population. AD is one of the most common neurodegenerative diseases in the elderly [Citation36]. While the pathogenesis is still unclear, AD is likely the result of environmental factors in interaction with genetic susceptibility. Identification of the involved genes would help us discover the pathogenesis of AD and find some efficient interventions to reduce the occurrence of the disease [Citation36]. MTHFR is a vital enzyme for DNA synthesis and homocysteine remethylation [Citation16]. Therefore, dysfunction of this gene might be a harmful factor for the development of AD. The correlation of the MTHFR A1298C mutation with susceptibility to AD was previously reported in a published meta-analysis [Citation37]. However, the former work only included four studies [Citation21,22,24,27], which were all searched based on the Alzgene database on 1 December 2005. The earlier meta-analysis performed the allelic genetic model only and suggested that the C allele of MHTFR A1298C polymorphism might play a protective role in the process of AD risk (OR = 0.85, 95% CI: 0.73–1, P = 0.05).
Table 3. Meta-analysis of the association between MTHFR A1298C polymorphism and the risk of AD.
Over the last ten years, six more publications investigated the association between MTHFR A1298C and AD risk, and these studies displayed controversial results. To give a more precise assessment of the correlation between this variation and susceptibility to AD, we collected all 10 eligible studies [Citation16,19,21–28] with a total of 1067 AD cases and 1679 controls to conduct this meta-analysis. No significant association between MTHFR A1298C polymorphism and the risk of AD was found in all five genetic models, revealing that this polymorphism might not add to the risk of developing AD. The result was not changed when the study reported by Ravaglia et al. [Citation23] was excluded due to the disagreement of genotype distribution in its control group with the HWE. In a cumulative meta-analysis, the accumulating evidence of the earlier publications before 2005 could suggest that the C allele was correlated with decreased AD risk, which was in accordance with the results of the previous meta-analysis [Citation37]. However, we speculated that the pooled ORs and CIs based on those earlier studies might be false positive results due to their relatively small number of samples. When more studies were included, the result of cumulative meta-analysis detected a dynamic tendency of no association between MTHFR A1298C polymorphism and AD risk. This trend is in good accordance with a speculation that some unpublished studies earlier, which are more likely to show lack of association according to the principle of publication bias, may not be included in the previous meta-analysis [Citation37] and also in this meta-analysis. This coincidence might also strengthen the overall finding of our work.
Sensitivity analysis was conducted because heterogeneity was observed in this meta-analysis, and the study conducted by Mansouri et al. in 2013 [Citation16] was found to account for a proportion of the heterogeneity. It has been acknowledged that some potential factors, including age, gender, ethnicity, and ApoE ε4 status, could contribute to the heterogeneity and the different results among the studies. Therefore, we tried to extract the data of these potential factors of AD to perform subgroup analysis. However, we could not carry out the subgroup analysis by age or gender due to insufficient data. All the studies, except for the study performed by Anello et al. [Citation21], lack independent genotype data at different age onsets, such as early-onset (<65 years) and late-onset (>65). Participants were only men in one study [Citation26] but with a certain proportion of different genders in all other studies. On the other hand, we performed subgroup analysis by Caucasian ethnicity because eight of the ten studies involved Caucasian. The results did not give much difference as those in the general population (data not shown). Finally, we also attempted to conduct a subgroup and stratified analysis by ApoE ε4 allele but unfortunately failed because all included studies lacked independent genotype data based on the classification of ApoE ε4 status.
Our meta-analysis had four advantages. First, this retrospective study was performed based on more studies with a larger sample size as compared with the previous meta-analysis. Second, we used five genetic models in this meta-analysis, which might clarify the potentially different roles that the varied genotypes of the polymorphism might play in AD risk. Third, publication bias was not only assessed by Begg’s and Egger’s tests, but also employed by the trim and fill method to identify and amend asymmetry arising from publication bias. Fourth, we also performed a cumulative meta-analysis by publication year to investigate a tendency of the correlation between MTHFR A1298C polymorphism and AD risk, and the result indicated that our conclusion was robust when more new studies were added.
AD is the most prevalent neurodegenerative disease in the elderly and has a great impact on today’s aging society [Citation38,39]. Social as well as economical costs of AD prevention and cure require improved methods of diagnosis and treatment. There are currently no effective treatment for AD [Citation40], no biomarkers for early diagnosis of AD and the pathogenesis of AD is uncertain even now [Citation22,41]. Therefore, it is vital to expand our knowledge of AD pathogenic mechanisms or we will nearly fail to develop efficient therapies for AD. An intense search for the association between the polymorphisms of some genes, such as MTHFR A1298C and the risk of AD could be very important for better understanding the pathomorphological changes of the disease, and it could also help to identify the patients which might undergo special AD prophylactics. It might also locate new biomarkers of the disease after the detailed diagnostics of these patients. In addition, such an analysis would also offer good support in evidence-based medicine for the advance of personalized gene therapy by summing up the positive, negative, or no correlation between AD susceptibility and gene polymorphisms.
Several potential limitations should also be recognized in this meta-analysis. The included studies were limited to just English and Chinese literature, which might bias the results. The sample sizes of cases and controls were relatively small, which may result in false negative or false positive results. Owing to the inadequate data, we did not perform subgroup analyses regarding other factors, such as age, gender, and lifestyle habits, which may contribute to AD development. Finally, significant heterogeneity was detected in this retrospective analysis. The study designs, populations, ethnicity, and habits may play a role in the heterogeneity.
Conclusion
This meta-analysis indicated that MTHFR A1298C polymorphism might not be related to genetic susceptibility of AD in the general population based on existing studies. Additional studies with large sample sizes, gene–gene, and gene–environment interactions are necessary to provide a reliable assessment of the association between MTHFR A1298C polymorphism and AD risk.
Authors contribution
Shumin Liu and Yongfu Wu carried out the experiments, collected the data, and drafted the manuscript. Xu Liu made additional experiments, including cumulative meta-analysis and sensitivity analysis. Jiahui Zhou and Ziyou Wang participated in reviewing potential articles, abstract data, and used software to create tables and figures. Zunnan Huang and Zhiwei He designed this study, revised this manuscript, and helped in analyzing and interpreting data.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the National Natural Science Foundation of Guangdong Province, China [grant number 2015A030313518]; Scientific Research Foundation for Returned Overseas Scholars of Guangdong Medical University, China [grant number B2012082]; Sail Plan ‘the Introduction of the Shortage of Top-Notch Talent’ Project of Guangdong Province, China [grant number YueRenCaiBan [2014] 1].
Acknowledgments
The authors thank Dr. Guoliang Huang from Dongguan Scientific Research Center, Guangdong Medical University for putting forward useful suggestions in the manuscript and Timothy H. Click from National Chiao Tung University for assisting in the revision of this paper.
References
- Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 2012;8:131–168.
- Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006239.
- Alzheimer’s Association. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10:e47–e92.
- Baumgart M, Snyder HM, Carrillo MC, et al. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: a population-based perspective. Alzheimers Dement. 2015;11:718–726.10.1016/j.jalz.2015.05.016
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. 10.1016/j.bbadis.2015.12.023
- Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712.
- Hooper C, Lovestone S, Sainz-Fuertes R. Alzheimer’s disease, diagnosis and the need for biomarkers. Biomark Insights. 2008;3:317–323.
- Cross DS, Ivacic LC, Stefanski EL, et al. Population based allele frequencies of disease associated polymorphisms in the Personalized Medicine Research Project. BMC Genet. 2010;11:51.10.1186/1756-0500-7-194
- McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269.10.1016/j.jalz.2011.03.005
- Bekris LM, Yu CE, Bird TD, et al. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213–227.10.1177/0891988710383571
- Alonso Vilatela ME, López-López M, Yescas-Gómez P. Genetics of Alzheimer’s disease. Arch Med Res. 2012;43:622–631.10.1016/j.arcmed.2012.10.017
- Lambert JC, Amouyel P. Genetic heterogeneity of Alzheimer’s disease: complexity and advances. Psychoneuroendocrinology. 2007;32(Suppl 1):S62–S70.10.1016/j.psyneuen.2007.05.015
- Gao Y, Tan MS, Wang HF, et al. ZCWPW1 is associated with late-onset Alzheimer’s disease in Han Chinese: a replication study and meta-analyses. Oncotarget. 2016;7:20305–20311.
- Li Y, Song D, Jiang Y, et al. CR1 rs3818361 polymorphism contributes to Alzheimer’s disease susceptibility in Chinese population. Mol Neurobiol. 2016;53:4054–4059.10.1007/s12035-015-9343-7
- Wang M, Yang J, Su J. Relationship between the polymorphism in exon 5 of BACE1 gene and Alzheimer’s disease. Aging Clin Exp Res. 2016; [ Epub ahead of print]. DOI:10.1007/s40520-016-0539-0
- Mansouri L, Fekih-Mrissa N, Klai S, et al. Association of methylenetetrahydrofolate reductase polymorphisms with susceptibility to Alzheimer’s disease. Clin Neurol Neurosurg. 2013;115:1693–1696.
- Goyette P, Sumner JS, Milos R, et al. Human methylenetetrahydrofolate reductase: isolation of cDNA mapping and mutation identification. Nat Genet. 1994;7:551.
- Nishiyama M, Kato Y, Hashimoto M, et al. Apolipoprotein E, methylenetetrahydrofolate reductase (MTHFR) mutation and the risk of senile dementia – an epidemiological study using the polymerase chain reaction (PCR) method. J Epidemiol. 2000;10:163–172.
- Silva VC, Ramos FJ, Freitas EM, et al. Alzheimer’s disease in Brazilian elderly has a relation with homocysteine but not with MTHFR polymorphisms. Arq Neuropsiquiatr. 2006;64:941–945.10.1590/S0004-282X2006000600010
- Andreassi MG, Botto N, Cocci F, et al. Methylenetetrahydrofolate reductase gene C677T polymorphism, homocysteine, vitamin B12, and DNA damage in coronary artery disease. Hum Genet. 2003;112:171–177.
- Anello G, Guéant-Rodríguez RM, Bosco P, et al. Homocysteine and methylenetetrahydrofolate reductase polymorphism in Alzheimer’s disease. NeuroReport. 2004;15:859–861.10.1097/00001756-200404090-00025
- Wakutani Y, Kowa H, Kusumi M, et al. Genetic analysis of vascular factors in Alzheimer’s disease. Ann N Y Acad Sci. 2002;977:232–238.10.1111/nyas.2002.977.issue-1
- Ravaglia G, Forti P, Maioli F, et al. Common polymorphisms in methylenetetrahydrofolate reductase (MTHFR): relationships with plasma homocysteine concentrations and cognitive status in elderly northern italian subjects. Arch Gerontol Geriatr Suppl. 2004;9:339–348.10.1016/j.archger.2004.04.044
- Bosco P. Association of IL-1 RN*2 allele and methionine synthase 2756 AA genotype with dementia severity of sporadic Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75:1036–1038.10.1136/jnnp.2003.025866
- Mansoori N, Tripathi M, Luthra K, et al. MTHFR (677 and 1298) and IL-6-174 G/C genes in pathogenesis of Alzheimer’s and vascular dementia and their epistatic interaction. Neurobiol Aging. 2012;33:1003.e1–1003.e8.
- Giedraitis V, Kilander L, Degerman-Gunnarsson M, et al. Genetic analysis of Alzheimer’s disease in the Uppsala Longitudinal Study of Adult Men. Dement Geriatr Cogn Disord. 2009;27:59–68.10.1159/000191203
- Linnebank M, Linnebank A, Jeub M, et al. Lack of genetic dispositions to hyperhomocysteinemia in Alzheimer disease. Am J Med Genet A. 2004;131A:101–102.10.1002/ajmg.a.30295
- Dorszewska J, Florczak J, Rozycka A, et al. Oxidative DNA damage and level of thiols as related to polymorphisms of MTHFR, MTR, MTHFD1 in Alzheimer’s and Parkinson’s diseases. Acta Neurobiol Exp (Wars). 2007;67:113–129.
- Crow JF. Hardy, Weinberg and language impediments. Genetics. 1999;152:821–825.
- Ziegler A, Van Steen K, Wellek S. Investigating Hardy–Weinberg equilibrium in case-control or cohort studies or meta-analysis. Breast Cancer Res Tr. 2011;128:197–201.10.1007/s10549-010-1295-z
- Wells G, Shea B, O’connell D, et al. The Newcastle–Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses [ Internet]. [ cited 2017 Jan 22]. Available from: www.ohri.ca/programs/clinical_epidemiology/oxford.htm
- Copas J, Shi JQ. Meta-analysis, funnel plots and sensitivity analysis. Biostatistics (Oxford, England). 2000;1:247–262.10.1093/biostatistics/1.3.247
- Bagos PG, Nikolopoulos GK. Generalized least squares for assessing trends in cumulative meta-analysis with applications in genetic epidemiology. J Clin Epidemiol. 2009;62:1037–1044.10.1016/j.jclinepi.2008.12.008
- Muellerleile P, Mullen B. Sufficiency and stability of evidence for public health interventions using cumulative meta-analysis. Am J Public Health. 2006;96:515–522.10.2105/AJPH.2003.036343
- Peters JL, Sutton AJ, Jones DR, et al. Contour-enhanced meta-analysis funnel plots help distinguish publication bias from other causes of asymmetry. J Clin Epidemiol. 2008;61:991–996.10.1016/j.jclinepi.2007.11.010
- Bird TD. Genetic factors in Alzheimer’s disease. N Engl J Med. 2005;352:862–864.10.1016/j.gene.2015.03.014
- Bertram L, McQueen MB, Mullin K, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23.10.1038/ng1934
- Ballard C, Gauthier S, Corbett A, et al. Alzheimer’s disease. Lancet. 2011;377:1019–1031.10.1016/S0140-6736(10)61349-9
- Chan KY, Wang W, Wu JJ, et al. Epidemiology of Alzheimer’s disease and other forms of dementia in China, 1990–2010: a systematic review and analysis. Lancet. 2013;381:2016–2023.10.1016/S0140-6736(13)60221-4
- Bredesen DE. Inhalational Alzheimer’s disease: an unrecognized – and treatable – epidemic. Aging. 2016;8:304–313.
- Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7:137–152.