
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
High-throughput experimental data are accumulating exponentially in public databases. Unfortunately, however, mining valid scientific discoveries from these abundant resources is hampered by technical artifacts and inherent biological heterogeneity. The former are usually termed “batch effects,” and the latter is often modeled by subtypes. Existing methods either tackle batch effects provided that subtypes are known or cluster subtypes assuming that batch effects are absent. Consequently, there is a lack of research on the correction of batch effects with the presence of unknown subtypes. Here, we combine a location-and-scale adjustment model and model-based clustering into a novel hybrid one, the batch-effects-correction-with-unknown-subtypes model (BUS). BUS is capable of (a) correcting batch effects explicitly, (b) grouping samples that share similar characteristics into subtypes, (c) identifying features that distinguish subtypes, (d) allowing the number of subtypes to vary from batch to batch, (e) integrating batches from different platforms, and (f) enjoying a linear-order computation complexity. We prove the identifiability of BUS and provide conditions for study designs under which batch effects can be corrected. BUS is evaluated by simulation studies and a real breast cancer dataset combined from three batches measured on two platforms. Results from the breast cancer dataset offer much better biological insights than existing methods. We implement BUS as a free Bioconductor package BUScorrect. Supplementary materials for this article are available online.
1. Introduction
To date, more than 1.7 million samples have been deposited into the Gene Expression Omnibus (Edgar et al. Citation2002), which provides unprecedented opportunities to decipher the gene regulation machinery (Pickrell et al. Citation2010), understand disease mechanisms (Chahrour et al. Citation2008), and develop personalized treatments (Suárez-Fariñas et al. Citation2010). Nevertheless, digging in this gold mine is a daunting task. On the one hand, raw data from high-throughput experiments suffer from various types of technical artifacts. On the other hand, biological samples are inherently heterogenous. How to tease out meaningful biological variations from technical artifacts is the major complication in fully using the abundant data sources available for downstream analysis.
Researchers have long been aware that samples generated on different days are not directly comparable. Samples processed at the same time are usually referred to as coming from the same “batch.” Even when the same biological conditions are measured, data from different batches can present very different patterns. The variation among different batches may be due to changes in laboratory conditions, preparation time, reagent lots, and experimenters (Leek et al. Citation2010). The effects caused by these systematic factors are called “batch effects.” Batch effects are prevalent among all types of high-throughput technologies, ranging from microarray (Irizarry et al. Citation2005) and next-generation sequencing (Taub et al. Citation2010) to single-cell sequencing experiments (Hicks et al. Citation2015).
Various “batch effects” correction methods have been proposed when the subtype information for each sample is known. Here, we adopt a “broad” definition for “subtype.” “Subtype” is defined as a set of samples that share the same underlying genomic profile, in other words biological variability, when measured with no technical artifacts. For instance, groupings such as “case” and “control” can be viewed as two subtypes. Given subtypes that are known, Johnson et al. (Citation2007) adjusted batch effects for microarray data in a model-based location-and-scale (L/S) scheme via an empirical Bayesian approach called ComBat. Their model also allows the incorporation of known confounding factors such as age and gender. However, in many experiments, researchers have no measurements of these confounding factors. To adjust for “unmodeled factors,” Leek and Storey (Citation2007) introduced surrogate variable analysis (SVA) for microarray data. Although unmodeled factors cannot be reconstructed exactly, the surrogate variables, whose expanded linear space is the same as that of unmodeled factors, can be learned directly from the data. Accordingly, introducing surrogate variables into the model sidesteps spurious variation due to unmodeled factors and provides accurate estimation of the association between disease status and gene expression. Later, Leek (Citation2014) extends SVA to deal with batch effects in next generation sequencing datasets.
Unfortunately, both ComBat and SVA require the subtype information for each sample and are infeasible when the subtypes are unknown. The frozen robust multi-array analysis (fRMA; McCall et al. Citation2010) was developed to normalize microarray samples individually so that samples from different batches are comparable after normalization. fRMA relies on training a large database of samples from the same microarray platform. As a result, fRMA is only available for a very limited number of array platforms and cannot handle batches measured on different platforms. The single-channel array normalization (SCAN) proposed by Piccolo et al. (Citation2012) can normalize microarray samples measured by different platforms. SCAN fits a two-component normal mixture to the probes of a single sample, one component for background noise and the other for signals. However, SCAN only looks at one sample at a time and thus fails to identify systematic biases associated with the batch. To borrow information and estimate the correlation of measurements from different platforms, Franks et al. (Citation2015) proposed a hierarchical model and applied it to quantify the coordination between transcription and translation level in yeast. Nevertheless, the model assumes a single biological condition without subtype heterogeneity. Therefore, to the best of our knowledge, the general problem of batch effects correction in the presence of unknown subtypes remains an open problem.
Meanwhile, assuming the absence of batch effects, there is extensive research on subtype discovery. Pan and Shen (Citation2007) proposed a model-based clustering algorithm using penalized Expectation Maximization (EM). The approach appends a Lasso penalty (Tibshirani Citation1996) to the Q function in the EM algorithm (Dempster et al. Citation1977). The missing data formulation in the EM algorithm clusters samples into subtypes. The penalty term helps to distinguish the genes whose expression levels remain the same across all subtypes from those genes with varying means among different subtypes. This latter set of genes characterizing distinct subtypes is called the intrinsic gene set (Huo et al. Citation2016). Wang and Zhu (Citation2008) improved the penalized approach by replacing the Lasso penalty with an L∞-norm penalty or a hierarchical penalty. Consequently, parameters belonging to one gene are treated as a single group and penalized together, thus reducing the number of falsely included noninformative genes. Intrinsic gene selection for subtype discovery is also discussed in sparse K-means (Witten and Tibshirani Citation2012). Note that the traditional K-means can be viewed as maximizing Between Cluster Sum of Squares (BCSS), which can be further partitioned into the summation of gene-specific BCSS. Sparse K-means give nonnegative weights to each gene-specific BCSS term and then optimize the weighted BCSS under L1 and L2 constraints on weights. Finally, genes with nonzero weights remain in the intrinsic gene set, and clustering is achieved simultaneously. However, none of the aforementioned methods consider batch effects. As a result, direct application of the above methods to data combined from several batches will lead to problematic scientific conclusions. However, clustering subtypes within each batch separately suffers from small sample size and fails to use shared information among the different batches. Therefore, methods for removing batch effects from datasets with unknown subtypes are important and in urgent demand.
Recently, building upon sparse K-means, Huo et al. (Citation2016) proposed MetaSparseKmeans to discover subtypes using samples from multiple batches. MetaSparseKmeans designs a pattern-matching reward function to encourage the matching of the same subtype across batches. Nevertheless, MetaSparseKmeans requires each batch to contain all subtypes, which is not feasible for many experimental designs. Moreover, the computational complexity of calculating the pattern-matching reward function is O((K!)B − 1) for K subtypes and B batches. As admitted by the authors, even when K = 5 and B = 5, an accurate exhaustive search is prohibitive for 207.36 million comparisons. Furthermore, most importantly, MetaSparseKmeans does not explicitly characterize and correct for batch effects. Consequently, it is only limited to the clustering problem and cannot benefit other analyses such as differential gene expression detection and gene regulatory network construction.
In this article, we integrate the L/S model with model-based clustering and propose the batch-effects-correction-with-unknown-Subtypes (BUS) approach. BUS simultaneously corrects batch effects, discovers subtypes, and selects features that discriminate different subtypes. After correcting the batch effects with BUS, the corrected value can be used for other analysis as if all samples are measured in a single batch. BUS can integrate batches measured from different platforms and allow subtypes to be measured in some but not all of the batches. We prove the identifiability of BUS. Based on the theoretical results, we provide conditions for experimental designs under which batch effects can be corrected.
We conduct statistical inference under the Bayesian framework and develop a Gibbs sampler to draw samples from the posterior distribution. BUS can compute efficiently with the computational complexity growing linearly as subtype number K and batch number B increase. Our simulation studies demonstrate that BUS accurately estimates batch effects and subtype effects, clusters subtypes, as well as selects intrinsic genes. Finally, BUS is applied to three batches of breast cancer microarray data with five subtypes measured on two different platforms (Huo et al. Citation2016). Although we focus on gene expression microarray data here, the same framework can be adapted to DNA methylation microarrays, RNA-seq data, and single-cell sequencing data.
2. Model Formulation
2.1. Location-and-Scale Adjustments
In this subsection, we review the classic location-and-scale adjustment model (L/S) for batch effects correction when the subtypes are known (Johnson et al. Citation2007). The L/S model characterizes two types of batch effects, the additive effects influencing the means and the multiplicative effects affecting the variances of gene expression. Specifically, let us denote the gene expression level of gene g for the sample j in the batch b by Ybjg, then the L/S model is formulated as follows:
(2.1)
(2.1) Here, Xbj encodes the study design, that is, which subtype each sample comes from. Suppose there are K subtypes in the dataset, then Xbj is a K-dimensional binary vector with its kth element being one and the others being zeros if sample j in the batch b belongs to subtype k. Consequently,
records the subtype effects for gene g. To make the model identifiable, in the following analysis, we set μg1 = 0. Subsequently, αg represents the mean gene expression level of gene g in subtype one.
In terms of batch effects, the “location” term γbg captures the shift in the mean due to additive effects, and the “scale” term δbg quantifies the variability of the gene expression caused by the multiplicative batch effects. For model identifiability, without loss of generality, we always refer to batch one as the “reference batch,” where the location batch effects and the scale batch effects are assumed to be absent. In other words, γ1g = 0 and δ1g = 1 for all 1 ⩽ g ⩽ G. Finally, the noise term ϵbjg is assumed to follow a normal distribution N(0, σ20g) with mean zero and variance σ20g.
For the convenience of future discussion, we absorb δbgϵbjg into εbjg and arrive at the equivalent model:
(2.2)
(2.2) where εbjg follows N(0, σ2bg), and note that the ratio of σ2bg to σ21g (= δ2bg) is the squared multiplicative batch effect.
2.2. The BUS Model
Nevertheless, the L/S model assumes that the subtype information is known to the investigator. To handle the case in which subtype information is unknown, we propose the following Batch-effects-correction-with-Unknown-Subtypes (BUS) model built upon the L/S model. The main challenge is that the subtype indicators Xbj in Equation (Equation2.2(2.2)
(2.2) ) now become missing data, and hence, we need to infer the subtype information for each sample as well. Here, we employ ideas from the literature on model-based clustering (Banfield and Raftery Citation1993; Yeung et al. Citation2001; Fraley and Raftery Citation2002; McLachlan and Peel Citation2004).
Let us first assume that all of the samples come from K subtypes and are measured in a single batch. Therefore, we temporarily drop the subscript b for batch indicators and collect all of the gene expression values for sample j into Yj = (Yj1, …, YjG). If sample j belongs to subtype k, then we assume Yj follows a multivariate Gaussian distribution N(mk, Σ) with a subtype mean mk = (m1k, …, mGk) and a common diagonal covariance matrix across the subtypes (Fraley and Raftery Citation2002; McLachlan and Peel Citation2004; Bickel and Levina Citation2004). The resulting model is a Gaussian mixture:
(2.3)
(2.3) where “iid” stands for “independent and identically distributed,” and πk indicates the proportion of subtype k in the samples, satisfying πk ⩾ 0, ∑Kk = 1πk = 1. Bringing in a subtype indicator Zj for each sample j, in which Zj = k if sample j comes from subtype k, an equivalent model can be formulated as follows: for 1 ⩽ j ⩽ n and 1 ⩽ g ⩽ G
(2.4)
(2.4) To align with model 2.2, Zj = k can be mapped to Xj with its kth element equal to one and all of the others being zero and vice versa.
Going one step further, we now consider the multi-batch case in which heterogenous samples from multiple subtypes are measured in multiple batches. We assume that there are in total B batches and K subtypes, and batch b contains nb samples. For each sample, gene expression levels in all G genes are measured. As in Section 2.1, we denote the gene expression level of gene g for the sample j in the batch b by Ybjg. We allow the proportion of subtypes to vary from one batch to another. Therefore, we use πbk to denote the proportion of subtype k in the batch b; thus, ∑Kk = 1πbk = 1. Consequently, the integration of Model 2.2 and Model 2.3 gives rise to: for 1 ⩽ b ⩽ B, 1 ⩽ j ⩽ nb, 1 ⩽ g ⩽ G
(2.5)
(2.5) Accordingly, the complete likelihood function for the observed data
and missing data
becomes:
where Θ is composed of all unknown parameters {πbk: 1 ⩽ b ⩽ B, 1 ⩽ k ⩽ K}, {αg: 1 ⩽ g ⩽ G}, {μgk: 1 ⩽ g ⩽ G, 2 ⩽ k ⩽ K}, {γbg: 2 ⩽ b ⩽ B, 1 ⩽ g ⩽ G}, and {σ2bg: 1 ⩽ b ⩽ B, 1 ⩽ g ⩽ G}. Recall that μg1 and γ1g (1 ⩽ g ⩽ G) are constrained to zero for identifiability.
As a side note, BUS can also handle known confounding factors such as age, gender, and BMI by modifying mgk to , where Ubj corresponds to the confounding variables.
3. Identifiability
In this section, we first prove that BUS is identifiable when every subtype presents on every batch. Next, we extend the results to more general study design and provide guidelines on the experimental design so that batch effects can be corrected.
3.1. With Complete Subtypes
We first investigate the scenario as proposed by MetaSparseKmeans (Huo et al. Citation2016) in which all subtypes are required to occur in all the batches. We refer to this setting as the “complete subtypes” case. In BUS model, the likelihood function for the observed data is
where Θ consists of all unknown parameters
with
,
,
, and
. Recall that
,
.
Theorem 1.
If for any (k1, k2) ≠ (k3, k4) (Assumption I) and πbk > 0 for every b and k, then BUS is identifiable (up to label switching) in the sense that Lo(Θ|Y) = Lo(Θ*|Y) for any Y implies that πbk = π*bρ(k),
,
, and
, where ρ is a permutation of {1, 2, …, K}.
The condition on πbk > 0 corresponds to the “complete subtypes” experimental design. Assumption I is a very mild one. It actually only asks for the existence of one gene g whose mean expression differences between subtypes satisfy for any (k1, k2) ≠ (k3, k4), which is always easily met for high-throughput biology data.
3.2. With Missing Subtypes
Next, we investigate the scenario in which subtypes are measured in some but not all of the batches, which is the general study design usually encountered in real life. We call this setting as “with missing subtypes.” In other words, we allow πbk = 0 for some b and k. In the following, we use Cb to denote the subtypes that are present in batch b and Db to denote all of the data measured on batch b. We provide two types of experimental design that guarantee the identifiability of BUS.
Theorem 2.
Given (A) , (B) Kb = |Cb|, the cardinality of Cb, Kb ⩾ 2 for every batch b, (C) C1 = {1, 2, …, K} and Assumption I is satisfied, then BUS is identifiable (up to label switching).
Condition (C) assumes that there exist a reference batch, without loss of generality taken as batch one, that contains samples from all subtypes. In practice, this assumption often holds if we have a large database built upon previous experiments. In a new experiment consisting of batches two to B (B ⩾ 2), if for each batch b (2 ⩽ b ⩽ B) we collect samples that are from at least two subtypes present in batch one, then Theorem 2 holds. Therefore, we can apply BUS to the dataset {Db: b = 1, …, B} to separate the confounding batch effects from the true biological variations in the new experiment.
Theorem 3.
Given (A) , (B) Kb = |Cb|, the cardinality of Cb, Kb ⩾ 2 for every batch b, (D)
for every b ⩾ 2 and Assumption I is satisfied, then BUS is identifiable (up to label switching).
Theorem 3 tells us how to design a valid study when we do not have a large database that contains every subtype. In detail, we can begin with batch one whose samples are known to be taken from at least two subtypes. When preparing samples for batch two, we recommend collecting samples that are from at least two known subtypes in batch one. Based on Theorem 3, we can then apply BUS to {Db: b = 1, 2} to learn the subtype for each sample in batch two. In the same spirit, we require the samples for batch three to be from at least two learned subtypes in batch two, and we then apply BUS to {Db: b = 1, 2, 3} and so on and so forth. This chain-type experimental design satisfies the conditions of Theorem 3, so batch effects are estimable.
In a nutshell, both the conditions for Theorems 2 and 3 can be executed and implemented in planning real multi-batch experiments. Therefore, from the perspective of practitioners, these conditions help to guide experimental designs which can simultaneously filter out batch effects and keep true biological variations.
4. Statistical Inference
4.1. Prior Specification
We adopt a full Bayesian approach to conduct statistical inference (Hein et al. Citation2005; Gelman et al. Citation2014). We assign independent conjugate priors to each component of Θ as follows: ; αg ∼ N(m, τ2m), 1 ⩽ g ⩽ G; γbg ∼ N(0, τ2γ), 2 ⩽ b ⩽ B, 1 ⩽ g ⩽ G;
with hyper-parameters
.
Regarding the subtype effect μgk, we assign a spike-and-slab prior (George and McCulloch Citation1993) using a normal mixture. Specifically, one component of the mixture concentrates near zero with a small variance, and the other is more dispersed with a larger variance. To represent the mixture distribution, we introduce the latent variable Lgk to indicate which component of the mixture distribution μgk comes from. When Lgk = 0, gene g is believed to have the same expression level in subtype k and subtype one. Therefore, μgk is assumed to be close to zero, following the normal component with a small variance. When Lgk = 1, gene g is differentially expressed (DE) between subtype k and subtype one. Consequently, μgk tends to largely deviate from zero, following the normal component with a large variance. As a result, the expression of gene g does not hold constant across subtypes if and only if Dg≐∑Kk = 2Lgk > 0. We call such genes intrinsic genes following Huo et al. (Citation2016) and name Dgs intrinsic gene indicators. Scientifically, Lgks identify the set of genes that define and differentiate subtypes. Denoting the proportion of Lgks being one by p, the relationship between μgk and Lgk for g = 1, …, G; k = 2, …, K is as follows:
τ2μ1 is set to a large number, and τ2μ0 follows an inverse-gamma prior InvΓ(aτ, bτ) with a small prior mean. Meanwhile, p has the conjugate prior
.
With all the priors, the full posterior distribution f(Θ, Z, L|Y) is proportional to
4.2. Posterior Inference
To explore the posterior distribution, we develop a Gibbs sampler algorithm to draw samples (Geman and Geman Citation1984; Robert and Casella Citation2013). At iteration t:
| 1. | Update the inclusion probability p[t] for Lgks from
| ||||
| 2. | Sample the variance of the spike component of the spike-and-slab prior (τ[t]μ0)2 from
| ||||
| 3. | For each gene g and for 2 ⩽ k ⩽ K, update indicator L[t]gk from
| ||||
| 4. | For each batch b, sample subtype proportions | ||||
| 5. | For each sample j in every batch b, update its subtype indicator according to
| ||||
| 6. | For each gene g, sample its baseline expression level α[t]g from
| ||||
| 7. | For each gene g in subtype two to K, sample its subtype effect μ[t]gk from
| ||||
| 8. | For each gene in batch two to B, sample the additive “location” batch effects γ[t]bg from
| ||||
| 9. | Sample the multiplicative “scale” batch effects from the inverse-Gamma distribution
| ||||
To determine the number of iterations for the Gibbs sampler, we adopt the estimated potential scale reduction (EPSR) factors criterion (Gelman et al. Citation2014) (see supplementary materials Section S1). Based on the collected samples from the Gibbs sampler, we conduct posterior inferences. For the underlying subtype effects (μgks), location batch effects (αbgs), and scale batch effects (σ2bgs), which take continuous values, we use the means of their posterior samples for estimation, since the posterior mean minimizes the Bayes risk (Casella and Berger Citation2002). Regarding clustering, we take the posterior mode of samples for Zbj as the subtype for sample j in batch b.
Compared to single-batch-based methods, BUS can borrow information across all batches and all genes. For example, in step 7 of the Gibbs sampler, updating subtype effects μ[t]gk depends on data related to gene g and subtype k in all of the batches, which offers more robust and accurate estimation. On the other hand, in step 5, the subtype determination for a given sample uses information across the genes. The two-way information sharing across genes and batches improves the statistical power of BUS.
Recall that gene g is an intrinsic gene if Lgk = 1 for some k (2 ⩽ k ⩽ K). To reduce the errors in inferring Lgk’s, we control the Bayesian false discovery rate (FDR; Newton et al. Citation2004; Peterson et al. Citation2015). We denote by the posterior marginal probability for gene g to be DE in subtype k compared to subtype one and let
; then according to Newton et al. (Citation2004) and Peterson et al. (Citation2015), the expected Bayesian FDR for inferring DE indicators becomes
(4.1)
(4.1) From the posterior samples of Lgk,
, we can estimate
and further approximate
. If we want to control the FDR at a prespecified threshold α, such as 0.1, we can select κ0 such that estimated
. In other words, if the estimated
, we claim Lgk as one and zero otherwise.
So far, we have focused on the case where investigators have prior knowledge of the number of subtypes. When K is unknown, the Bayesian information criterion (BIC; Schwarz et al. Citation1978) can be used to select the optimal number of subtypes. The Bayesian information criterion (BIC) formula for BUS is
(4.2)
(4.2)
(4.3)
(4.3) where the first term (Equation4.2
(4.2)
(4.2) ) approximates two times the negative observed-data log likelihood and the second term (Equation4.3
(4.3)
(4.3) ) is the product of the parameter number and the logarithm of the observation number.
,
,
,
, and
are the posterior mean estimates. We choose the subtype number K such that BIC attains its minimum. Once the optimal number of K is determined, all of the aforementioned analyses follow.
Compared to MetaSparseKmeans (Huo et al. Citation2016), BUS has two advantages for clustering. First, BUS allows only some of the total K subtypes to appear in each batch. In contrast, MetaSparseKmeans requires all subtypes to be measured on every batch, which imposes very strong constraints on the datasets that can be used for meta-analysis and is often infeasible for many experimental designs. Second, BUS avoids the combinatorial matching encountered by MetaSparseKmeans. The computation complexity of BUS for every round of updates is only O(∑Bb = 1nb · GK), both linear in the number of batches B and in the number of subtypes K, thus significantly outperforming the computational complexity of MetaSparseKmeans O((K!)B − 1), which grows exponentially in the number of batches B and factorially in the number of subtypes K.
4.3. Downstream Analysis
In addition to clustering, BUS also provides an explicit characterization of batch effects and enables correction of the raw input data. To correct batch effects, we can follow a similar approach as the L/S model (Johnson et al. Citation2007). As batch one is always taken as the reference batch, no correction is needed. For batches two to B, the corrected gene expression value after removing the “location” and “scale” batch effects can be calculated as
(4.4)
(4.4)
The corrected expression values will be free from nonbiological effects and serve as valid data sources for downstream analysis such as differential gene expression detection and gene regulatory network construction.
5. Simulation
In this section, we evaluate the performance of BUS in correcting batch effects, clustering subtypes as well as selecting intrinsic genes via simulation studies. We compare BUS to MetaSpaseKmeans (Huo et al. Citation2016) and a two-stage approach coupling ComBat (Johnson et al. Citation2007) with SparseKmeans (Witten and Tibshirani Citation2012).
5.1. With Complete Subtypes
Following theterminology in Section 3, we first investigate the “complete subtypes” case. In Simulation I, we simulate expression level for G = 10, 000 genes from K = 3 subtypes measured in B = 3 batches. The sample sizes for each batch are (n1, n2, n3) = (100, 110, 120). The subtype proportions for each batch are ,
and
, respectively. For each batch b, we assume the first πb1 · 100% of simulated samples are from subtype one, the second πb2 · 100% of simulated samples from subtype two, and the rest of the samples from subtype three.
We then specify the underlying mean gene expression level. αg is set to two for all of the genes. Recall that subtype effect μg1 is constrained to zero for all genes in subtype one. The top 500 genes are chosen to be DE in subtype two compared to subtype one with a mean shift of two: μg2 = 2, g = 1…, 500. Similarly, genes 501–1000 are up-regulated in subtype three with a mean difference of two: μg3 = 2, g = 501…, 1000. Other than that, none of the remaining genes are DE. In other words, μg2 = 0, g = 501…, 1000; μg3 = 0, g = 1…, 500; and μgk = 0, k = 2, 3 for the rest of the genes. (a) shows the underlying true gene expression level αg + μgk for each gene in each subtype.
Figure 1. Patterns for Simulation I. (a) True subtype mean. Each row represents a gene, and each column corresponds to a subtype. There are in total 10,000 genes and three subtypes. (b) True batch effects. Each row represents a gene and each column corresponds to a batch. There are three batches in total. (c) Observed gene expression. Each row represents a gene and each column is a sample. There are 330 samples. (d) BIC plot. (e) Estimated subtype mean. (f) Estimated batch effects. (g) Corrected gene expression grouped by batches. The samples are first ordered by batch (the top bar) and then ordered by subtype (the bottom bar). (h) Corrected gene expression grouped by subtypes. The samples are first ordered by subtype (the bottom bar) and then ordered by batch (the top bar).

Now, we determine the batch effects. For the additive batch effects in batch 2, as illustrated in (b), we set γ2g = 3 for genes 1–2000; γ2g = 2 for genes 2001–4000; γ2g = 1 for genes 4001–6000; γ2g = 2 for genes 6001–8000; and γ2g = 3 for genes 8001–10,000. If we abbreviate such pattern for 2000 consecutive genes as (3,2,1,2,3), then γ3g are specified to (1,2,3,2,1) in the same fashion for batch three. The multiplicative batch effects are σ21g = 0.1, σ22g = 0.2, and σ23g = 0.15 for all genes.
Finally, we generate all of the raw gene expression values from Model Equation2.5(2.5)
(2.5) . (c) displays the observed data. Obviously, a naive application of traditional clustering methods to the raw data without considering batch effects would fail to identify the true subtypes.
For the prior distributions discussed in Section 4.1, we set the hyper-parameters as follows: m = 1, ,
, α = 2,
,
, aτ = 2, bτ = 0.005, (ap, bp) = (1, 3), and τμ1 = 10. We conduct analysis for K from 2 to 10. BIC correctly chooses K = 3 ((d)). The Markov chain produced by the Gibbs sampler converges very quickly according to the EPSR factors (see supplementary materials Section S1 for details and supplementary Figure S1 for trace plots). After 150 iterations of the burn-in period, we collect 150 iterations for posterior inference. (e) and (f) shows the posterior means for subtype means and additive batch effects. Comparing to (a) and (b), BUS accurately estimates the true parameters. Figure 1(g) and 1(h) is the heatmaps for batch-effects-corrected
, and it can be seen that the corrected gene expression values now no longer suffer from technical noise and reveal their true biological variation.
Besides correcting batch effects, BUS can cluster all samples automatically with the posterior modes of their subtype indicators . Samples with the same value of
are grouped together. To measure how close the estimates
are to the underlying truth Z, we adopt the adjusted Rand index (ARI; Hubert and Arabie Citation1985). The ARI is bounded above by one, and the higher the value is, the more consistent the two categorical vectors are. The ARI between
and Z is exactly one, showing that BUS perfectly recovers the underlying true subtypes for all samples.
Next, we study the performance of BUS in identifying intrinsic genes that reflect the difference among subtypes. The intrinsic gene indicator Dg is estimated as , where
are estimated by setting κ = 0.5 to control FDR in Equation (Equation4.1
(4.1)
(4.1) ) below 0.1. Genes with
are regarded as intrinsic genes. BUS claims all of the 1000 assumed intrinsic genes correctly with only one false discovery.
In comparison, for this small-scale dataset, MetaSparseKmeans also has an ARI = 1 and identifies all of the intrinsic genes correctly. Both MetaSparseKmeans and BUS have comparably fast computation time here around 2 min. Nevertheless, noteworthy, the samples from the Markov chain Monte Carlo (MCMC) method (Robert and Casella Citation2013) obtained by BUS can provide the full posterior distribution for all of the parameters, especially quantifying the uncertainties in parameter estimations.
We also test a two-stage approach that we abbreviate as “ComSpa.” ComSpa first applies ComBat to the three batches, then treats the corrected values from all of the batches as a single batch and uses SparseKmeans to cluster samples. However, even after the correction by ComBat, the expression profiles of the same subtype show very distinct patterns in different batches. For example, in supplementary Figure S2, subtype two (colored by blue in the lower color bar) samples on batch one show significantly higher expression values than subtype two samples on batch two for the top 500 intrinsic genes. The issue is that ComBat requires known subtype information or homogenous biological samples to tease out batch effects. When multiple unknown subtypes exist within each batch, ComBat can easily over-correct the biological variations as artificial noise. The problematic batch effects correction in the first-stage leads to the poor clustering performance of ComSpa with a low ARI of 0.546.
5.1.1. Sensitivity Analysis
We conduct sensitivity analysis to check the influence of prior distributions and find that the choices of hyper-parameters have little effect on the posterior inference (see supplementary materials Section S2.1 and supplementary Figure S3), so we fix the same set of hyperparameters as in Simulation I throughout the article.
In addition to the choices of hyper-parameters, we investigate the effects of sample misclassification on batch effects estimation (see supplementary materials Section S2.2). According to supplementary Figure S4, the batch effects estimations are not sensitive to sample misclassification for intrinsic genes and are always accurate for nonintrinsic genes. Therefore, even in cases where some subtypes are wrongly claimed, Equation (Equation4.4(4.4)
(4.4) ) still provides robust corrected expression values.
5.1.2. Varied Signal Strengths
We further test the robustness of BUS under different signal strengths. Notice that there are two ways to vary signals: (a) keep the sample size fixed and change the strengths of the subtype effects and (b) fix the strengths of the subtype effects and vary the sample size.
For scenario a, we fix the sample size as (100, 110, 120) for the three batches, respectively. For each gene g, we set σ2g1 = 0.1 on batch one, σ2g2 = 0.2 on batch two, and σ2g3 = 0.15 on batch three. We represent the signal matrix that encodes the subtype effects as
Each column of S(v) corresponds to a subtype, and each row refers to a gene. Specifically, the kth column of S(v), S(v)k = (μ1k, …, μGk)T, represents the subtype effects of all of the genes for subtype k. Consequently, v indicates the signal strength of the subtype effects: the lower the v is, the weaker the signals are. We vary v from 0 to 3 taking values at {0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.4, 0.5, 1, 1.5, 2, 2.5, 3}. For each setting, we apply BUS and calculate the corresponding ARI (see supplementary Figure S5(a)). When v < 0.15, the signals are overwhelmed by noise. However, as long as v ⩾ 0.4 which is about the size of
, BUS can precisely cluster all of the samples and infer the underlying parameters.
For scenario b, keeping the subtype effect strength v = 2 and the other settings the same as above, we reduce the sample size from (100, 110, 120) to (100, 110, 120) · ω for ω = 0.1, 0.2, …, 0.8, 0.9, 1. For each generated dataset, we apply BUS and calculate the corresponding ARI (see supplementary Figure S5(b)). As long as ω ⩾ 0.2, even with a small-sample size (20, 22, 24) for ω = 0.2, BUS is able to correctly cluster all of the samples and estimate the parameters precisely.
5.1.3. Model Misspecification
BUS assumes that given a subtype the expression levels of different genes are independent, which leads to a diagonal covariance matrix in Equation (Equation2.3
(2.3)
(2.3) ). However, as is often the case, the expression levels of many genes are correlated, so
is no longer diagonal. We incorporate correlation structures into gene expression following a similar simulation setting of Huo et al. (Citation2016), assuming that the expression values for every 20 consecutive intrinsic genes follow a multivariate normal distribution with the covariance matrix sampled from an inverse Wishart distribution W− 1(Φ, 60), where Φ = 0.5I20 × 20 + 0.5120 × 11T20 × 1. The expressions for the remaining genes, that is, the nonintrinsic genes, are independent. We apply BUS to this model-misspecified setting, and it turns out that BUS successfully identifies the true subtypes for all of the samples (
) and estimates subtype effects and location batch effects very well (see supplementary materials Section S3.1 and supplementary Figure S6).
In real-world datasets, some samples might be purely noise or outliers and cannot be grouped into any cluster, which are defined as “scattered samples” (Tseng and Wong Citation2005; Maitra and Ramler Citation2009). Although Tseng and Wong (Citation2005) proposed an algorithm under the K-means framework, in principle their resampling ideas are generalizable to detection of scattered samples for other clustering methods. We adapt algorithm A of Tseng and Wong (Citation2005) to BUS and propose an algorithm to detect scattered samples with a given cluster number (see supplementary materials Section S3.2). To illustrate, we add one scattered sample to each batch in Simulation I. In batch b, the expression level of gene g in the scattered sample is drawn from N(4 + γgb, σ2gb). We then carry out the “scattered sample-finding” algorithm. As shown in supplementary Figure S7(a)– S7(c), the scattered sample is successfully identified for each batch.
All in all, BUS is robust to the choices of hyper-parameters, sample misclassification, noise levels, correlation structures, and scattered samples.
For the “complete subtypes” setting, we also test a large-scale dataset, Simulation II, which consists of 10 batches and 5 subtypes. The performance of BUS remains the same, taking 1.32 hr on a 2.6GHz processor (the same processor used across the article) and resulting in zero FDR (κ = 0.5) and an ARI of one, whereas the original MetaSparseKmeans algorithm fails to work using its exhaustive version. Therefore, we run an approximate version MetaSparseMeans using simulated annealing (MetaSparseMeans-SA), which takes 1.73 hr and also has an ARI of one. Thus, MetaSparseMeans-SA has performance comparable to BUS on Simulation II. Nevertheless, as discussed above, BUS can provide the full posterior distribution for all of the parameters. Please see supplementary materials Section S4 and supplementary Table S1 for the details of Simulation II.
5.2. With Missing Subtypes
Now we move on to the scenario “with missing subtypes” as defined in Section 3. We consider a large-scale case, Simulation III, in which there are 10 batches. The sample sizes of the 10 batches are set to (300, 110, 220, 100, 180, 110, 100, 180, 150, 110), respectively. The total number of subtypes across all samples, K, is set to be 5. However, let Kb denote the number of subtypes in batch b, then Kb ⩽ K changes across batches. For illustration, subtype one appears only in batch one and batch eight; subtype five is present only in batch one and two. Supplementary Table S2 shows the values of all of the parameters, and (a)–(c) depicts the assumed subtype means, batch effects as well as the observed data, respectively.
Figure 2. Patterns for Simulation III. (a) True subtype mean. Each row represents a gene, and each column corresponds to a subtype. There are in total 10,000 genes and five subtypes. (b) True batch effects. Each row represents a gene and each column corresponds to a batch. There are 10 batches in total. (c) Observed gene expression. Each row represents a gene and each column is a sample. There are 1560 samples. (d) BIC plot. (e) Estimated subtype mean. (f) Estimated batch effects. (g) Corrected gene expression grouped by batches. The samples are first ordered by batch (the top bar) and then ordered by subtype (the bottom bar). (h) Corrected gene expression grouped by subtypes. The samples are first ordered by subtype (the bottom bar) and then ordered by batch (the top bar).
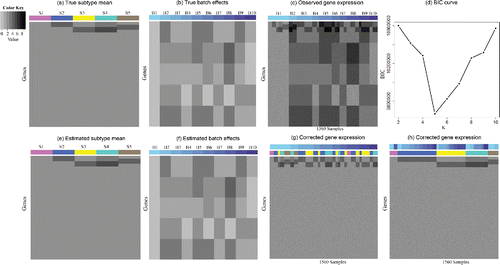
BIC values ((d)) are calculated for K = 2, 3, …, 10, respectively, and the optimal subtype number is five, in accord with the underlying truth. We conduct the same posterior inference as before, run 3000 MCMC iterations which achieve convergence based on EPSR factors, and collect samples from the last 1500 iterations. The Gibbs Sampler cost 1.73 hr.
Subtype means ((e)) and batch effects ((f)) are correctly recovered once again. The corrected expression data are free from technical artifacts and demonstrate their underlying true biological variability (Figure 2(g) and 2(h)). The ARI between the results produced by BUS and the underlying truth is one again, which indicates the perfect sample clustering of BUS. BUS also finds all the intrinsic genes with no false discovery (κ = 0.5 in Equation (Equation4.1(4.1)
(4.1) )).
In comparison, we run MetaSparseKmeans-SA, SparseKemans, and ComSpa with the prespecified total cluster number set to 5. MetaSparseKmeans-SA and ComSpa are applied to the whole dataset combined from 10 batches, and SparseKmeans is applied to each single batch separately. Finally, we calculate their ARIs with the underlying truth. Across all samples from all batches, the MetaSparseKmeans-SA’s ARI is 0.289 and cost 2.43 hr; ComSpa has an ARI of 0.43 and takes 13.3 hr; and the SparseKmeans consumes less than 1 hr but its ARI is 0.193. All of them have ARIs much lower than BUS’ ARI of one. The main issues are: (a) MetaSparseKmeans and SparseKmeans force all of the K subtypes to be present in all of the batches, as a result, they ignore the fact that Kb’s can be less than K in each individual batch and vary across batches; (b) ComBat, the first stage of ComSpa, cannot filter out batch effects correctly when there are unknown biological variations.
6. Application
In this section, we apply BUS to a breast cancer dataset that is preprocessed by Huo et al. (Citation2016) and used to apply the MetaSparseKmeans model. The study design of the dataset is summarized in . In total, B = 3 batches are analyzed and they come from two different platforms. The batch generated by The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Network Citation2012) was measured using the Agilent platform, and consequently, the measurement unit is log ratio intensity. The two batches generated by Wang et al. (Citation2005) and Desmedt et al. (Citation2007) used the Affymetrix array, and hence, the measurement unit is log intensity. From , we can see that the expression values from the two platforms are of very different ranges and medians. (a) is the heatmap of the raw gene expression combined directly from the three batches. Not only do the batches measured on different platform (batch 1 vs. batches 2 and 3) show distinct patterns due to artifacts, but also the batches measured on the same platform (batch 2 vs. 3) demonstrate strong batch effects. The subtype differences are completely overwhelmed by the batch effects.
Figure 3. Heatmaps for breast cancer datasets. (a) Observed gene expression, where rows represent 11,058 genes and columns represent 533 (TCGA)+260 (Wang et al.)+164 (Desmedt et al.) = 957 samples; (b) Corrected gene expression by BUS.
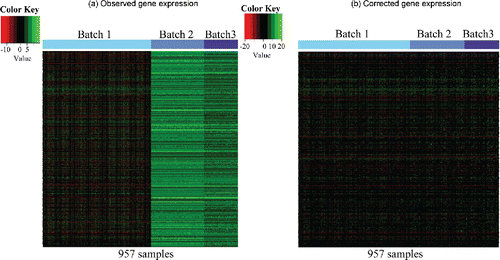
Table 1. Breast cancer datasets information. Part of the table is reproduced from Table 1 of Huo et al. (Citation2016).
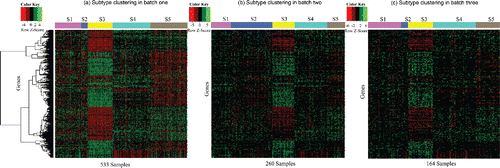
According to BIC (see supplementary Figure S8), BUS identifies four subtypes, which is consistent with conclusions from existing biomedical literature (Carey et al. Citation2006; Onitilo et al. Citation2009) that there are four main breast cancer subtypes: basal-like, HER2+/ER-, luminal A, and luminal B. However, to have a direct comparison with MetaSparseKmeans (Huo et al. Citation2016) which specifies five subtypes for the same dataset, we deliberately fix K at five instead of using the BIC. After carrying out Gibbs sampling for 4000 iterations which meets the convergence criterion by the EPSR factors (see supplementary materials Section S1), we treat the first 2000 iterations as burn-in and keep the last 2000 iterations. It takes in total 1.81 hr. (a)–(c) plots the three heatmaps for the gene expression values of each batch with samples ordered by learned subtypes, respectively. (b) corresponds to the corrected expression values by BUS, and the heatmap illustrates that the corrected values can be viewed as being measured in the same batch.
Figure 4. Heatmaps for intrinsic genes selected by BUS from the breast cancer dataset. (a) Row-scaled gene expression values from TCGA. The rows are clustered by hierarchical clustering, and the columns are clustered based on the BUS model. (b) Row-scaled gene expression values from Wang et al. The row order is the same as that in (a), and columns are clustered based on the BUS model; (c) Row-scaled gene expression values from Desmedt et al. The row order is the same as that in (a), and columns are clustered based on the BUS model.
Now, for each subtype k and each gene g, we calculate by controlling the Bayesian FDR in Equation (Equation4.1
(4.1)
(4.1) ) at 0.1, which leads to κ = 0.4, and estimate
accordingly. Subsequently, 391 genes are selected as the intrinsic genes, which potentially explain the differences among breast cancer subtypes. From the heatmaps for the intrinsic genes (), the subtype patterns are observable for each batch.
In contrast, 203 intrinsic genes are obtained using MetaSparseKmeans (Huo et al. Citation2016). We note that BUS associates each intrinsic gene g with the intrinsic gene indicator Dg ≔ ∑Kk = 2Lgk > 0. Thus, genes with the highest Dg’s show the strongest signals to be intrinsic genes. Therefore, to select the same number of intrinsic genes as that identified by MetaSparseKmeans, we order the intrinsic genes by Dg’s and choose the top 203 intrinsic genes to conduct pathway analysis on the same website http://software.broadinstitute.org/gsea/msigdb/annotate.jsp using the same BioCarta database. The top 203 intrinsic genes produced by BUS lead to 21 significant (q-value less than 0.05) BIOCARTA pathways (see the column “BUS 203” in ). In contrast, the intrinsic gene set on the same dataset selected by MetaSparseKmeans gives seven significant BIOCARTA pathways. Only three enriched BIOCARTA pathways, BIOCARTA_RANMS_PATHWAY, BIOCARTA_MCM_PATHWAY, and BIOCARTA_G1_PATHWAY, overlap between these two intrinsic gene sets. However, the rest of the pathways enriched in BUS analysis are closely related to breast cancer. Especially, the highest-ranked-enriched pathway from BUS analysis is the BIOCARTA_HER2_PATHWAY. Her2 is one of the most important biomarkers in breast cancer (Slamon et al. Citation1987). Actually, overexpression of Her2 is strongly associated with increased disease recurrence and a poor prognosis (Xia et al. Citation2004). Subtype-specific therapy, trastuzumab, targets Her2 and is only effective in Her2-positive patients (Piccart-Gebhart et al. Citation2005). The efficacy of trastuzumab is a classic example of subtype-specific diagnosis and treatment. However, this very important breast cancer pathway—BIOCARTA_HER2_PATHWAY—is completely mis-sed by MetaSparseKmeans. Moreover, IGF-1 (Wolf et al. Citation2008), MTA-3 (Fujita et al. Citation2003), and STATHMIN (Alli et al. Citation2007) are all key pathways for breast cancer. They are significant for BUS but missed by MetaSparseKmeans. These facts strongly support that the intrinsic gene set from BUS offers scientifically meaningful subtyping.
Table 2. Significant (q-value less than 0.05) BIOCARTA pathways identified by different studies. Results from joint analyses of three batches by BUS are compared to that from MetaSparseKmeans on three batches and sparse K-means on each batch separately. “BUS 391” indicates that we use all of the 391 intrinsic genes identified by BUS to carry out pathway analysis. “BUS 203” corresponds to using the top 203 genes with the highest intrinsic gene indicators learned by BUS. “-” represents that the corresponding BIOCARTA pathway is not significant for the corresponding method. The numbers displayed in the table are q-values.
It is noteworthy that the two-stage approach ComSpa learn more than 4000 intrinsic genes for the same dataset. Such a large number of intrinsic genes tends to include many false discoveries, and it exceeds the largest number of genes allowed for the pathway analysis on http://software.broadinstitute.org/gsea/msigdb/annotate.jsp.
We also use the original 391 intrinsic genes called by BUS to conduct pathway analyses on the BioCarta (see the column “BUS 391” in ), KEGG and GO biological processes, respectively (see supplementary Tables S3 to S5). For the KEGG database, there are 58 significant pathways including the KEGG_ERBB_SIGNALING_PATHWAY (q-value: 6.02 × 10− 3), which plays a crucial role in the development of breast cancer, KEGG_MAPK_SIGNALING_PATHWAY (q-value: 5.76 × 10− 5), and KEGG_PHOSPHATIDYLINOSITOL_SIGNALING_SYSTEM (q-value: 0.0181), which are activated by most ErbBs receptors, and the KEGG_PATHWAYS_IN_CANCER (q-value: 1.33 × 10− 11). Meanwhile, more than 100 GO biological processes are significantly enriched. Most of them pertain to the cell cycle, cell proliferation, cell death, cell differentiation, cell communication, all of which are potential factors related to the induction of breast cancer.
The pathway analyses very convincingly show that BUS provides biologically and clinically valid subtyping and outperforms MetaSparseKmeans, let alone sparse K-means applied to individual batch separately.
7. Discussion
To the best of our knowledge, BUS is the first method to explicitly model batch effects and discover underlying subtypes at the same time. Scientifically, BUS is able to integrate batches measured from different platforms in which subtypes can be present on some but not all batches. Statistically, BUS leverages information across batches to estimate subtype effects and borrows strength across genes to cluster subtypes, thus providing robust and legitimate inferences. Computationally, BUS overcomes the factorial growth of computation time, and its computational complexity only grows linearly with the batch number and subtype number. Moreover, BUS models the additive and multiplicative batch effects explicitly. Consequently, we can easily filter the two types of batch effects directly from the raw input data. The corrected data are robust to sample misclassification and can be used for downstream analysis as if they originate from a single batch.
Theoretically, we prove that BUS is identifiable when all subtypes are measured on each batch. In addition, we offer two very convenient experimental designs where subtypes are allowed to be measured on a subset of batches, and we prove the model identifiability under each scenario. We hope these results will provide researchers more freedom and flexibility in designing valid studies that can be protected from batch effects.
During the review period for this article, Jacob et al. (Citation2016) proposed approaches to remove unwanted factors with unknown information of batches or biological phenotypes. However, their approaches require control genes whose expressions are known to be irrelevant to the factors of interest and/or control samples that have replicates in all batches, and their methods can work only when the control genes or control samples are available. Practically, incorporating control genes or control samples can be challenging. In contrast, BUS does not require any control genes, and the experimental designs suggested by Theorems 2 and 3 are more flexible than those that use control samples. BUS only asks for batch information. In real datasets, batch information can often be obtained directly or indirectly such as by tracking series numbers of the assayed arrays or sequencing experiments. Therefore, the requirement of known batch information is not very restrictive. In addition, as mentioned above, BUS explicitly models the batch effects and offers the capability of sample clustering.
BUS serves as a general framework for batch effects correction when unknown subtypes are present. It can be further tailored to adapt to the distributions from other types of high-throughput datasets such as DNA methylation microarrays, next-generation bulk sequencing data, and single-cell sequencing data. In BUS, we assume that given a batch the gene expression profile for a sample follows a mixture of multivariate normal distributions (see Equation (Equation2.3(2.3)
(2.3) )). In the same spirit, we can adopt a mixture of beta distributions (Ji et al. Citation2005) to model the DNA methylation values that are between zero and one, a mixture of multivariate Poisson distributions (Karlis and Meligkotsidou Citation2007) to fit RNA-seq count data, and a mixture of zero-inflated Poisson distributions to account for dropout events in the single-cell RNA-seq experiments. Nevertheless, the exact implementations of BUS for each of the above data types, such as computation with nonconjugate priors, are beyond the scope of this article, and they will be our future research directions. For now, to apply the current version of BUS to RNA-seq data and DNA methylation data for convenience, we can first transform the data and then apply BUS to the transformed values. We provide an example of RNA-seq data as a proof of principle in the supplementary materials Section S5.
Given the statistical power and computation efficiency of BUS, we believe that BUS will become a powerful tool. On the one hand, its ability to correct batch effects will substantially facilitate preprocessing of the enormous amount of noisy, heterogenous data in public databases and thus speed up mining of such rich data resources for valid scientific discoveries. On the other hand, its capability to identify subtypes will also help identify subgroups of patients. We provide BUS as a user-friendly free Bioconductor package BUScorrect, and we envision it will be widely adopted in the era of personalized medicine.
Supplementary Materials
The supplementary materials provide technical details and figures referred in the article as well as the datasets used in simulation studies and real application.
UASA_A_1497494_Supplement.zip
Download Zip (235.4 MB)Related Research Data
References
- Alli, E., Yang, J., and Hait, W. (2007), “Silencing of Stathmin Induces Tumor-Suppressor Function in Breast Cancer Cell Lines Harboring Mutant p53,” Oncogene 26, 1003–1012.
- Banfield, J. D., and Raftery, A. E. (1993), “Model-Based Gaussian and Non-Gaussian Clustering,” Biometrics, 49, 803–821.
- Bickel, P. J., and Levina, E. (2004), “Some Theory for Fisher’s Linear Discriminant Function, ’Naive Bayes’, and Some Alternatives When there are Many More Variables than Observations,” Bernoulli, 10, 989–1010.
- Carey, L. A., Perou, C. M., Livasy, C. A., Dressler, L. G., Cowan, D., Conway, K., Karaca, G., Troester, M. A., Tse, C. K., Edmiston, S., Deming, S. L., Geradts, J., Cheang, M. C. U., Nielsen, T. O., Moorman, P. G., Earp, H. S., and Millikan, R. C. (2006), “Race, Breast Cancer Subtypes, and Survival in the Carolina Breast Cancer Study,” Journal of the American Medical Association, 295, 2492–2502.
- Casella, G., and Berger, R. L. (2002), “Statistical Inference (Vol. 2), Pacific Grove, CA: Duxbury.
- Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., and Zoghbi, H. Y. (2008), “Mecp2, A Key Contributor to Neurological Disease, Activates and Represses Transcription,” Science, 320, 1224–1229.
- Dempster, A. P., Laird, N. M., and Rubin, D. B. (1977), “Maximum Likelihood from Incomplete Data via the EM Algorithm,” Journal of the Royal Statistical Society, Series B, 39, 1–38.
- Desmedt, C., Piette, F., Loi, S., Wang, Y., Lallemand, F., Haibe-Kains, B., Viale, G., Delorenzi, M., Zhang, Y., Saghatchian d'Assignies, M., Bergh, J., Lidereau, R., Ellis, P., Harris, A. L., Klijn, J. G. M., Foekens, J. A., Cardoso, F., Piccart, M. J., Buyse, M., and Sotiriou, C. (2007), “Strong Time Dependence of the 76-gene Prognostic Signature for Node-Negative Breast Cancer Patients in the Transbig Multicenter Independent Validation Series,” Clinical Cancer Research, 13, 3207–3214.
- Edgar, R., Domrachev, M., and Lash, A. E. (2002), “Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository,” Nucleic Acids Research, 30, 207–210.
- Fraley, C., and Raftery, A. E. (2002), “Model-Based Clustering, Discriminant Analysis, and Density Estimation,” Journal of the American Statistical Association, 97, 611–631.
- Franks, A. M., Csárdi, G., Drummond, D. A., and Airoldi, E. M. (2015), “Estimating A Structured Covariance Matrix from Multilab Measurements in High-Throughput Biology,” Journal of the American Statistical Association, 110, 27–44.
- Fujita, N., Jaye, D. L., Kajita, M., Geigerman, C., Moreno, C. S., and Wade, P. A. (2003), “Mta3, a Mi-2/NuRD Complex Subunit, Regulates An Invasive Growth Pathway in Breast Cancer,” Cell, 113, 207–219.
- Gelman, A., Carlin, J. B., Stern, H. S., and Rubin, D. B. (2014), Bayesian Data Analysis, (Vol. 2), Boca Raton, FL: Chapman & Hall/CRC.
- Geman, S., and Geman, D. (1984), “Stochastic Relaxation, Gibbs Distributions, and the Bayesian Restoration of Images,” IEEE Transactions on Pattern Analysis and Machine Intelligence, PAMI-6, 721–741.
- George, E. I., and McCulloch, R. E. (1993), “Variable Selection via Gibbs Sampling,” Journal of the American Statistical Association, 88, 881–889.
- Hein, A.-M. K., Richardson, S., Causton, H. C., Ambler, G. K., and Green, P. J. (2005), “Bgx: A Fully Bayesian Integrated Approach to the Analysis of Affymetrix GeneChip Data,” Biostatistics, 6, 349–373.
- Hicks, S. C., Teng, M., and Irizarry, R. A. (2015), “On the Widespread and Critical Impact of Systematic Bias and Batch Effects in Single-Cell RNA-seq Data,” BioRxiv, 025528.
- Hubert, L., and Arabie, P. (1985), “Comparing Partitions,” Journal of Classification, 2, 193–218.
- Huo, Z., Ding, Y., Liu, S., Oesterreich, S., and Tseng, G. (2016), “Meta-analytic Framework for Sparse k-Means to Identify Disease Subtypes in Multiple Transcriptomic Studies,” Journal of the American Statistical Association, 111, 27–42.
- Irizarry, R. A., Warren, D., Spencer, F., Kim, I. F., Biswal, S., Frank, B. C., Gabrielson, E., Garcia, J. G. N., Geoghegan, J., Germino, G., Griffin, C., Hilmer, S. C., Hoffman, E., Jedlicka, A. E., Kawasaki, E., Martínez-Murillo, F., Morsberger, L., Lee, H., Petersen, D., Quackenbush, J., Scott, A., Wilson, M., Yang, Y., Ye, S. Q., and Yu, W. (2005), “Multiple-Laboratory Comparison of Microarray Platforms,” Nature Methods, 2, 345–350.
- Jacob, L., Gagnon-Bartsch, J. A., and Speed, T. P. (2016), “Correcting Gene Expression Data When Neither the Unwanted Variation Nor the Factor of Interest are Observed,” Biostatistics, 17, 16–28.
- Ji, Y., Wu, C., Liu, P., Wang, J., and Coombes, K. R. (2005), “Applications of Beta-Mixture Models in Bioinformatics,” Bioinformatics, 21, 2118–2122.
- Johnson, W. E., Li, C., and Rabinovic, A. (2007), “Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods,” Biostatistics, 8, 118–127.
- Karlis, D., and Meligkotsidou, L. (2007), “Finite Mixtures of Multivariate Poisson Distributions with Application,” Journal of Statistical Planning and Inference, 137, 1942–1960.
- Leek, J. T. (2014), “svaseq: Removing Batch Effects and Other Unwanted Noise from Sequencing Data,” Nucleic Acids Research, gku864, e161.
- Leek, J. T., Scharpf, R. B., Bravo, H. C., Simcha, D., Langmead, B., et al. (2010), “Tackling the Widespread and Critical Impact of Batch Effects in High-Throughput Data,” Nature Reviews Genetics, 11, 733–739.
- Leek, J. T., and Storey, J. D. (2007), “Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis,” PLoS Genetics,3, e161.
- Maitra, R., and Ramler, I. P. (2009), “Clustering in the Presence of Scatter,” Biometrics, 65, 341–352.
- McCall, M. N., Bolstad, B. M., and Irizarry, R. A. (2010), “Frozen Robust Multiarray Analysis (FRMA),“ Biostatistics, 11, 242–253.
- McLachlan, G., and Peel, D. (2004), Finite Mixture Models, New York: Wiley.
- Newton, M. A., Noueiry, A., Sarkar, D., and Ahlquist, P. (2004), “Detecting Differential Gene Expression with a Semiparametric Hierarchical Mixture Method,” Biostatistics, 5, 155–176.
- Onitilo, A. A., Engel, J. M., Greenlee, R. T., and Mukesh, B. N. (2009), “Breast Cancer Subtypes based on ER/PR and Her2 Expression: Comparison of Clinicopathologic Features and Survival,” Clinical Medicine & Research, 7, 4–13.
- Pan, W., and Shen, X. (2007), “Penalized Model-Based Clustering with Application to Variable Selection,” The Journal of Machine Learning Research, 8, 1145–1164.
- Peterson, C., Stingo, F. C., and Vannucci, M. (2015), “Bayesian Inference of Multiple Gaussian Graphical Models,” Journal of the American Statistical Association, 110, 159–174.
- Piccart-Gebhart, M. J., Procter, M., Leyland-Jones, B., Goldhirsch, A., Untch, M., et al. (2005), “Trastuzumab after Adjuvant Chemotherapy in Her2-Positive Breast Cancer,” New England Journal of Medicine, 353, 1659–1672.
- Piccolo, S. R., Sun, Y., Campbell, J. D., Lenburg, M. E., Bild, A. H., and Johnson, W. E. (2012), “A Single-Sample Microarray Normalization Method to Facilitate Personalized-Medicine Workflows,” Genomics, 100, 337–344.
- Pickrell, J. K., Marioni, J. C., Pai, A. A., Degner, J. F., Engelhardt, B. E., Nkadori, E., Veyrieras, J.-B., Stephens, M., Gilad, Y., and Pritchard, J. K. (2010), “Understanding Mechanisms Underlying Human Gene Expression Variation with RNA Sequencing,” Nature, 464, 768–772.
- Ritter, G. (2014), Robust Cluster Analysis and Variable Selection, Boca Raton, FL: CRC Press.
- Robert, C., and Casella, G. (2013), Monte Carlo Statistical Methods, New York: Springer Science & Business Media.
- Schwarz, G. (1978), “Estimating the Dimension of a Model,” The Annals of Statistics, 6, 461–464.
- Slamon, D. J., Clark, G. M., Wong, S. G., Levin, W. J., Ullrich, A., and McGuire, W. L. (1987), “Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the Her-2/Neu Oncogene,” Science, 235, 177–182.
- Suárez-Fariñas, M., Shah, K. R., Haider, A. S., Krueger, J. G., and Lowes, M. A. (2010), “Personalized Medicine in Psoriasis: Developing a Genomic Classifier to Predict Histological Response to Alefacept,” BMC Dermatology, 10, 1–8.
- Taub, M. A., Corrada Bravo, H., and Irizarry, R. A. (2010), “Overcoming Bias and Systematic Errors in Next Generation Sequencing Data,” Genome Medicine, 2, 87.
- The Cancer Genome Atlas Network (2012), “Comprehensive Molecular Portraits of Human Breast Tumours,” Nature, 490, 61–70.
- Tibshirani, R. (1996), “Regression Shrinkage and Selection via the Lasso,” Journal of the Royal Statistical Society, Series B, 58, 267–288.
- Tseng, G. C., and Wong, W. H. (2005), “Tight Clustering: A Resampling-Based Approach for Identifying Stable and Tight Patterns in Data,” Biometrics, 61, 10–16.
- Wang, S., and Zhu, J. (2008), “Variable Selection for Model-Based High-Dimensional Clustering and its Application to Microarray Data,” Biometrics, 64, 440–448.
- Wang, Y., Klijn, J. G., Zhang, Y., Sieuwerts, A. M., Look, M. P., Yang, F., Talantov, D., Timmermans, M., Meijer-van Gelder, M. E., Yu, J., Jatkoe, T., Berns, Els M. J. J., Atkins, D., and Foekens, J. A. (2005), “Gene-Expression Profiles to Predict Distant Metastasis of Lymph-Node-Negative Primary Breast Cancer,” The Lancet, 365, 671–679.
- Witten, D. M., and Tibshirani, R. (2012), “A Framework for Feature Selection in Clustering,” Journal of the American Statistical Association, 105, 713–726.
- Wolf, I., Levanon-Cohen, S., Bose, S., Ligumsky, H., Sredni, B., Kanety, H., Kuro-o, M., Karlan, B., Kaufman, B., Koeffler, H. P., and Rubinek, T. (2008), “Klotho: A Tumor Suppressor and A Modulator of the igf-1 and fgf Pathways in Human Breast Cancer,” Oncogene, 27, 7094–7105.
- Xia, W., Chen, J.-S., Zhou, X., Sun, P.-R., Lee, D.-F., Liao, Y., Zhou, B. P., and Hung, M.-C. (2004), “Phosphorylation/Cytoplasmic Localization of p21cip1/waf1 is Associated with Her2/neu Overexpression and Provides a Novel Combination Predictor for Poor Prognosis in Breast Cancer Patients,” Clinical Cancer Research, 10, 3815–3824.
- Yakowitz, S. J., and Spragins, J. D. (1968), “On the Identifiability of Finite Mixtures,” The Annals of Mathematical Statistics, 39, 209–214.
- Yeung, K. Y., Fraley, C., Murua, A., Raftery, A. E., and Ruzzo, W. L. (2001), “Model-Based Clustering and Data Transformations for Gene Expression Data,” Bioinformatics, 17, 977–987.
Appendix: Proofs
A1. Theorem 1
Proof.
Writing , then the marginal distribution of
reduces to a Gaussian mixture model on batch b. Therefore, the BUS model can be viewed as a combination of B Gaussian mixture models sharing common parameters
(
). In other words, we can consider it as B Gaussian mixture models each with parameter sets
under the constraint that
for each k and
.
Within Gaussian mixture model b, it is identifiable (up to label switching) (Yakowitz and Spragins Citation1968; Ritter Citation2014), so there exists a permutation of {1, 2, …, K}, ρ(b), such that Because
and
, we have
(A.1)
(A.1) Recall that
, let k = 1 in Equation (EquationA.1
(A.1)
(A.1) ), then we have
(A.2)
(A.2) Combining Equation (EquationA.1
(A.1)
(A.1) ) and Equation (EquationA.2
(A.2)
(A.2) ) leads to
for every b, k. Specifically,
for every b and k. By Assumption I, ρ(b) = ρ(1) for each b. Now denote ρ = ρ(1) = ⋅⋅⋅ = ρ(B), we have πbk = π*bρ(k), and
for each b, k. In Equation (EquationA.1
(A.1)
(A.1) ), let (k, b) = (1, 1) and k = 1, respectively, then
Consequently, we have
. Therefore, BUS is identifiable (up to label switching).
A2. Theorem 2
Proof.
We just need to prove that ρ(b)(k) = ρ(1)(k) for each k ∈ Cb (b ⩾ 2). Note that Equation (EquationA.1(A.1)
(A.1) ) still holds for k ∈ Cb. In Equation (EquationA.1
(A.1)
(A.1) ), let b = 1 and since
, then
(A.3)
(A.3) Equation (EquationA.1
(A.1)
(A.1) ) minus Equation (EquationA.3
(A.3)
(A.3) ) gives
for every k ∈ Cb. Therefore, for any distinct k1, k2 ∈ Cb, we have
, which implies that ρ(b)(k) = ρ(1)(k) for each k ∈ Cb by Assumption I.
A3. Theorem 3
Proof.
Our goal is to prove that, given a batch b, for each , assuming k also belongs to
, the equation
always holds. We separate the proof into two steps.
We first prove ρ(b)(k) = ρ(b − 1)(k) for each . Note that
(A.4)
(A.4)
(A.5)
(A.5) Let Equation (EquationA.4
(A.4)
(A.4) ) minus (EquationA.5
(A.5)
(A.5) ) for
, and then we have
(A.6)
(A.6) For each distinct
, Equation (EquationA.6
(A.6)
(A.6) ) implies that
. By Assumption I, we proved ρ(b)(k) = ρ(b − 1)(k) provided that
.
Second, for any , there exists a
such that
. Without loss of generality, we assume
. From the two equations
we have that
(A.7)
(A.7) Meanwhile, ρ(b)(k) = ρ(b − 1)(k) for
proven in the first step turns Equation (EquationA.6
(A.6)
(A.6) ) into
for each b ⩾ 2. As a result,
. Consequently,
according to Equation (EquationA.7
(A.7)
(A.7) ). By Assumption I,
. Thus, we have proved the identifiability in Theorem 3.